
Core Proteomics and Immunoinformatic Approaches to Design a Multiepitope Reverse Vaccine Candidate against Chagas Disease
 , , , , and
, , , , and
Abstract
:1. Introduction
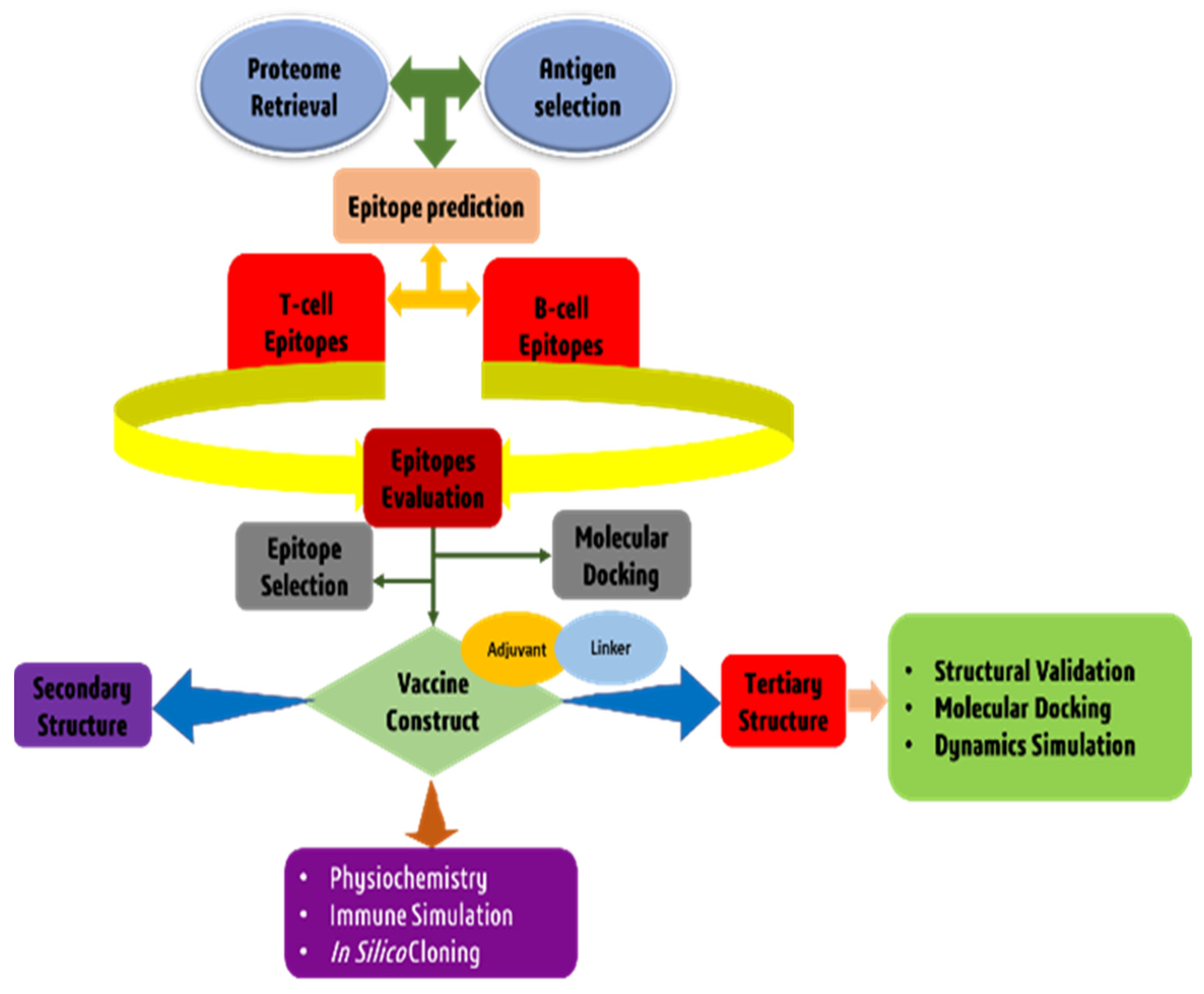
2. Materials and Methods
2.1. T. cruzi Core Proteome Identification
2.2. Subtractive Proteomics Approach
2.3. Epitopes Prediction and Assessment
2.3.1. Cytotoxic T-Cell Lymphocytes (CTLs)
2.3.2. Helper T-Lymphocytes (HTLs)
2.3.3. Linear B-Lymphocytes (LBLs)
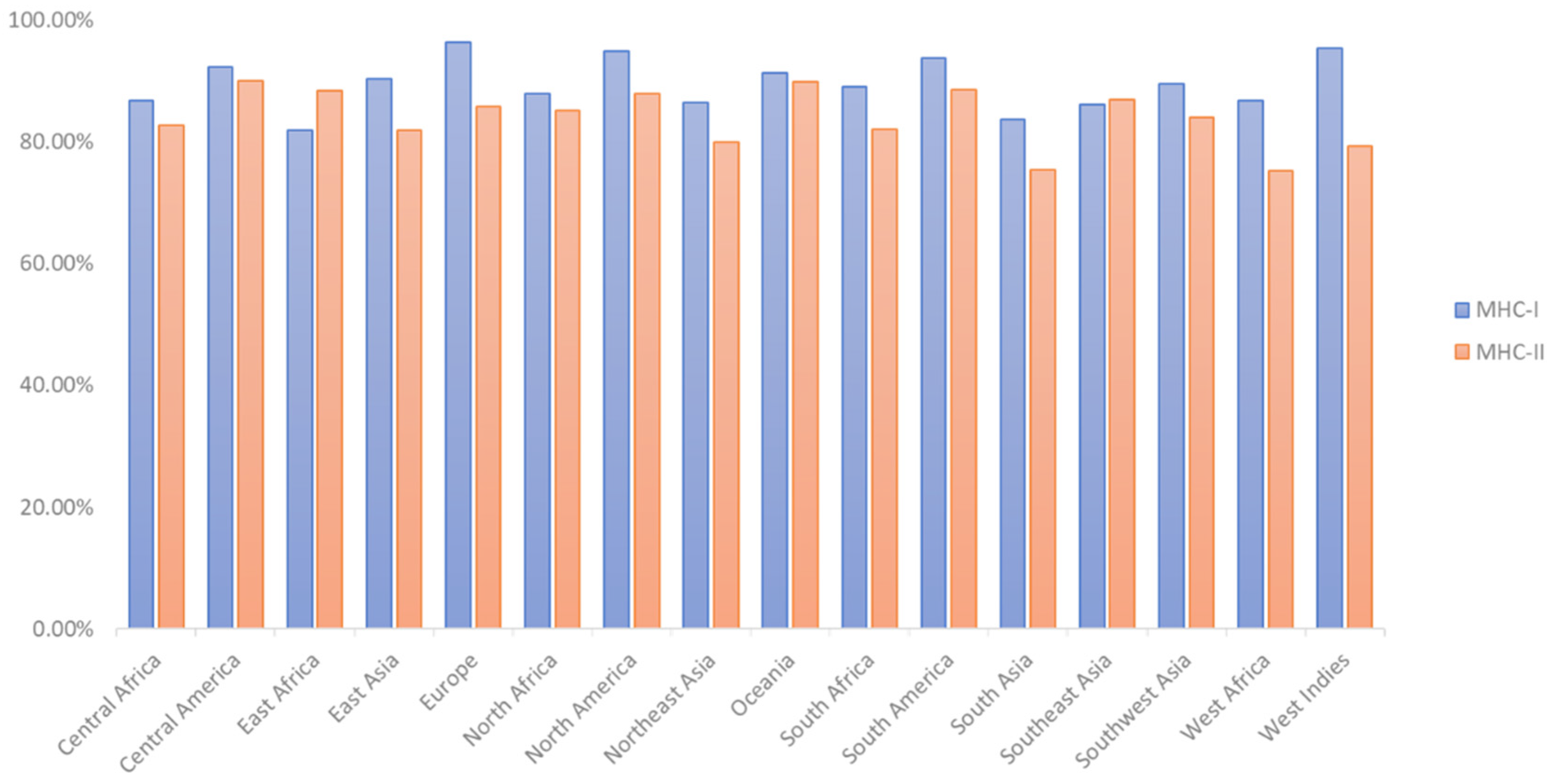
2.4. T-Cell Epitope Population Coverage and Conservation Analysis
2.5. Modeling of Peptides and Molecular Docking
2.6. Construction of a Multi-Epitope Vaccine Candidate
2.7. Structural Analysis of Multi Epitope Reverse Vaccine (MERV) Construct
2.8. 3D Structure Prediction and Confirmation
2.9. Discontinuous B Cell Epitope Prediction
2.10. Disulfide Engineering for Vaccine Candidate
2.11. Molecular Docking
2.12. MD Simulation and Analysis in Normal Mode
2.13. Modeling the Immune System
2.14. Codon Optimization and In-Silico Cloning
3. Results
3.1. Examination of the Core Proteome
3.2. Identification of Interest Proteins
3.3. Prediction of Epitopes
3.4. T-Cell Epitope Population Coverage and Conservation Analysis
3.5. Epitope and Allele Docking Studies
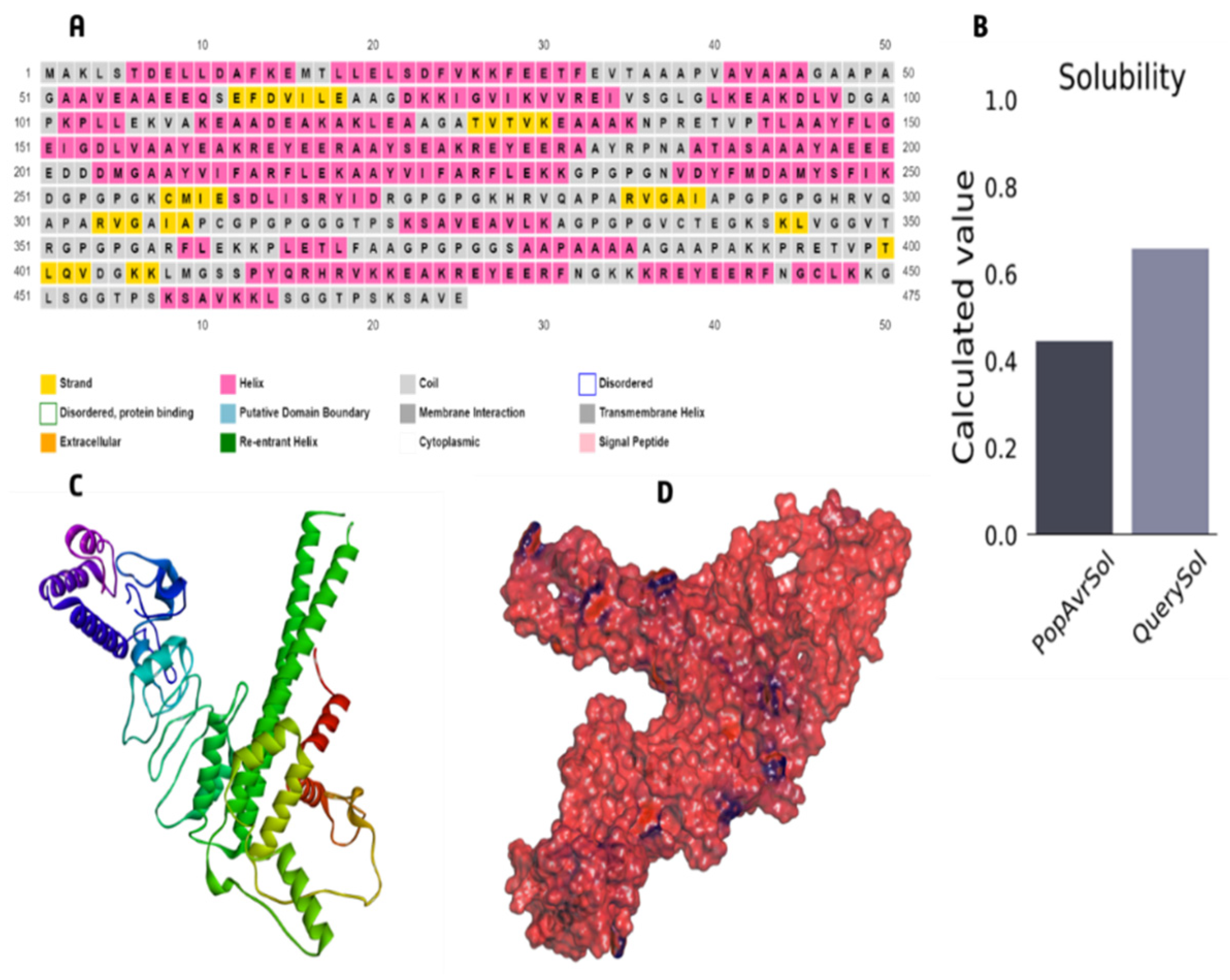
3.6. The Core Properties and Structure of the Vaccine Candidate
3.7. Immunological Evaluation and Physicochemical Properties
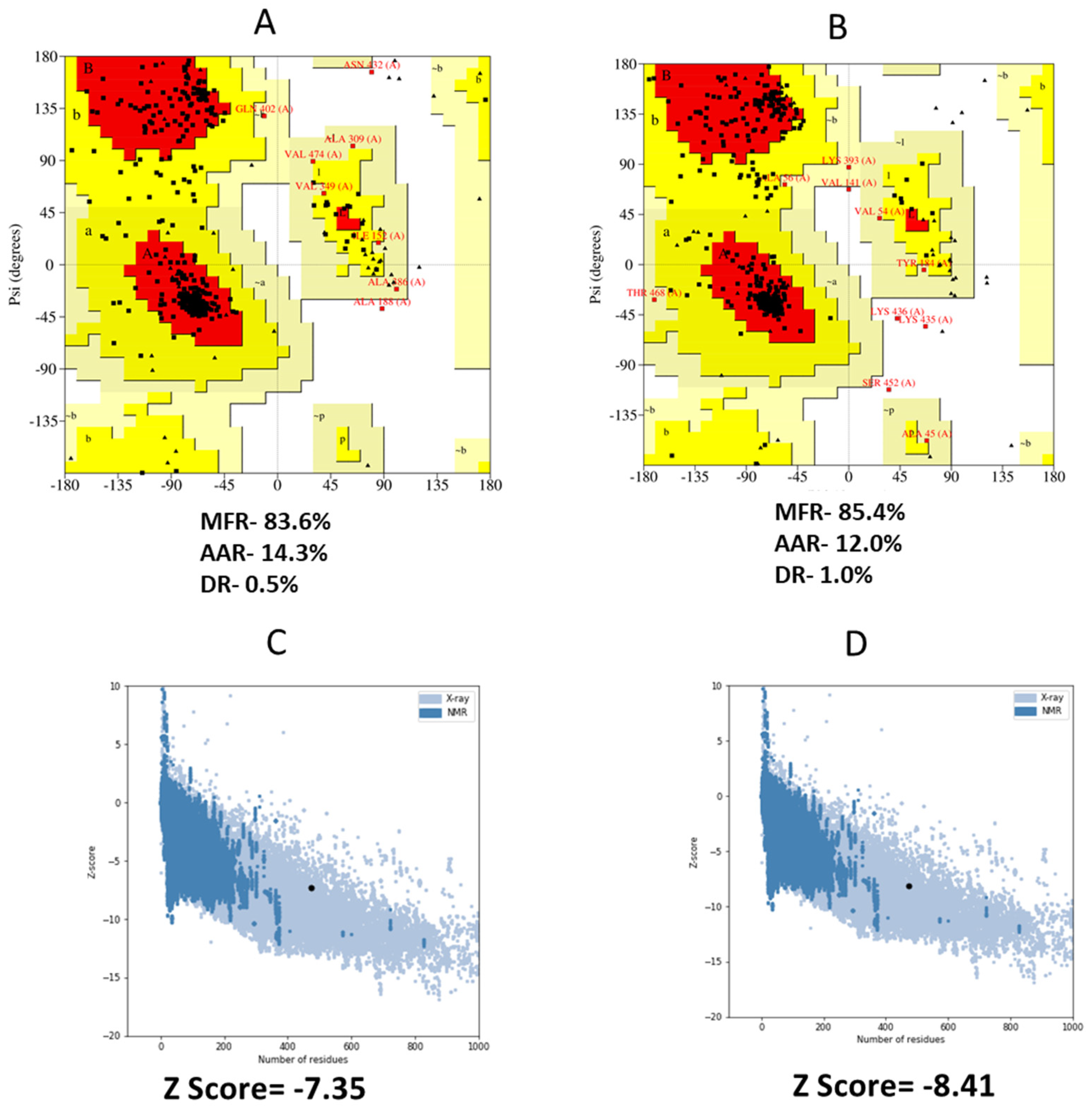
3.8. 3D Structure Refinement
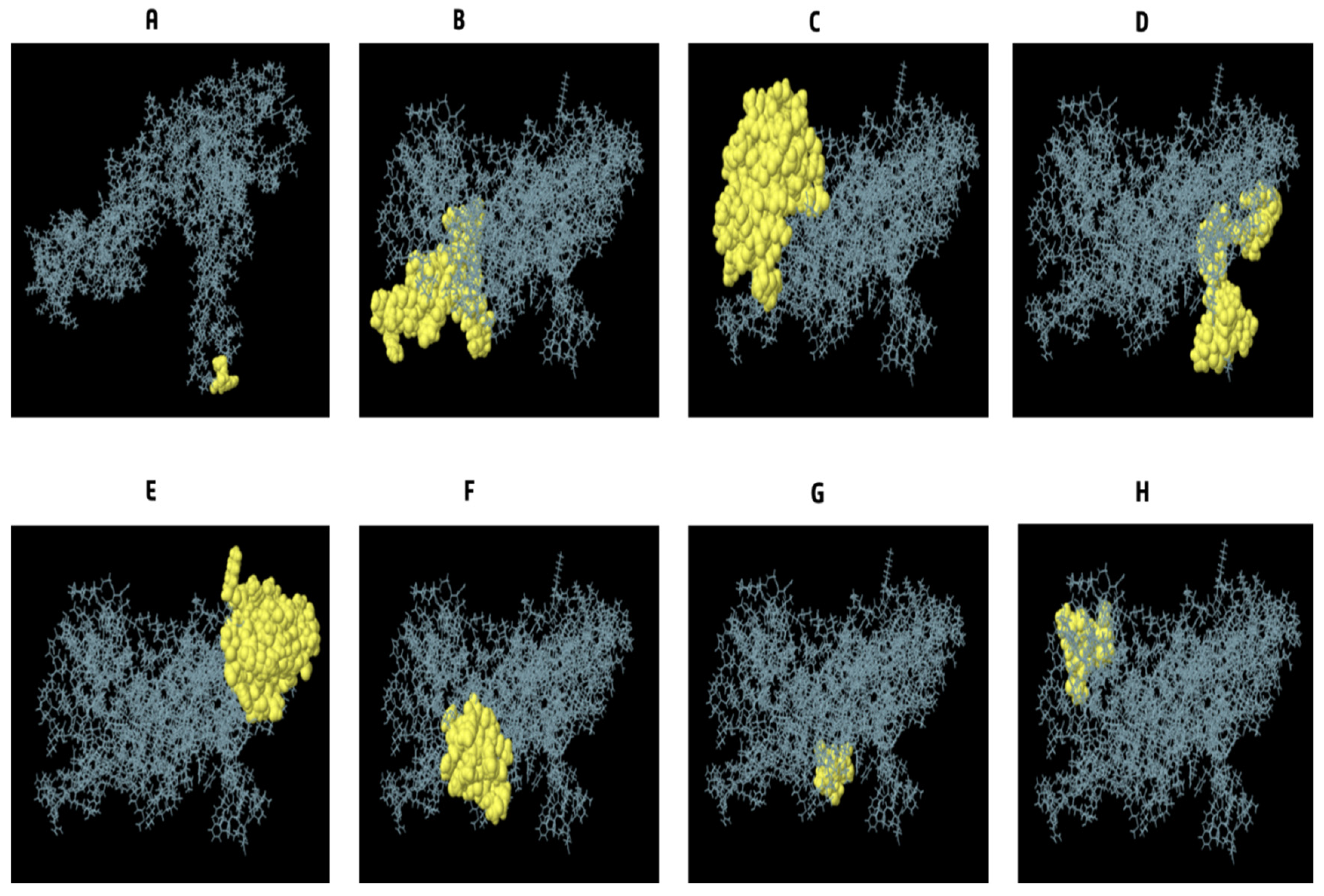
3.9. Conformational B Cell Epitopes Prediction
3.10. Disulfide Engineering for Vaccines
