Assessing the Effects of Cytoprotectants on Selective Neuronal Loss, Sensorimotor Deficit and Microglial Activation after Temporary Middle Cerebral Occlusion

Abstract
:1. Introduction
2. Material and Methods
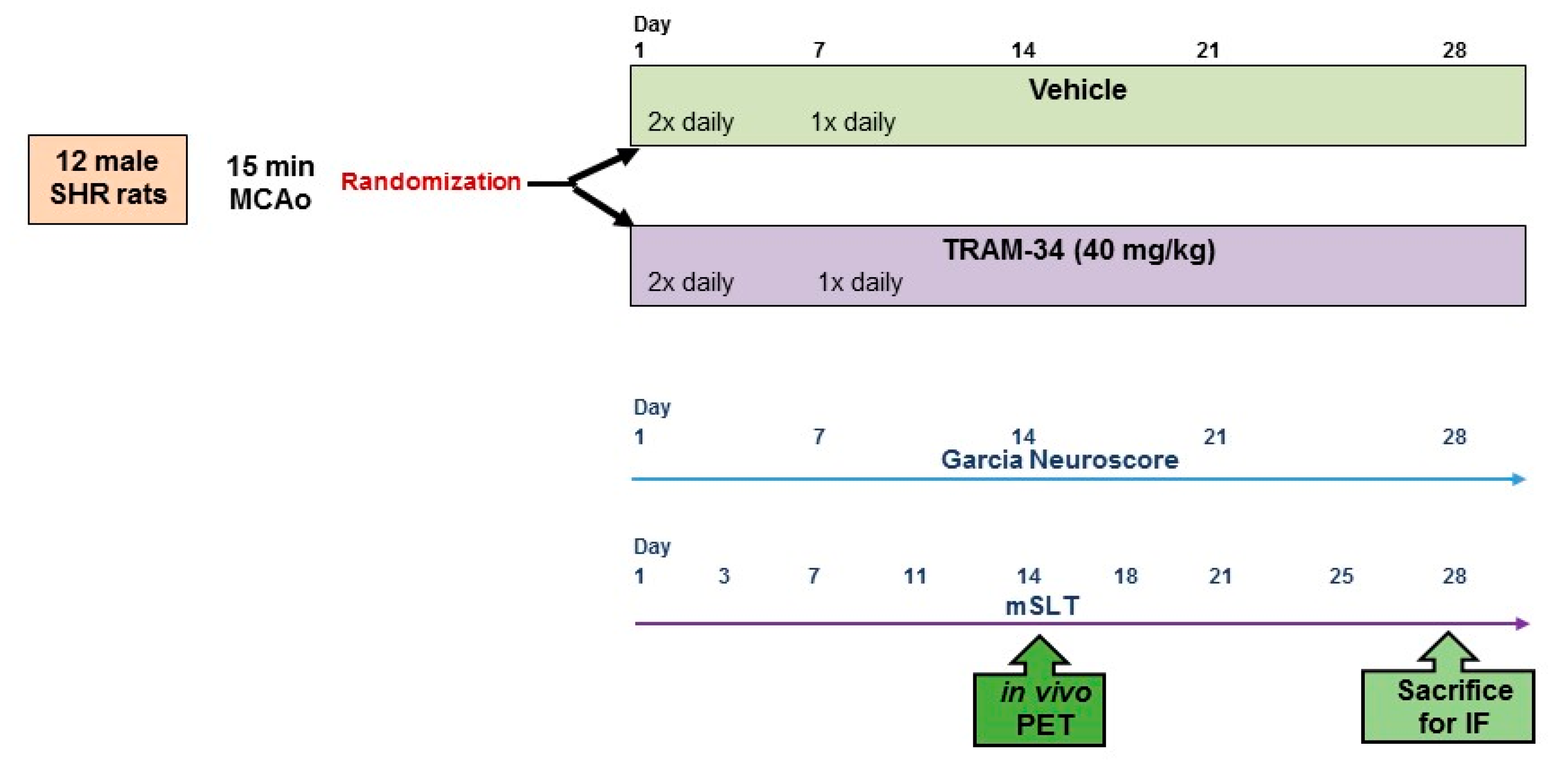
2.1. Overall Study Design
2.2. Animals
2.3. Anaesthesia
2.4. Middle Cerebral Artery Occlusion (MCAo)
2.5. Treatment Groups
2.6. Behavioural Testing
2.7. [11C]-PK11195 PET
2.7.1. PET Scanning Procedure
2.7.2. MRI Acquisition
2.7.3. PET Data Post-Processing
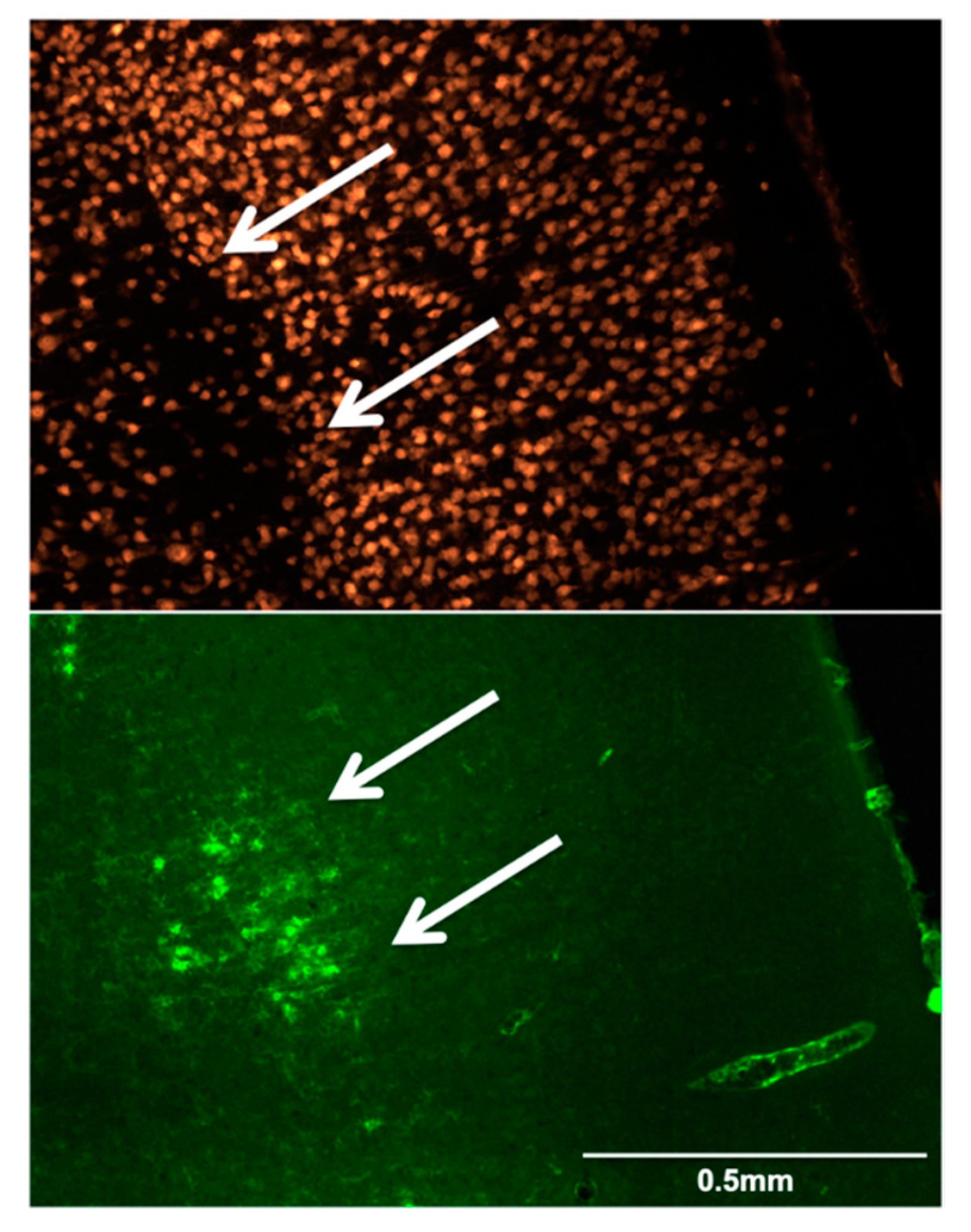
2.8. Immunofluorescence (IF)
2.9. Histopathological Evaluation of Ischemic Damage
2.10. PET Data Analysis
2.11. Statistical Analysis
3. Results
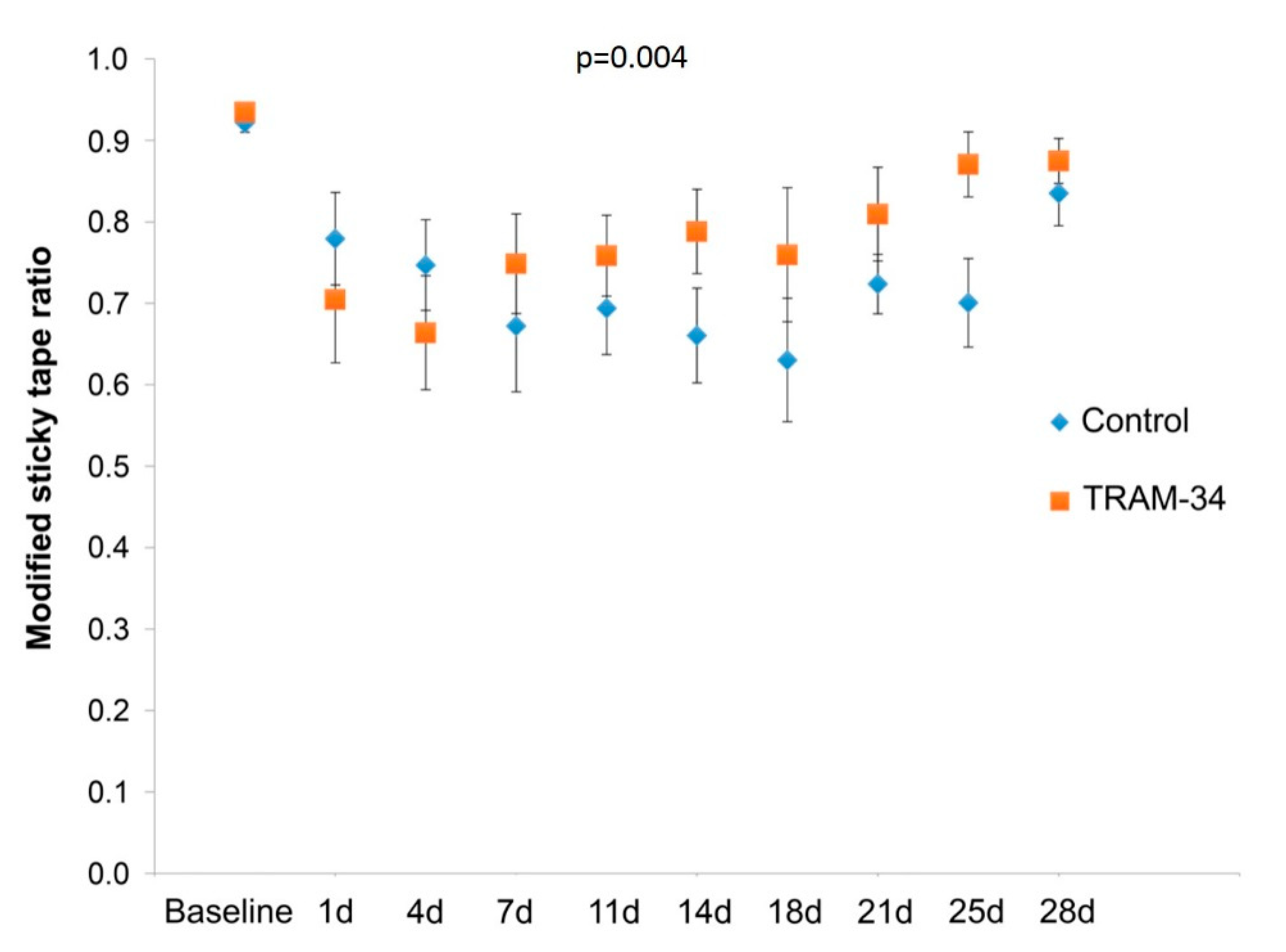
3.1. Behaviour
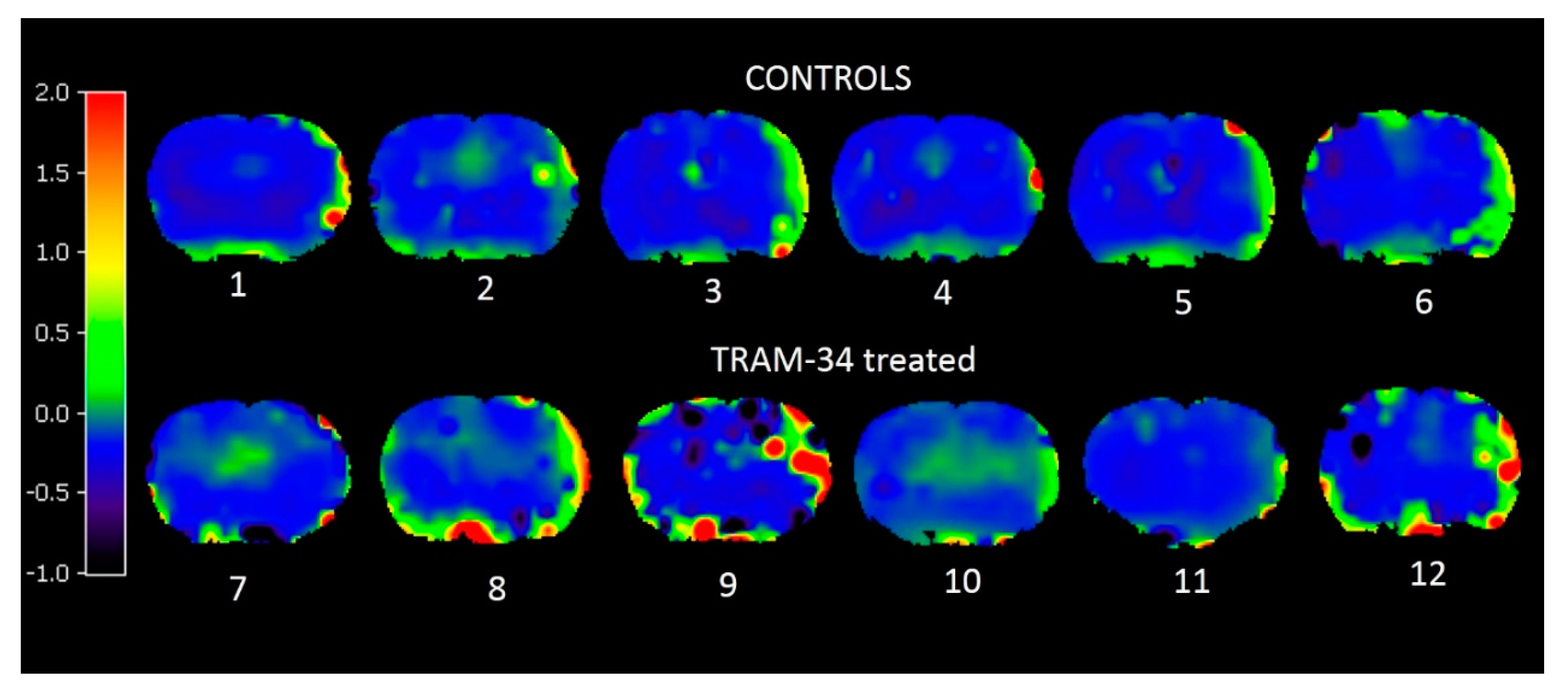
3.2. PET Results
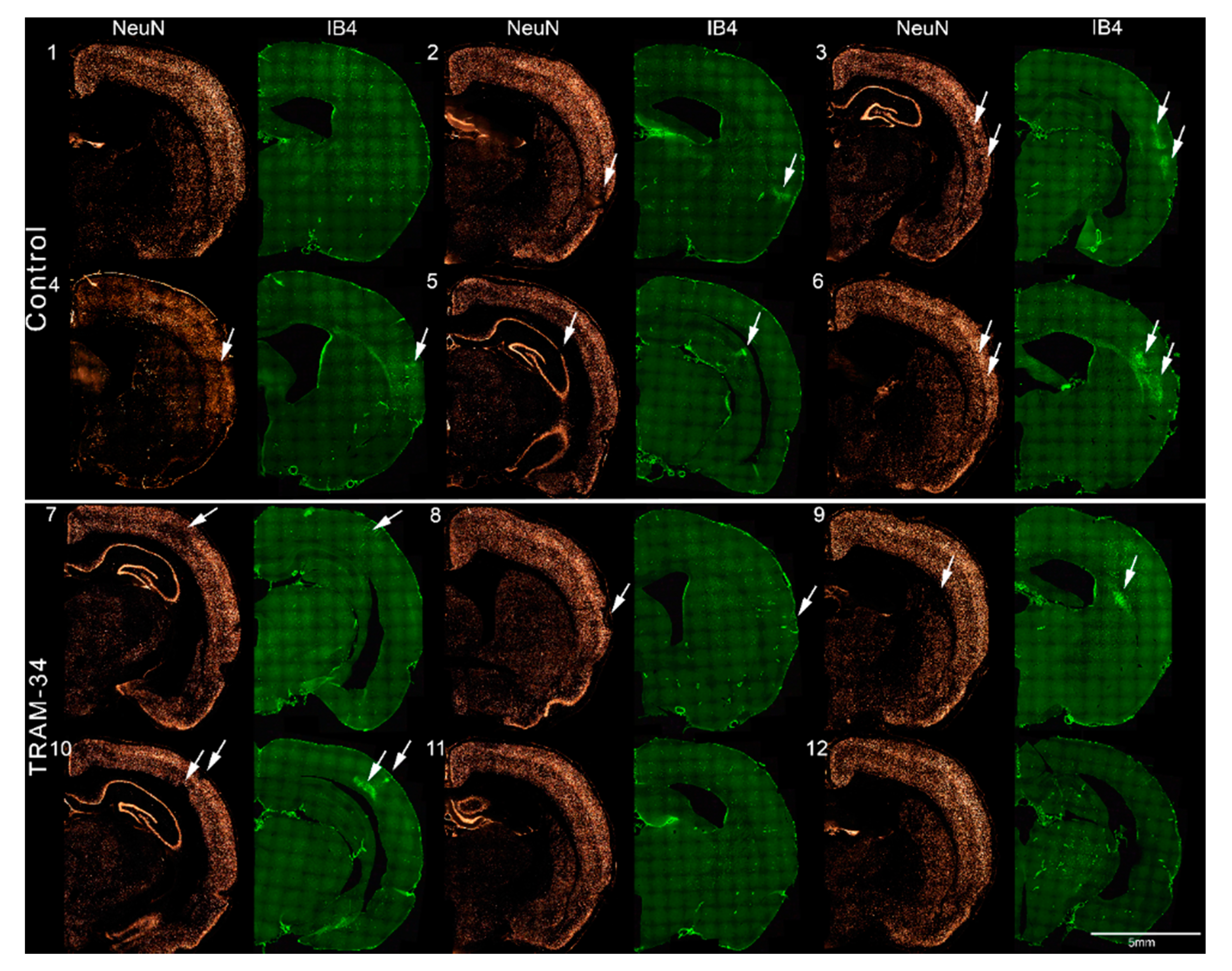
3.3. Immunofluorescence Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Powers, W.J.; Derdeyn, C.P.; Biller, J.; Coffey, C.S.; Hoh, B.L.; Jauch, E.C.; Johnston, K.C.; Johnston, S.C.; Khalessi, A.A.; Kidwell, C.S.; et al. American Heart Association Stroke C: 2015 american heart association/american stroke association focused update of the 2013 guidelines for the early management of patients with acute ischemic stroke regarding endovascular treatment: A guideline for healthcare professionals from the american heart association/american stroke association. Stroke 2015, 46, 3020–3035. [Google Scholar] [PubMed]
- Goyal, M.; Hill, M.D.; Saver, J.L.; Fisher, M. Challenges and opportunities of endovascular stroke therapy. Ann. Neurol. 2016, 79, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.; Saver, J.L. Future directions of acute ischaemic stroke therapy. Lancet Neurol. 2015, 14, 758–767. [Google Scholar] [CrossRef]
- Savitz, S.I.; Baron, J.C.; Fisher, M.; Consortium, S.X. Stroke treatment academic industry roundtable x: Brain cytoprotection therapies in the reperfusion era. Stroke 2019, 50, 1026–1031. [Google Scholar] [CrossRef]
- Baron, J.C. How healthy is the acutely reperfused ischemic penumbra? Cerebrovasc. Dis. 2005, 20 (Suppl. 2), 25–31. [Google Scholar] [CrossRef]
- Bai, J.; Lyden, P.D. Revisiting cerebral postischemic reperfusion injury: New insights in understanding reperfusion failure, hemorrhage, and edema. Int. J. Stroke 2015, 10, 143–152. [Google Scholar] [CrossRef]
- Baron, J.C.; Yamauchi, H.; Fujioka, M.; Endres, M. Selective neuronal loss in ischemic stroke and cerebrovascular disease. J. Cereb. Blood Flow Metab. 2014, 34, 2–18. [Google Scholar] [CrossRef]
- Baron, J.C. Mapping neuronal density in peri-infarct cortex with pet. Hum. Brain Mapp. 2017, 38, 5822–5824. [Google Scholar] [CrossRef]
- Ejaz, S.; Emmrich, J.V.; Sawiak, S.J.; Williamson, D.J.; Baron, J.C. Cortical selective neuronal loss, impaired behavior, and normal magnetic resonance imaging in a new rat model of true transient ischemic attacks. Stroke 2015, 46, 1084–1092. [Google Scholar] [CrossRef]
- Sicard, K.M.; Henninger, N.; Fisher, M.; Duong, T.Q.; Ferris, C.F. Long-term changes of functional mri-based brain function, behavioral status, and histopathology after transient focal cerebral ischemia in rats. Stroke 2006, 37, 2593–2600. [Google Scholar] [CrossRef]