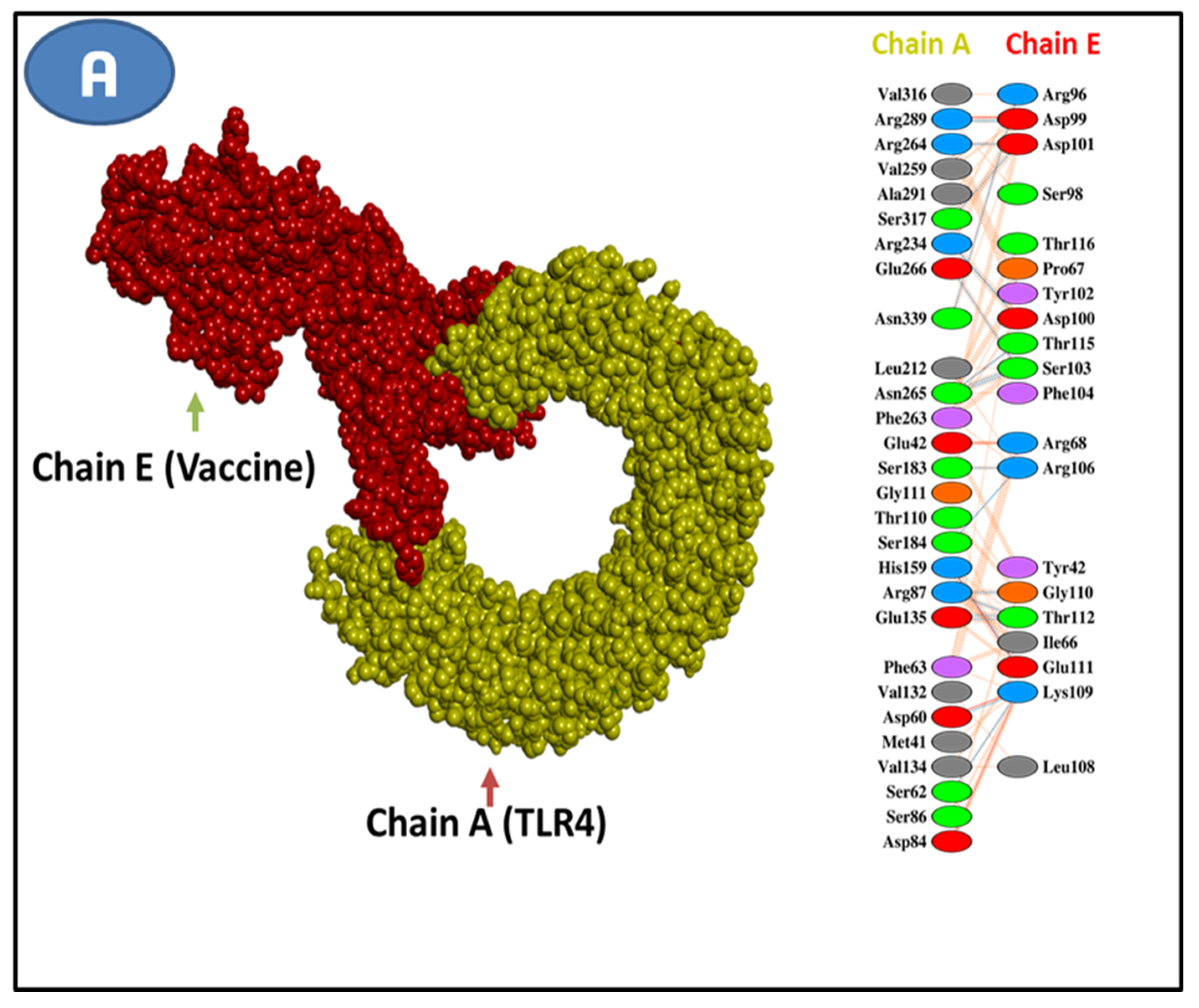
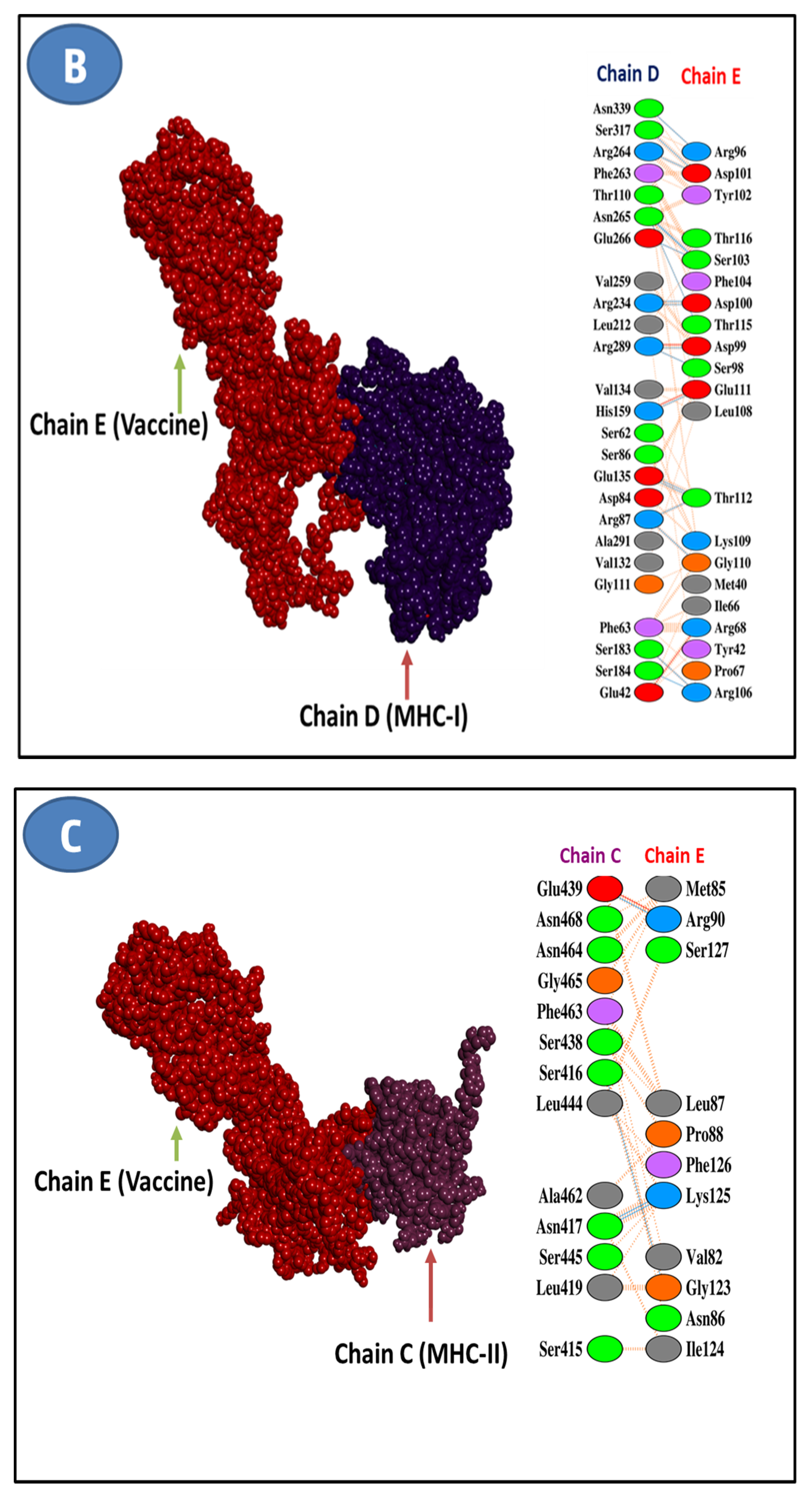
3.11. Molecular Docking Research
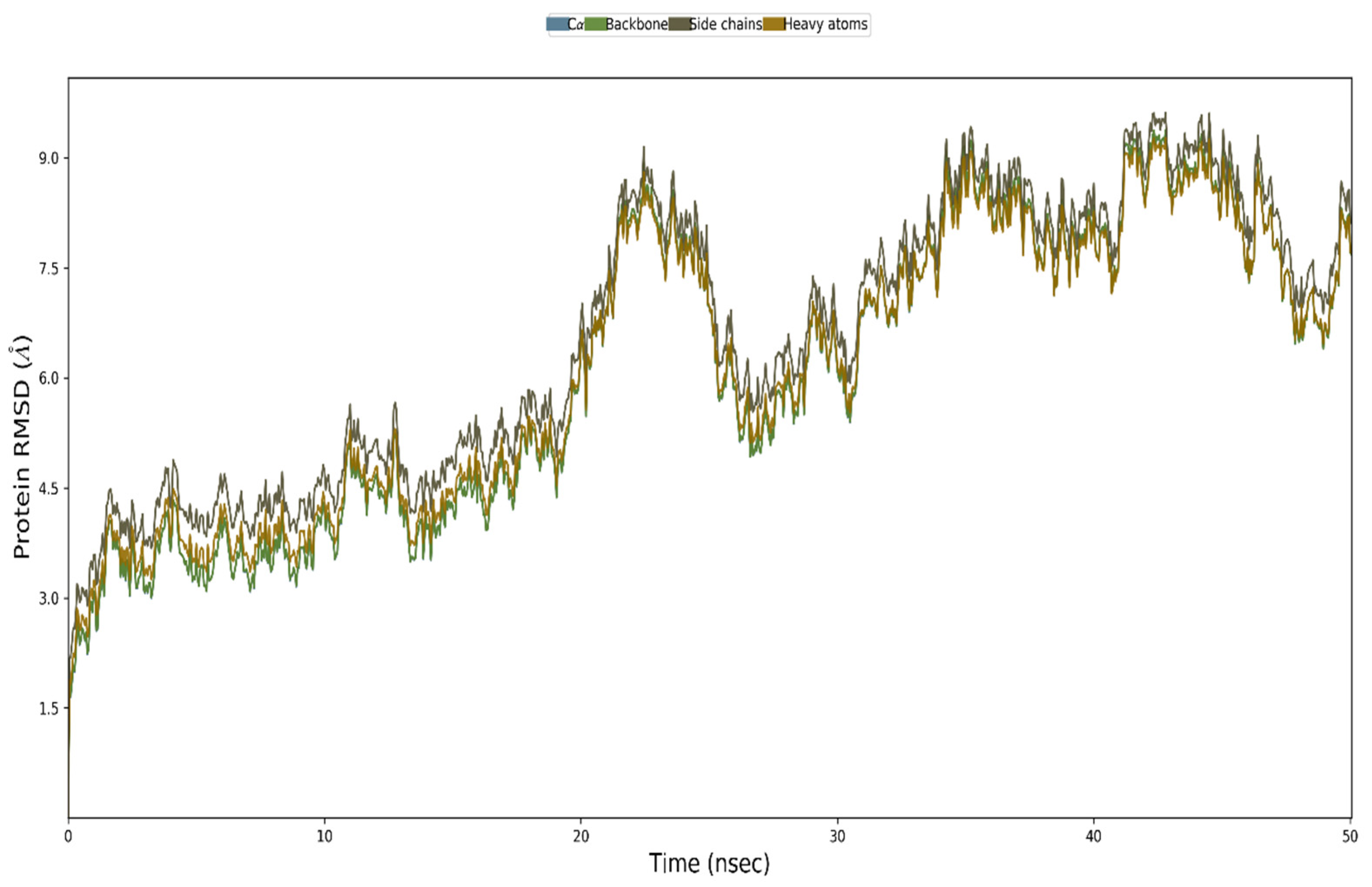
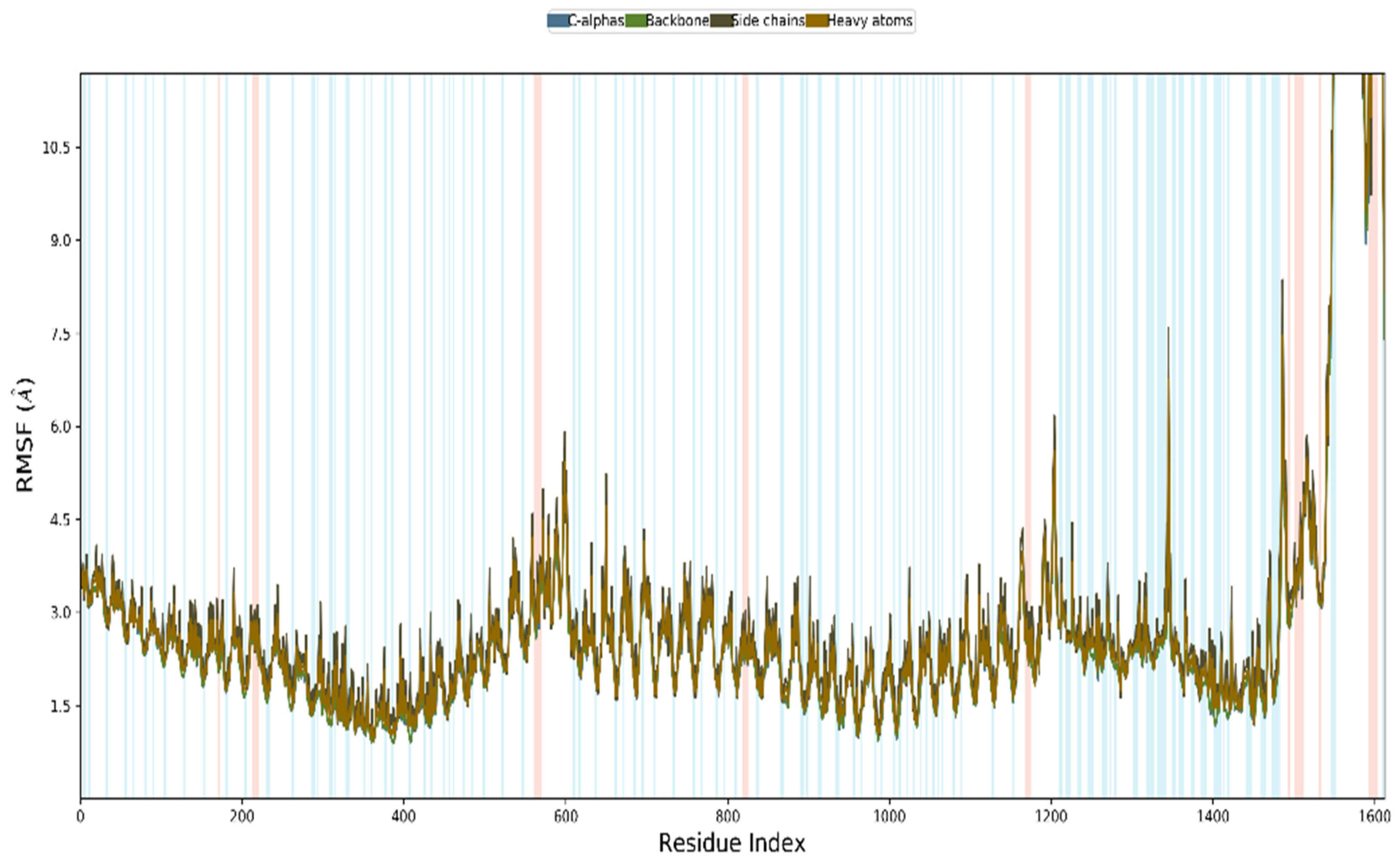
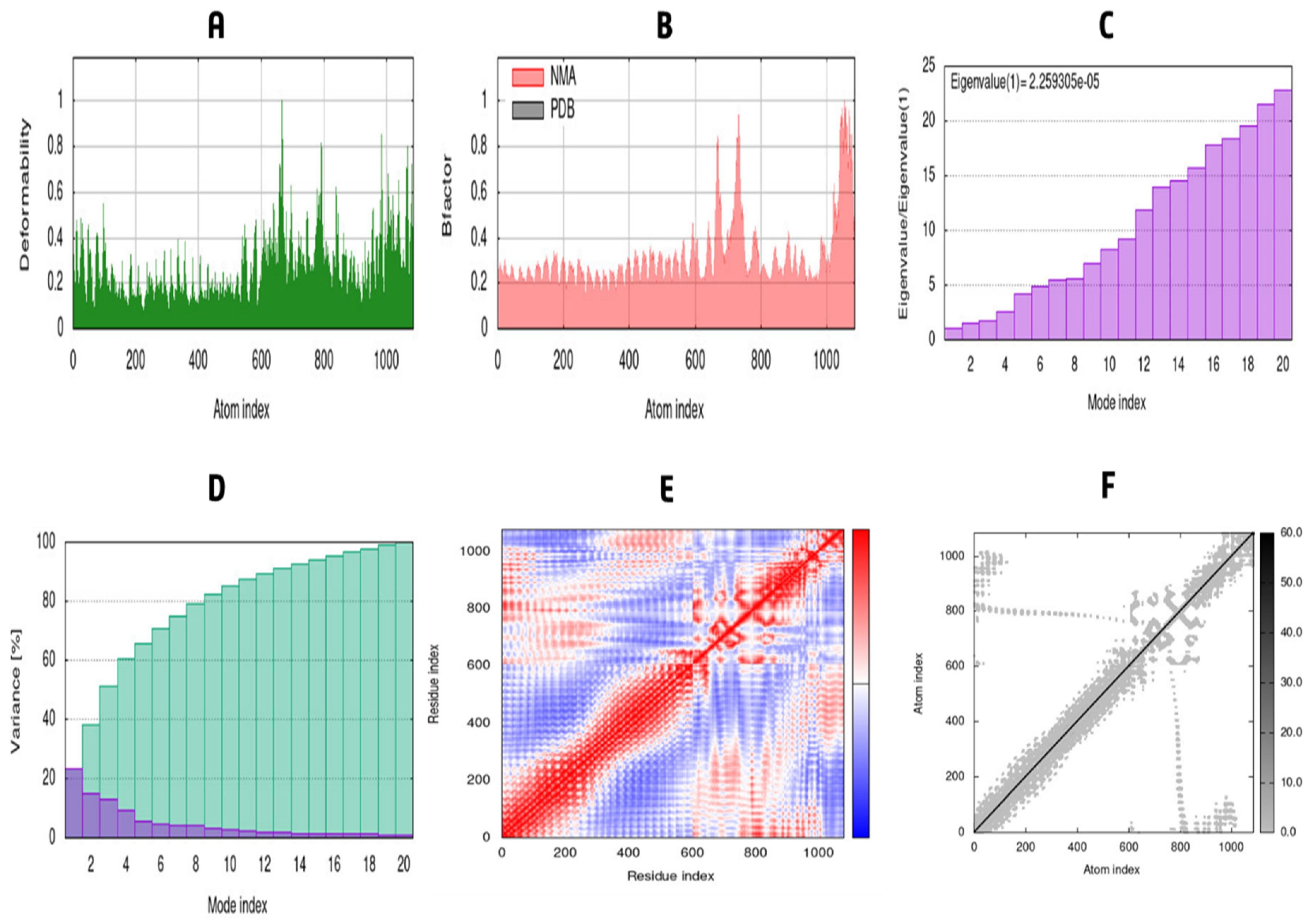
3.12. MD Simulation
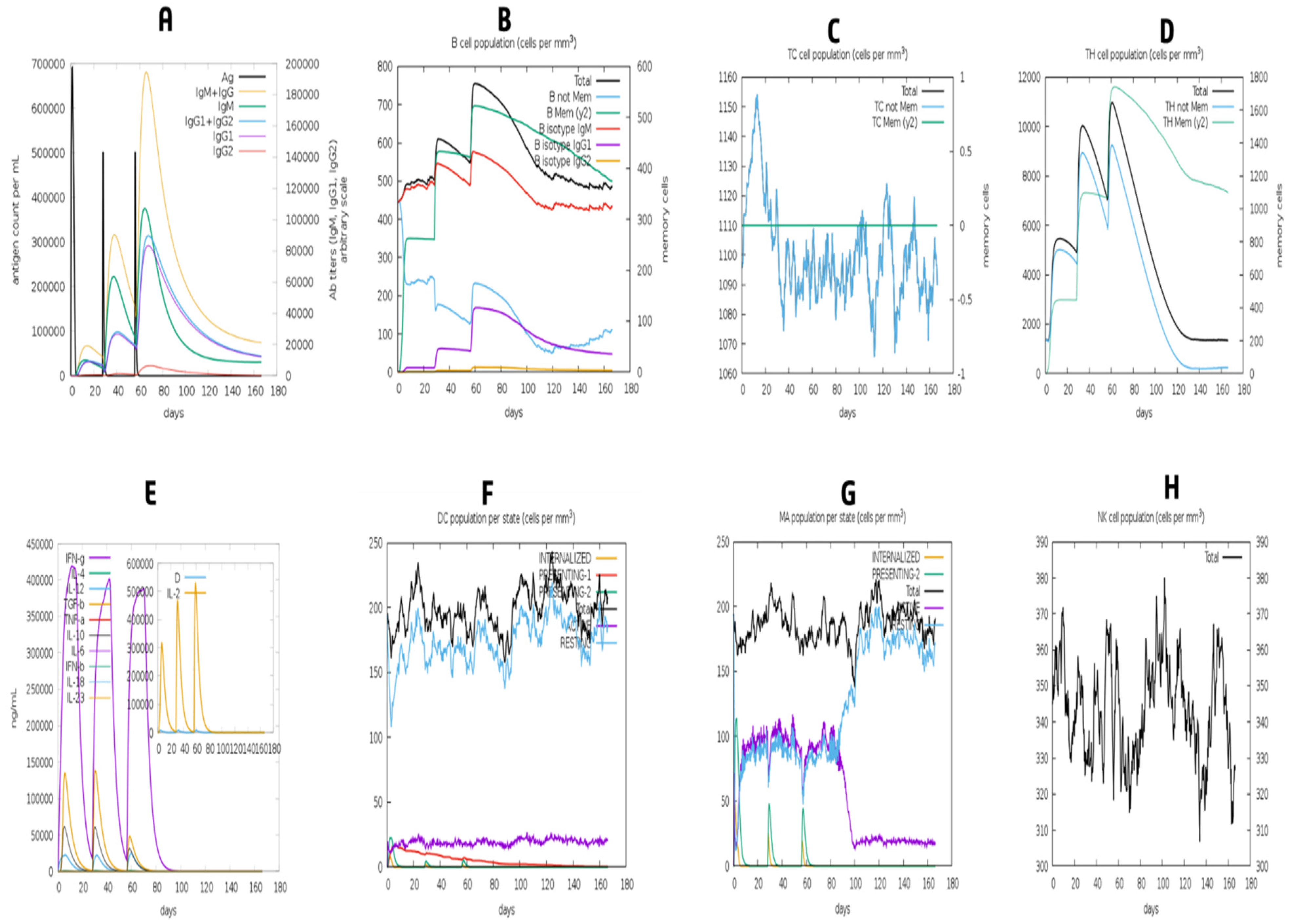
3.13. Simulation of Immune Response
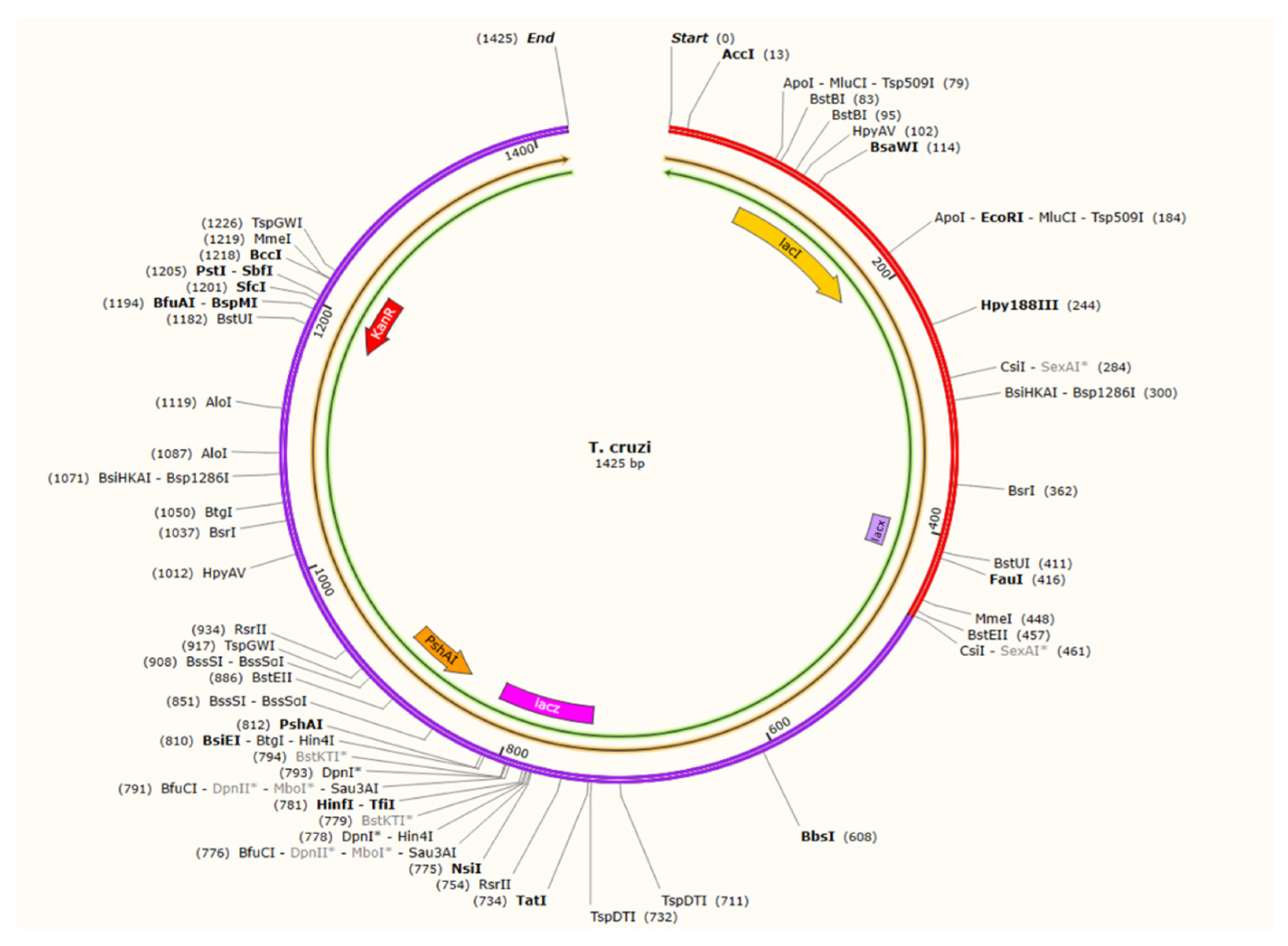
3.14. Codon Adaptation and In Silico Cloning
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bern, C.; Kjos, S.; Yabsley, M.J.; Montgomery, S.P. Trypanosoma cruzi and Chagas’ Disease in the United States. Clin. Microbiol. Rev. 2011, 24, 655–681. [Google Scholar] [CrossRef] [Green Version]
- Lidani, K.C.F.; Andrade, F.A.; Bavia, L.; Damasceno, F.S.; Beltrame, M.H.; Messias-Reason, I.J.; Sandri, T.L. Chagas Disease: From Discovery to a Worldwide Health Problem. Front. Public Health 2019, 7, 166. [Google Scholar] [CrossRef]
- Moncayo, A.; Yanine, M.I.O. An update on Chagas disease (human American trypanosomiasis). Ann. Trop. Med. Parasitol. 2006, 100, 663–677. [Google Scholar] [CrossRef]
- Gascon, J.; Bern, C.; Pinazo, M.-J. Chagas disease in Spain, the United States and other non-endemic countries. Acta Trop. 2010, 115, 22–27. [Google Scholar] [CrossRef]
- Hotez, P.J.; Molyneux, D.H.; Fenwick, A.; Kumaresan, J.; Sachs, S.E.; Sachs, J.D.; Savioli, L. Control of neglected tropical diseases. N. Engl. J. Med. 2007, 357, 1018–1027. [Google Scholar] [CrossRef] [Green Version]
- Schmunis, G.A.; Yadon, Z.E. Chagas disease: A Latin American health problem becoming a world health problem. Acta Trop. 2010, 115, 14–21. [Google Scholar] [CrossRef]