- Carrera, E.; Jones, P.S.; Morris, R.S.; Alawneh, J.; Hong, Y.T.; Aigbirhio, F.I.; Fryer, T.D.; Carpenter, T.A.; Warburton, E.A.; Baron, J.C. Is neural activation within the rescued penumbra impeded by selective neuronal loss? Brain 2013, 136, 1816–1829. [Google Scholar] [CrossRef] [PubMed]
- Guadagno, J.V.; Jones, P.S.; Aigbirhio, F.I.; Wang, D.; Fryer, T.D.; Day, D.J.; Antoun, N.; Nimmo-Smith, I.; Warburton, E.A.; Baron, J.C. Selective neuronal loss in rescued penumbra relates to initial hypoperfusion. Brain 2008, 131, 2666–2678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawara, J.; Sperling, B.; Lassen, N.A. Incomplete brain infarction of reperfused cortex may be quantitated with iomazenil. Stroke 1997, 28, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Saur, D.; Buchert, R.; Knab, R.; Weiller, C.; Rother, J. Iomazenil-single-photon emission computed tomography reveals selective neuronal loss in magnetic resonance-defined mismatch areas. Stroke 2006, 37, 2713–2719. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, S.R.; Choi, S.S.; Yeo, H.G.; Chang, K.T.; Lee, H.J. Therapeutically targeting neuroinflammation and microglia after acute ischemic stroke. Biomed Res. Int. 2014, 2014, 297241. [Google Scholar] [CrossRef]
- Kriz, J.; Lalancette-Hebert, M. Inflammation, plasticity and real-time imaging after cerebral ischemia. Acta Neuropathol. 2009, 117, 497–509. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization-new prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef]
- Ejaz, S.; Williamson, D.J.; Ahmed, T.; Sitnikov, S.; Hong, Y.T.; Sawiak, S.J.; Fryer, T.D.; Aigbirhio, F.I.; Baron, J.C. Characterizing infarction and selective neuronal loss following temporary focal cerebral ischemia in the rat: A multi-modality imaging study. Neurobiol. Dis. 2013, 51, 120–132. [Google Scholar] [CrossRef]
- Ejaz, S.; Williamson, D.J.; Jensen-Kondering, U.; Ahmed, T.; Sawiak, S.J.; Baron, J.C. What is the optimal duration of middle-cerebral artery occlusion consistently resulting in isolated cortical selective neuronal loss in the spontaneously hypertensive rat? Front. Neurol. 2015, 6, 64. [Google Scholar] [CrossRef]
- Hughes, J.L.; Beech, J.S.; Jones, P.S.; Wang, D.; Menon, D.K.; Baron, J.C. Mapping selective neuronal loss and microglial activation in the salvaged neocortical penumbra in the rat. Neuroimage 2010, 49, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Katchanov, J.; Waeber, C.; Gertz, K.; Gietz, A.; Winter, B.; Bruck, W.; Dirnagl, U.; Veh, R.W.; Endres, M. Selective neuronal vulnerability following mild focal brain ischemia in the mouse. Brain Pathol. 2003, 13, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.L.; Jones, P.S.; Beech, J.S.; Wang, D.; Menon, D.K.; Aigbirhio, F.I.; Fryer, T.D.; Baron, J.C. A micropet study of the regional distribution of [11C]-pk11195 binding following temporary focal cerebral ischemia in the rat. Correlation with post mortem mapping of microglia activation. Neuroimage 2012, 59, 2007–2016. [Google Scholar] [CrossRef] [PubMed]
- Wulff, H.; Miller, M.J.; Hansel, W.; Grissmer, S.; Cahalan, M.D.; Chandy, K.G. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: A potential immunosuppressant. Proc. Natl. Acad. Sci. USA 2000, 97, 8151–8156. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, V.; Schlichter, L.C. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J. Neurosci. 2008, 28, 2221–2230. [Google Scholar] [CrossRef]