- Burleigh, B.A.; Andrews, N.W. The Mechanisms of Trypanosoma Cruzi Invasion of Mammalian Cells. Annu. Rev. Microbiol. 1995, 49, 175–200. [Google Scholar] [CrossRef]
- Miles, M.A. Trypanosoma cruzi and Chagas disease: Diversity, progress and challenges. Mem. Inst. Oswaldo Cruz. 2022, 117, e210193chgsb. [Google Scholar] [CrossRef]
- Dumonteil, E.; Herrera, C. The Case for the Development of a Chagas Disease Vaccine: Why? How? When? Trop. Med. Infect. Dis. 2021, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Marin-Neto, J.A.; Cunha-Neto, E.; Maciel, B.C.; Simões, M.V. Pathogenesis of chronic Chagas heart disease. Circulation 2007, 115, 1109–1123. [Google Scholar] [CrossRef]
- Borges, M.; da Silva, A.C.; Sereno, D.; Ouaissi, A. Peptide-based analysis of the amino acid sequence important to the immunoregulatory function of Trypanosoma cruzi Tc52 virulence factor. Immunology 2003, 109, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Garzón, E.; Borges, M.C.; Cordeiro-Da-Silva, A.; Nacife, V.; Meirelles, M.D.N.; Guilvard, E.; Bosseno, M.F.; Guevara, A.G.; Brenière, S.F.; Ouaissi, A. Trypanosoma cruzi carrying a targeted deletion of a Tc52 protein-encoding allele elicits attenuated Chagas’ disease in mice. Immunol. Lett. 2003, 89, 67–80. [Google Scholar] [CrossRef]
- Gómez, J.A.; Aguilar, C. Chagas disease: A homology model for the three-dimensional structure of the Trypanosoma cruzi ribosomal P0 antigenic protein. Eur. Biophys. J. 2014, 43, 361–366. [Google Scholar] [CrossRef]
- Wayengera, M. Searching for new clues about the molecular cause of endomyocardial fibrosis by way of in silico proteomics and analytical chemistry. PLoS ONE 2009, 4, e7420. [Google Scholar] [CrossRef] [Green Version]