- Maezawa, I.; Zimin, P.I.; Wulff, H.; Jin, L.W. Amyloid-beta protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J. Biol. Chem. 2011, 286, 3693–3706. [Google Scholar] [CrossRef]
- Nguyen, H.M.; Grossinger, E.M.; Horiuchi, M.; Davis, K.W.; Jin, L.W.; Maezawa, I.; Wulff, H. Differential kv1.3, kca3.1, and kir2.1 expression in “classically” and “alternatively” activated microglia. Glia 2017, 65, 106–121. [Google Scholar] [CrossRef]
- Chen, Y.J.; Nguyen, H.M.; Maezawa, I.; Grossinger, E.M.; Garing, A.L.; Kohler, R.; Jin, L.W.; Wulff, H. The potassium channel kca3.1 constitutes a pharmacological target for neuroinflammation associated with ischemia/reperfusion stroke. J. Cereb. Blood Flow Metab. 2016, 36, 2146–2161. [Google Scholar] [CrossRef]
- Chen, Y.J.; Raman, G.; Bodendiek, S.; O’Donnell, M.E.; Wulff, H. The kca3.1 blocker tram-34 reduces infarction and neurological deficit in a rat model of ischemia/reperfusion stroke. J. Cereb. Blood Flow Metab. 2011, 31, 2363–2374. [Google Scholar] [CrossRef]
- Emmrich, J.V.; Ejaz, S.; Neher, J.J.; Williamson, D.J.; Baron, J.C. Regional distribution of selective neuronal loss and microglial activation across the mca territory after transient focal ischemia: Quantitative versus semiquantitative systematic immunohistochemical assessment. J. Cereb. Blood Flow Metab. 2015, 35, 20–27. [Google Scholar] [CrossRef]
- Amenta, F.; Tayebati, S.K.; Tomassoni, D. Spontaneously hypertensive rat neuroanatomy: Applications to pharmacological research. Ital. J. Anat. Embryol. 2010, 115, 13–17. [Google Scholar] [PubMed]