- Forsyth, C.J.; Hernandez, S.; Olmedo, W.; Abuhamidah, A.; Traina, M.I.; Sanchez, D.R.; Soverow, J.; Meymandi, S.K. Safety Profile of Nifurtimox for Treatment of Chagas Disease in the United States. Clin. Infect. Dis. 2016, 63, 1056–1062. [Google Scholar] [CrossRef] [Green Version]
- Castro, J.A.; de Mecca, M.M.; Bartel, L.C. Toxic side effects of drugs used to treat Chagas’ disease (American trypanosomiasis). Hum. Exp. Toxicol. 2006, 25, 471–479. [Google Scholar] [CrossRef]
- Arce-Fonseca, M.; Carbajal-Hernández, A.C.; Lozano-Camacho, M.; Carrillo-Sánchez, S.D.C.; Roldán, F.-J.; Aranda-Fraustro, A.; Rosales-Encina, J.L.; Rodríguez-Morales, O. DNA Vaccine Treatment in Dogs Experimentally Infected with Trypanosoma cruzi. J. Immunol. Res. 2020, 2020, 9794575. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Medina, J.E.; Sánchez-Vallejo, C.J.; Méndez-Tenorio, A.; Monroy-Muñoz, I.E.; Angeles-Martínez, J.; Santos Coy-Arechavaleta, A.; Santacruz-Tinoco, S.E.; González-Ibarra, J.; Anguiano-Hernández, Y.M.; González-Bonilla, C.R.; et al. In Silico Identification of Highly Conserved Epitopes of Influenza A H1N1, H2N2, H3N2, and H5N1 with Diagnostic and Vaccination Potential. Biomed. Res. Int. 2015, 2015, 813047. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.T.; Morshed, M.M.; Hassan, F. A Computational Approach for Designing a Universal Epitope-Based Peptide Vaccine Against Nipah Virus. Interdiscip. Sci. Comput. Life Sci. 2015, 7, 177–185. [Google Scholar] [CrossRef]