- Leoni, R.F.; Paiva, F.F.; Henning, E.C.; Nascimento, G.C.; Tannus, A.; de Araujo, D.B.; Silva, A.C. Magnetic resonance imaging quantification of regional cerebral blood flow and cerebrovascular reactivity to carbon dioxide in normotensive and hypertensive rats. Neuroimage 2011, 58, 75–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchan, A.M.; Xue, D.; Slivka, A. A new model of temporary focal neocortical ischemia in the rat. Stroke 1992, 23, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Takasawa, M.; Beech, J.S.; Fryer, T.D.; Hong, Y.T.; Hughes, J.L.; Igase, K.; Jones, P.S.; Smith, R.; Aigbirhio, F.I.; Menon, D.K.; et al. Imaging of brain hypoxia in permanent and temporary middle cerebral artery occlusion in the rat using 18f-fluoromisonidazole and positron emission tomography: A pilot study. J. Cereb. Blood Flow Metab. 2007, 27, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Takasawa, M.; Beech, J.S.; Fryer, T.D.; Jones, P.S.; Ahmed, T.; Smith, R.; Aigbirhio, F.I.; Baron, J.C. Single-subject statistical mapping of acute brain hypoxia in the rat following middle cerebral artery occlusion: A micropet study. Exp. Neurol. 2011, 229, 251–258. [Google Scholar] [CrossRef]
- Fryer, T.D.; Ejaz, S.; Jensen-Kondering, U.; Williamson, D.J.; Sitnikov, S.; Sawiak, S.J.; Aigbirhio, F.I.; Hong, Y.T.; Baron, J.C. Effects of hyperoxia on 18f-fluoro-misonidazole brain uptake and tissue oxygen tension following middle cerebral artery occlusion in rodents: Pilot studies. PLoS ONE 2017, 12, e0187087. [Google Scholar] [CrossRef]
- Garcia, J.H.; Wagner, S.; Liu, K.F.; Hu, X.J. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Stroke 1995, 26, 627–635. [Google Scholar] [CrossRef]
- Komotar, R.J.; Kim, G.H.; Sughrue, M.E.; Otten, M.L.; Rynkowski, M.A.; Kellner, C.P.; Hahn, D.K.; Merkow, M.B.; Garrett, M.C.; Starke, R.M.; et al. Neurologic assessment of somatosensory dysfunction following an experimental rodent model of cerebral ischemia. Nat. Protoc. 2007, 2, 2345–2347. [Google Scholar] [CrossRef]
- Sughrue, M.E.; Mocco, J.; Komotar, R.J.; Mehra, A.; D’Ambrosio, A.L.; Grobelny, B.T.; Penn, D.L.; Connolly, E.S., Jr. An improved test of neurological dysfunction following transient focal cerebral ischemia in rats. J. Neurosci. Methods 2006, 151, 83–89. [Google Scholar] [CrossRef]
- Freret, T.; Bouet, V.; Leconte, C.; Roussel, S.; Chazalviel, L.; Divoux, D.; Schumann-Bard, P.; Boulouard, M. Behavioral deficits after distal focal cerebral ischemia in mice: Usefulness of adhesive removal test. Behav. Neurosci. 2009, 123, 224–230. [Google Scholar] [CrossRef]
- Bouet, V.; Boulouard, M.; Toutain, J.; Divoux, D.; Bernaudin, M.; Schumann-Bard, P.; Freret, T. The adhesive removal test: A sensitive method to assess sensorimotor deficits in mice. Nat. Protoc. 2009, 4, 1560–1564. [Google Scholar] [CrossRef] [PubMed]
- Schallert, T.; Upchurch, M.; Lobaugh, N.; Farrar, S.B.; Spirduso, W.W.; Gilliam, P.; Vaughn, D.; Wilcox, R.E. Tactile extinction: Distinguishing between sensorimotor and motor asymmetries in rats with unilateral nigrostriatal damage. Pharmacol. Biochem. Behav. 1982, 16, 455–462. [Google Scholar] [CrossRef]