- Anwar, S.; Mourosi, J.T.; Khan, F.; Hosen, M.J. Prediction of Epitope-Based Peptide Vaccine Against the Chikungunya Virus by Immuno-informatics Approach. Curr. Pharm. Biotechnol. 2020, 21, 325–340. [Google Scholar] [CrossRef]
- Dash, R.; Das, R.; Junaid; Akash, F.C.; Islam, A.; Hosen, S.Z. In silico-based vaccine design against Ebola virus glycoprotein. Adv. Appl. Bioinform. Chem. 2017, 10, 11–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Zhang, J.; Li, S.; Sun, J.; Teng, Y.; Wu, M.; Li, J.; Li, Y.; Hu, N.; Wang, H.; et al. Epitope-Based Vaccine Target Screening against Highly Pathogenic MERS-CoV: An In Silico Approach Applied to Emerging Infectious Diseases. PLoS ONE 2015, 10, e0144475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, M.; Nygren, P.-A.K.; Ståhl, S. Design and production of recombinant subunit vaccines. Biotechnol. Appl. Biochem. 2000, 32, 95–107. [Google Scholar] [CrossRef]
- Malonis, R.J.; Lai, J.R.; Vergnolle, O. Peptide-Based Vaccines: Current Progress and Future Challenges. Chem. Rev. 2020, 120, 3210–3229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Rahman, N.; Ajmal, A.; Ali, F.; Rastrelli, L. Core proteome mediated therapeutic target mining and multi-epitope vaccine design for Helicobacter pylori. Genomics 2020, 112, 3473–3483. [Google Scholar] [CrossRef]
- Sanober, G.; Ahmad, S.; Azam, S.S. Identification of plausible drug targets by investigating the druggable genome of MDR Staphylococcus epidermidis. Gene Rep. 2017, 7, 147–153. [Google Scholar] [CrossRef]
- Rédei, G. NCBI (National Center for Biotechnology Information). 2008. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 6 August 2022).
- Alzarea, S.I. Identification and construction of a multi-epitopes vaccine design against Klebsiella aerogenes: Molecular modeling study. Sci. Rep. 2022, 12, 14402. [Google Scholar] [CrossRef]
- Sancar, A.; Hack, A.M.; Rupp, W.D. Simple method for identification of plasmid-coded proteins. J. Bacteriol. 1979, 137, 692–693. [Google Scholar] [CrossRef] [Green Version]
- Shenoy, P.; Vin, H.M. Cello: A Disk Scheduling Framework for Next Generation Operating Systems. Real-Time Syst. 2002, 22, 9–48. [Google Scholar] [CrossRef]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting Transmembrane Protein Topology with a Hidden Markov Model: Application to Complete Genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meunier, M.; Guyard-Nicodème, M.; Hirchaud, E.; Parra, A.; Chemaly, M.; Dory, D. Identification of Novel Vaccine Candidates against Campylobacter through Reverse Vaccinology. J. Immunol. Res. 2016, 2016, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, V.K.; Avashthi, H.; Tiwari, A.; Jain, P.A.; Ramkete, P.W.; Kayastha, A.M.; Singh, V.K. MFPPI—Multi FASTA ProtParam Interface. Bioinformation 2016, 12, 74–77. [Google Scholar] [CrossRef]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2—A server for in silico prediction of allergens. J. Mol. Model 2014, 20, 2278. [Google Scholar] [CrossRef]
- Vita, R.; Overton, J.A.; Greenbaum, J.A.; Ponomarenko, J.; Clark, J.D.; Cantrell, J.R.; Wheeler, D.K.; Gabbard, J.L.; Hix, D.; Sette, A.; et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015, 43, D405–D412. [Google Scholar] [CrossRef] [PubMed]
- Calis, J.J.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; de Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S.; Open Source Drug Discovery Consortium. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e7395. [Google Scholar] [CrossRef] [Green Version]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP—A server for in silico prediction of allergens. BMC Bioinform. 2013, 14 (Suppl. 6), S4. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Jensen, K.K.; Andreatta, M.; Marcatili, P.; Buus, S.; Greenbaum, J.A.; Yan, Z.; Sette, A.; Peters, B.; Nielsen, M. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 2018, 154, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Dhanda, S.K.; Vir, P.; Raghava, G.P.S. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct 2013, 8, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagpal, G.; Usmani, S.S.; Dhanda, S.; Kaur, H.; Singh, S.; Sharma, M.; Raghava, G.P.S. Computer-aided designing of immunosuppressive peptides based on IL-10 inducing potential. Sci. Rep. 2017, 7, srep42851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nain, Z.; Abdullah, F.; Rahman, M.M.; Karim, M.M.; Khan, S.A.; Bin Sayed, S.; Mahmud, S.; Rahman, S.M.R.; Sheam, M.; Haque, Z.; et al. Proteome-wide screening for designing a multi-epitope vaccine against emerging pathogen Elizabethkingia anophelis using immunoinformatic approaches. J. Biomol. Struct. Dyn. 2020, 38, 4850–4867. [Google Scholar] [CrossRef]
- Manavalan, B.; Govindaraj, R.G.; Shin, T.H.; Kim, M.O.; Lee, G. iBCE-EL: A New Ensemble Learning Framework for Improved Linear B-Cell Epitope Prediction. Front. Immunol. 2018, 9, 1695. [Google Scholar] [CrossRef] [Green Version]
- Latysheva, N.S.; Babu, M.M. Discovering and understanding oncogenic gene fusions through data intensive computational approaches. Nucleic Acids Res. 2016, 44, 4487–4503. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zaro, J.L.; Shen, W.-C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Rana, A.; Akhter, Y. A multi-subunit based, thermodynamically stable model vaccine using combined immunoinformatics and protein structure based approach. Immunobiology 2016, 221, 544–557. [Google Scholar] [CrossRef]
- Dorosti, H.; Eslami, M.; Negahdaripour, M.; Ghoshoon, M.B.; Gholami, A.; Heidari, R.; Dehshahri, A.; Erfani, N.; Nezafat, N.; Ghasemi, Y. Vaccinomics approach for developing multi-epitope peptide pneumococcal vaccine. J. Biomol. Struct. Dyn. 2019, 37, 3524–3535. [Google Scholar] [CrossRef] [PubMed]
- Nain, Z.; Karim, M.M.; Sen, M.K.; Adhikari, U.K. Structural basis and designing of peptide vaccine using PE-PGRS family protein of Mycobacterium ulcerans-An integrated vaccinomics approach. Mol. Immunol. 2020, 120, 146–163. [Google Scholar] [CrossRef] [PubMed]
- Olejnik, J.; Hume, A.; Mühlberger, E. Toll-like receptor 4 in acute viral infection: Too much of a good thing. PLOS Pathog. 2018, 14, e1007390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, R.K.; Bhatt, T.K.; Prajapati, V.K. Novel Immunoinformatics Approaches to Design Multi-epitope Subunit Vaccine for Malaria by Investigating Anopheles Salivary Protein. Sci. Rep. 2018, 8, 1125. [Google Scholar] [CrossRef] [Green Version]
- Islam, S.I.; Mou, M.J.; Sanjida, S. Application of reverse vaccinology for designing of an mRNA vaccine against re-emerging marine birnavirus affecting fish species. Inform. Med. Unlocked 2022, 30, 100948. [Google Scholar] [CrossRef]
- Abdellrazeq, G.S.; Fry, L.M.; Elnaggar, M.M.; Bannantine, J.P.; Schneider, D.A.; Chamberlin, W.M.; Mahmoud, A.H.; Park, K.-T.; Hulubei, V.; Davis, W.C. Simultaneous cognate epitope recognition by bovine CD4 and CD8 T cells is essential for primary expansion of antigen-specific cytotoxic T-cells following ex vivo stimulation with a candidate Mycobacterium avium subsp. paratuberculosis peptide vaccine. Vaccine 2020, 38, 2016–2025. [Google Scholar] [CrossRef]
- Wilkins, M.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein Identification and Analysis Tools in the ExPASy Server. In The Proteomics Protocols Handbook. Springer Protocols Handbooks; Humana Press: Totowa, NJ, USA, 2008; pp. 531–552. [Google Scholar]
- Geourjon, C.; Deléage, G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 1995, 11, 681–684. [Google Scholar] [CrossRef]
- Hebditch, M.; Carballo-Amador, M.A.; Charonis, S.; Curtis, R.; Warwicker, J. Protein–Sol: A web tool for predicting protein solubility from sequence. Bioinformatics 2017, 33, 3098–3100. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; McPartlon, M.; Li, J. Improved protein structure prediction by deep learning irrespective of co-evolution information. Nat. Mach. Intell. 2021, 3, 601–609. [Google Scholar] [CrossRef]
- Nugent, T.; Cozzetto, D.; Jones, D.T. Evaluation of predictions in the CASP10 model refinement category. Proteins Struct. Funct. Bioinform. 2014, 82 (Suppl. 2), 98–111. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajighahramani, N.; Nezafat, N.; Eslami, M.; Negahdaripour, M.; Rahmatabadi, S.S.; Ghasemi, Y. Immunoinformatics analysis and in silico designing of a novel multi-epitope peptide vaccine against Staphylococcus aureus. Infect. Genet. Evol. 2017, 48, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, S.I.; Mahfuj, S.; Islam, M.J.; Mou, M.J.; Sanjida, S. Use of Integrated Core Proteomics, Immuno-Informatics, and In Silico Approaches to Design a Multiepitope Vaccine against Zoonotic Pathogen Edwardsiella tarda. Appl. Microbiol. 2022, 2, 414–437. [Google Scholar] [CrossRef]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [Green Version]
- Dombkowski, A.A.; Sultana, K.Z.; Craig, D.B. Protein disulfide engineering. FEBS Lett. 2014, 588, 206–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, S.J.; van Dijk, M.; Bonvin, A.M. The HADDOCK web server for data-driven biomolecular docking. Nat Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLano, W.L. PyMOL: An Open-Source Molecular Graphics Tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Pokhrel, S.; Bouback, T.A.; Samad, A.; Nur, S.M.; Alam, R.; Abdullah-Al-Mamun, M.; Nain, Z.; Imon, R.R.; Talukder, M.E.K.; Tareq, M.M.I.; et al. Spike protein recognizer receptor ACE2 targeted identification of potential natural antiviral drug candidates against SARS-CoV-2. Int. J. Biol. Macromol. 2021, 191, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Orti, E.S.; Chacón, P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef]
- Awan, F.M.; Obaid, A.; Ikram, A.; Janjua, H.A. Mutation-structure-function relationship based integrated strategy reveals the potential impact of deleterious missense mutations in autophagy-related proteins on hepatocellular carcinoma (HCC): A comprehensive informatics approach. Int. J. Mol. Sci. 2017, 18, 139. [Google Scholar] [CrossRef]
- Akhand, M.R.N.; Azim, K.F.; Hoque, S.F.; Moli, M.A.; Joy, B.D.; Akter, H.; Afif, I.K.; Ahmed, N.; Hasan, M. Genome-based evolutionary lineage of SARS-CoV-2 towards the development of novel chimeric vaccine. Infect. Genet. Evol. 2020, 85, 104517. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [Green Version]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Goldberg, M.F.; Roeske, E.K.; Ward, L.N.; Pengo, T.; Dileepan, T.; Kotov, D.I.; Jenkins, M.K. Salmonella Persist in Activated Macrophages in T Cell-Sparse Granulomas but Are Contained by Surrounding CXCR3 Ligand-Positioned Th1 Cells. Immunity 2018, 49, 1090–1102. [Google Scholar] [CrossRef] [Green Version]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.F.; Neuböck, R.; Hofacker, I.L. The Vienna RNA websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef] [Green Version]
- Sakharkar, K.R.; Sakharkar, M.K.; Chow, V.T. A novel genomics approach for the identification of drug targets in pathogens, with special reference to Pseudomonas aeruginosa. Silico Biol 2004, 4, 355–360. [Google Scholar]
- Azim, K.F.; Lasker, T.; Akter, R.; Hia, M.M.; Bhuiyan, O.F.; Hasan, M.; Hossain, N. Combination of highly antigenic nucleoproteins to inaugurate a cross-reactive next-generation vaccine candidate against Arenaviridae family. Heliyon 2021, 7, e07022. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Joshi, M.D.; Singhania, S.; Ramsey, K.H.; Murthy, A.K. Peptide Vaccine: Progress and Challenges. Vaccines 2014, 2, 515–536. [Google Scholar] [CrossRef] [Green Version]
- Bol, K.F.; Aarntzen, E.H.J.G.; Pots, J.M.; Nordkamp, M.A.M.O.; van de Rakt, M.W.M.M.; Scharenborg, N.M.; de Boer, A.J.; van Oorschot, T.G.M.; Croockewit, S.A.J.; Blokx, W.A.M.; et al. Prophylactic vaccines are potent activators of monocyte-derived dendritic cells and drive effective anti-tumor responses in melanoma patients at the cost of toxicity. Cancer Immunol. Immunother. 2016, 65, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Shamriz, S.; Ofoghi, H.; Moazami, N. Effect of linker length and residues on the structure and stability of a fusion protein with malaria vaccine application. Comput. Biol. Med. 2016, 76, 24–29. [Google Scholar] [CrossRef]
- Bonam, S.R.; Partidos, C.D.; Halmuthur, S.K.M.; Muller, S. An Overview of Novel Adjuvants Designed for Improving Vaccine Efficacy. Trends Pharmacol. Sci. 2017, 38, 771–793. [Google Scholar] [CrossRef] [PubMed]
- Khatoon, N.; Pandey, R.K.; Prajapati, V.K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 2017, 7, 8285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, S.; Mou, M.J.; Sanjida, S.; Tariq, M.; Nasir, S.; Mahfuj, S. Designing a novel mRNA vaccine against Vibrio harveyi infection in fish: An immunoinformatics approach. Genom. Inform. 2022, 20, e11. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
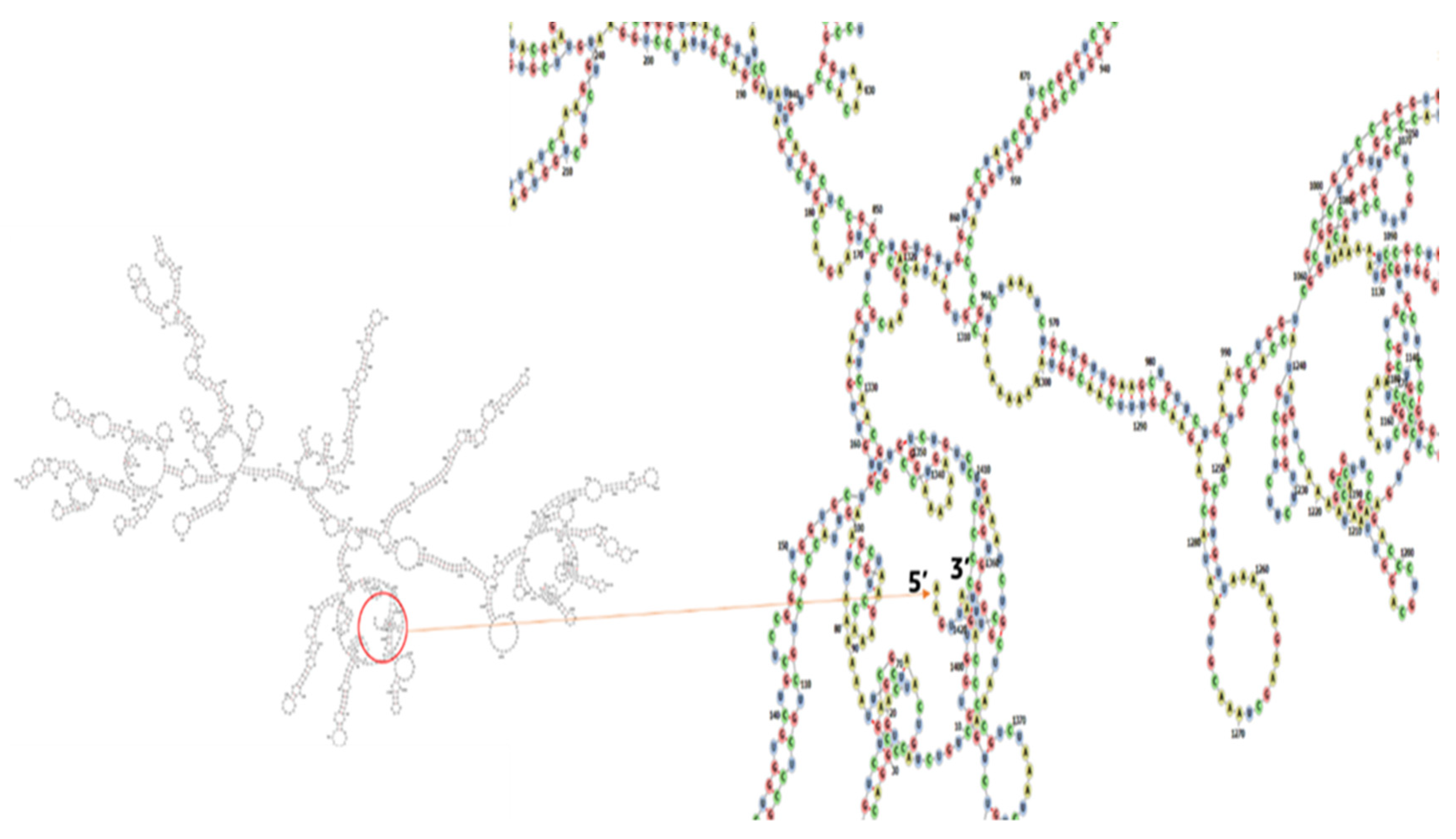{kind=link}
| Name of Protein | Accession No. | Sub-Cellular Localization | Transmembrane Helices | Antigenicity | Molecular Weight (kDa) |
|---|---|---|---|---|---|
| Thiol transferase Tc52 | AAO63160.1 | Cytoplasmic | 0 | 0.4473 | 48.12 |
| Ribosomal protein P0 | AAA30236.1 | Mitochondrial | 0 | 0.4933 | 34.95 |
| TcP2beta | CAA52941.1 | Mitochondrial | 0 | 0.6152 | 10.57 |
| Ribosomal protein P1 | AAT37631.1 | Plasma membrane | 0 | 0.6218 | 11.41 |
| Protein Name | Epitopes | Interacting HLAs Number | Immunogenicity | Allergenicity | Antigenicity | Toxicity | Conservancy (Identity ≤ 100) | Remarks |
|---|---|---|---|---|---|---|---|---|
| Thiol transferase Tc52 | NPRETVPTL | 54 | Positive | Non-allergen | 0.8667 | Non-toxic | 100.00% (1/1) | Selected |
| RVLITAKEK | 27 | Positive | Non-allergen | 0.8488 | Non-toxic | 100.00% (1/1) | ||
| ESQLIVHYL | 27 | Positive | Non-allergen | 0.8358 | Non-toxic | 100.00% (1/1) | ||
| FLGEIGDLV | 81 | Positive | Non-allergen | 1.5065 | Non-toxic | 100.00% (1/1) | Selected | |
| Ribosomal protein P0 | SLGAGIPTA | 81 | Positive | Non-allergen | 0.9863 | Non-toxic | 100.00% (1/1) | |
| EAKREYEER | 81 | Positive | Non-allergen | 1.1030 | Non-toxic | 100.00% (1/1) | Selected | |
| YGRVLFCLM | 27 | Positive | Non-allergen | 0.7849 | Non-toxic | 100.00% (1/1) | ||
| SEAKREYEER | 27 | Positive | Non-allergen | 1.0618 | Non-toxic | 100.00% (1/1) | Selected | |
| TcP2beta | RPNAATASA | 54 | Positive | Non-allergen | 1.1933 | Non-toxic | 100.00% (1/1) | Selected |
| TASAPTAAA | 27 | Positive | Non-allergen | 0.9171 | Non-toxic | 100.00% (1/1) | ||
| AEEEEDDDMG | 27 | Positive | Non-allergen | 1.1749 | Non-toxic | 100.00% (1/1) | Selected | |
| EEEDDDMGFG | 27 | Positive | Non-allergen | 0.8471 | Non-toxic | 100.00% (1/1) | ||
| Ribosomal Protein P1 | VIFARFLEK | 27 | Positive | Non-allergen | 1.3362 | Non-toxic | 100.00% (1/1) | Selected |
| LPVIFARFL | 27 | Positive | Non-allergen | 1.2247 | Non-toxic | 100.00% (1/1) | ||
| VIFARFLEKK | 54 | Positive | Non-allergen | 1.3986 | Non-toxic | 100.00% (1/1) | Selected | |
| TKEEEEDDDM | 27 | Positive | Non-allergen | 1.1196 | Non-toxic | 100.00% (1/1) |
| Protein Name | Epitopes | No. of Interacting HLAs | IL10 | IL4 | Antigenicity | IFN-γ | Conservancy (Identity ≤ 100) | Remarks |
|---|---|---|---|---|---|---|---|---|
| Thiol transferase Tc52 | NVDYFMDAMYSFIKD | 81 | Inducer | Inducer | 0.8484 | Positive | 100.00% (1/1) | Selected |
| SNVDYFMDAMYSFIK | 27 | Inducer | Inducer | 0.5137 | Positive | 100.00% (1/1) | ||
| KCMIESDLISRYIDR | 27 | Inducer | Inducer | 0.7380 | Positive | 100.00% (1/1) | Selected | |
| SYHVRFVESNVDYFM | 54 | Inducer | Inducer | 0.5797 | Positive | 100.00% (1/1) | ||
| Ribosomal protein P0 | KHRVQAPARVGAIAP | 54 | Inducer | Inducer | 1.1816 | Positive | 100.00% (1/1) | Selected |
| HRVQAPARVGAIAPC | 27 | Inducer | Inducer | 1.0619 | Positive | 100.00% (1/1) | Selected | |
| PCDVIVPAGNTGMEP | 54 | Inducer | Inducer | 0.6644 | Positive | 100.00% (1/1) | ||
| FKTLLGASVATEYEF | 27 | Inducer | Inducer | 0.4773 | Positive | 100.00% (1/1) | ||
| TcP2beta | EGKSKLVGGVTRPNA | 54 | Inducer | Inducer | 0.8056 | Positive | 98.54% (38/39) | |
| VGLSGGTPSKSAVEA | 27 | Inducer | Inducer | 1.2365 | Positive | 98.54% (38/39) | ||
| GGTPSKSAVEAVLKA | 54 | Inducer | Inducer | 0.8458 | Positive | 100.00% (1/1) | Selected | |
| VCTEGKSKLVGGVTR | 27 | Inducer | Inducer | 0.8138 | Positive | 100.00% (1/1) | Selected | |
| Ribosomal Protein P1 | EGAAAAPAAGSAAPA | 27 | Inducer | Inducer | 0.8360 | Positive | 100.00% (1/1) | |
| ARFLEKKPLETLFAA | 54 | Inducer | Inducer | 1.1755 | Positive | 100.00% (1/1) | Selected | |
| GSAAPAAAAAGAAPA | 27 | Inducer | Inducer | 0.9713 | Positive | 100.00% (1/1) | Selected | |
| TLPVIFARFLEKKPL | 27 | Inducer | Inducer | 0.7751 | Positive | 100.00% (1/1) |
| Protein Name | Sequence | Score | Antigenicity | Allergenicity | Toxicity | Remarks |
|---|---|---|---|---|---|---|
| Thiol transferase Tc52 | PRETVPTLQVDG | 0.7874 | 1.1616 | Non-allergen | Non-toxic | Selected |
| LNPRETVPTLQV | 0.7025 | 0.4951 | Non-allergen | Non-toxic | ||
| SRYIDRISSPAN | 0.7828 | 0.5385 | Non-allergen | Non-toxic | ||
| LMGSSPYQRHRV | 0.7522 | 1.0283 | Non-allergen | Non-toxic | Selected | |
| Ribosomal protein P0 | EAKREYEERFNG | 0.8234 | 1.2682 | Non-allergen | Non-toxic | Selected |
| SEAKREYEERFN | 0.8269 | 0.7628 | Non-allergen | Non-toxic | ||
| ERFNGCLTKYGR | 0.8194 | 0.7035 | Non-allergen | Non-toxic | ||
| KREYEERFNGCL | 0.7704 | 1.5208 | Non-allergen | Non-toxic | Selected | |
| TcP2beta | GLSGGTPSKSAV | 0.6102 | 1.4848 | Non-allergen | Non-toxic | Selected |
| LSGGTPSKSAVE | 0.6932 | 1.2704 | Non-allergen | Non-toxic | Selected | |
| SGGTPSKSAVEA | 0.6109 | 1.1716 | Non-allergen | Non-toxic |
| Selected T-Cell Epitopes | PDB IDs of HLAs/Receptors | Epitope Affinity (kcal/mol) | Control Affinity (kcal/mol) | Number of Hydrogens Bonds (CHB) | Residues Involved in CHB Networks |
|---|---|---|---|---|---|
| NPRETVPTL | 1a6a (HLA-DR3) | −7.3 | −6.9 | 8 (7) | Ala49, Trp7, Ile87, Gly19, Ile11, Ala29, Trp17, Tyr74 |
| FLGEIGDLV | 1h15 (HLA-DRA1*0101) | −7.1 | −7.0 | 8 (7) | Thr80, Lys91, Val156, Tyr7, Lys84, Leu66, Thr77, Asn143 |
| EAKREYEER | 2q6w (HLA- DRB3*0101) | −7.3 | −7.0 | 9 (7) | Lys26, Asn17, Asn77,Lys89, Tyr84,Tyr99,Thr343, Lys146, Trp34 |
| SEAKREYEER | 2seb (HLA-DR4) | −6.9 | −7.1 | 7 (5) | Arg171, Ala12, Asn82, Val1, Glu6, Ser4, Thr77 |
| RPNAATASA | 3c5 (HLA- (DRA*0101) | −7.3 | −7.5 | 9 (7) | Tyr17,Asp92,Asp99,Ser241, Lys66, Tyr99, Glu152, Glu152, Gln155 |
| AEEEEDDDMG | 2fse (HLA-DRB1*0101) | −6.7 | −6.3 | 7 (6) | Glu80, Trp72, Asn326, Glu7, His145, Phe37, Ile17 |
| VIFARFLEK | 1YDP (HLA-G) | −6.9 | −6.6 | 8 (5) | Lys80, Tyr84, Thr146, Val7, Lys9, Val66, Tyr77, Asn143 |
| VIFARFLEKK | 2D31 (HLA-G) | −7.7 | −7.3 | 10 (8) | Lys80, Tyr84, Thr146, Val7, Lys9, Val66, Tyr77, Asn143, Val13, Thr14 |
| NVDYFMDAMYSFIKD | 3C5J (HLA DR52c) | −7.1 | −6.3 | 9 (5) | Met69, Ile149, Thr7, Asn8, Ala19, Ile1, Gla2, Tyr7, Trp74 |
| KCMIESDLISRYIDR | 1EU3 (HLA-E) | −7.0 | −6.8 | 8 (7) | Lys87, Tyr84, Tyr99, Lys149, Thr146, Ile147, Glu152, Glu154 |
| KHRVQAPARVGAIAP | 3LQZ (HLA-DP2) | −6.8 | −6.0 | 7 (4) | Ser53, Glu89, Asn72, Ile17, His7, Glu45, Phe17 |
| HRVQAPARVGAIAPC | 4GKZ (HA1.7) | −7.0 | −6.9 | 8 (7) | Arg71, Asn12, Ala82, Val17, Ser6, Glu4, Thr77, Thr13 |
| GGTPSKSAVEAVLKA | 6J1V (HLA-A*3003/RT313) | −6.7 | −6.1 | 8 (5) | Tyr80, Lys84, Val146, Thr7, Lys9, Val66, Thr77, Asn143 |
| VCTEGKSKLVGGVTR | 6J1V (HLA-A*3003/RT313) | −6.5 | −6.8 | 7 (5) | Asn82, Glu1, Glu6, Ser4, Thr79, Ile13, Val114 |
| ARFLEKKPLETLFAA | 1KPR (HLA-E) | −7.1 | −7.0 | 7 (4) | Glu85, Trp326, Thr78, Glu45, Phe8, Ile17 |
| GSAAPAAAAAGAAPA | 6Z9V(A02 allele) | −6.9 | −6.0 | 7 (5) | Lys146, Trp147, Glu15, Glu152, Tyr84, Tyr99, Thr143, |
| Characteristics | Finding | Remark |
|---|---|---|
| Number of amino acids | 475 | Suitable |
| Molecular weight | 49,607.54 | Suitable |
| Theoretical pI | 8.53 | Base |
| Chemical formula | C2201H3525N609O671S11 | - |
| Instability index of vaccine | 27.83 | Stable |
| Aliphatic index of vaccine | 71.89 | Thermostable |
| Grand average of hydropathicity (GRAVY) | −0.352 | Hydrophilic |
| Antigenicity | 0.6635 | Antigenic |
| Immunogenicity | 1.08175 | Immunogenic |
| Allergenicity | No | Non-allergen |
| Solubility | 0.658 | Soluble |
| Characters | SOPMA | PSIPRED Server | ||
|---|---|---|---|---|
| AA | % | AA | % | |
| α helix | 241 | 50.74 | 232 | 48.84 |
| β strand | 34 | 7.16 | 34 | 7.157 |
| Random coil | 145 | 30.53 | 209 | 44 |
| No. | Residues | Number of Residues | Score |
|---|---|---|---|
| 1 | A: A182, A:A183, A:Y184 | 3 | 0.994 |
| 2 | A:M1, A:A2, A:K3, A:E164, A:Y165, A:E166, A:E167, A:R168, A:A169, A:A170, A:Y171, A:S172, A:E173, A:A174, A:K175, A:R176, A:E177, A:Y178, A:E179, A:E180, A:R181, A:R185, A:P186, A:N187, A:A188, A:A189, A:T190, A:A191, A:S192, A:A193, A:A194, A:A195, A:Y196, A:A197, A:E198, A:E199, A:E200, A:E201, A:D203, A:D204 | 40 | 0.833 |
| 3 | A:K393, A:P394, A:R395, A:E422, A:A423, A:K424, A:R425, A:E426, A:Y427, A:E428, A:E429, A:R430, A:F431, A:N432, A:G433, A:K434, A:K435, A:K436, A:R437, A:E438, A:Y439, A:E440, A:E441, A:R442, A:F443, A:N444, A:G445, A:C446, A:L447, A:K448, A:K449, A:G450, A:L451, A:S452, A:G453, A:G454, A:T455, A:P456, A:S457, A:K458, A:S459, A:A460, A:V461, A:K462, A:K463, A:L464, A:S465, A:G466, A:G467, A:T468, A:P469, A:S470, A:K471, A:S472, A:A473, A:V474, A:E475 | 57 | 0.775 |
| 4 | A:V25, A:K26, A:F28, A:E29, A:E30, A:T31, A:F32, A:V34, A:T35, A:A36, A:A37, A:A38, A:P39, A:V40, A:A41, A:V42, A:A43, A:A44, A:A45, A:G46, A:A47, A:A48, A:P49, A:A50, A:G51, A:A52, A:A53, A:V54, A:E55, A:A56, A:A57, A:E58, A:E59, A:Q60, A:S61, A:E62, A:F63 | 37 | 0.701 |
| 5 | A:D64, A:V65, A:I66, A:L67, A:E68, A:A69, A:A70, A:G71, A:D72, A:K73, A:K74, A:I75, A:K102, A:P103, A:L104, A:L105, A:E106, A:K107, A:V108, A:A109, A:K110, A:E111, A:A112, A:A113, A:D114, A:E115, A:A116, A:K117, A:A118, A:K119, A:L120, A:E121, A:A122, A:A123, A:G124, A:A125, A:T126, A:V127, A:T128, A:V129, A:K130, A:E131, A:A132, A:A133, A:A134, A:K135, A:N136, A:P137, A:R138, A:E139, A:T140, A:G232, A:P233, A:G234, A:P235, A:G236, A:R271, A:G272, A:P273, A:G274, A:P275, A:G276, A:K277, A:H278, A:R279, A:P295, A:G296 | 67 | 0.691 |
| 6 | A:K344, A:V398, A:T400, A:L401, A:Q402, A:V403, A:D404, A:G405, A:K406, A:K407, A:L408, A:M409, A:G410, A:S411, A:S412, A:P413, A:Y414, A:Q415, A:R416, A:H417, A:R418 | 21 | 0.624 |
| 7 | A:L4, A:S5, A:T6, A:D7, A:E8, A:L10 | 6 | 0.622 |
| 8 | A:A371, A:G372, A:P373, A:G374, A:P375, A:G376, A:G377, A:S378, A:A379, A:A380, A:P381, A:A382, A:A383, A:K392 | 14 | 0.53 |
| Features | MERV-MHCI | MERV-MHCII | MERV-TLR4 |
|---|---|---|---|
| HADDOCK Score | 217.3 ± 14.2 | 179.4 ±27.3 | 213.6 ± 14.6 |
| Cluster Size | 5 | 3 | 7 |
| Van der Waals energy | −40.8 ± 3.9 | −71.1 ± 2.25 | −41.7 ± 1.3 |
| Desolvation energy | −1.41 ± 0.7 | −10.7 ± 4.81 | −0.57 ± 3.35 |
| Electrostatic energy | −61.8 ± 9.5 | −267.1 ± 24.8 | −65.1 ± 23.8 |
| RMSD from the overall lowest energy structure | 35.6 ± 0.3 | 10.3 ± 0.5 | 50.4 ± 0.1 |
| Buried surface area | 2211.9 ± 100.2 | 2871.6 ± 60.1 | 2171.9 ± 121.9 |
| Z-Score | −1.2 | −0.9 | −1.7 |
| Restraint violation energy | 2246.2 ± 162.9 | 3724.6 ± 152.4 | 2920.9 ± 179.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, S.I.; Sanjida, S.; Ahmed, S.S.; Almehmadi, M.; Allahyani, M.; Aljuaid, A.; Alsaiari, A.A.; Halawi, M. Core Proteomics and Immunoinformatic Approaches to Design a Multiepitope Reverse Vaccine Candidate against Chagas Disease. Vaccines 2022, 10, 1669. https://doi.org/10.3390/vaccines10101669
Islam SI, Sanjida S, Ahmed SS, Almehmadi M, Allahyani M, Aljuaid A, Alsaiari AA, Halawi M. Core Proteomics and Immunoinformatic Approaches to Design a Multiepitope Reverse Vaccine Candidate against Chagas Disease. Vaccines. 2022; 10(10):1669. https://doi.org/10.3390/vaccines10101669
Chicago/Turabian StyleIslam, Sk Injamamul, Saloa Sanjida, Sheikh Sunzid Ahmed, Mazen Almehmadi, Mamdouh Allahyani, Abdulelah Aljuaid, Ahad Amer Alsaiari, and Mustafa Halawi. 2022. "Core Proteomics and Immunoinformatic Approaches to Design a Multiepitope Reverse Vaccine Candidate against Chagas Disease" Vaccines 10, no. 10: 1669. https://doi.org/10.3390/vaccines10101669