- Gunn, R.N.; Lammertsma, A.A.; Hume, S.P.; Cunningham, V.J. Parametric imaging of ligand-receptor binding in pet using a simplified reference region model. Neuroimage 1997, 6, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Price, C.J.; Wang, D.; Menon, D.K.; Guadagno, J.V.; Cleij, M.; Fryer, T.; Aigbirhio, F.; Baron, J.C.; Warburton, E.A. Intrinsic activated microglia map to the peri-infarct zone in the subacute phase of ischemic stroke. Stroke 2006, 37, 1749–1753. [Google Scholar] [CrossRef]
- Gerhard, A.; Schwarz, J.; Myers, R.; Wise, R.; Banati, R.B. Evolution of microglial activation in patients after ischemic stroke: A [11C](r)-pk11195 pet study. Neuroimage 2005, 24, 591–595. [Google Scholar] [CrossRef]
- Williamson, D.J.; Ejaz, S.; Sitnikov, S.; Fryer, T.D.; Sawiak, S.J.; Burke, P.; Baron, J.C.; Aigbirhio, F.I. A comparison of four pet tracers for brain hypoxia mapping in a rodent model of stroke. Nucl. Med. Biol. 2013, 40, 338–344. [Google Scholar] [CrossRef]
- Boscia, F.; Esposito, C.L.; Casamassa, A.; de Franciscis, V.; Annunziato, L.; Cerchia, L. The isolectin ib4 binds ret receptor tyrosine kinase in microglia. J. Neurochem. 2013, 126, 428–436. [Google Scholar] [CrossRef]
- Neher, J.J.; Emmrich, J.V.; Fricker, M.; Mander, P.K.; Thery, C.; Brown, G.C. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, E4098–E4107. [Google Scholar] [CrossRef] [Green Version]
- Streit, W.J. An improved staining method for rat microglial cells using the lectin from griffonia simplicifolia (gsa i-b4). J. Histochem. Cytochem. 1990, 38, 1683–1686. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The rat Brain in Stereotaxic Coordinates, 3rd ed.; Academic Press: San Diego, CA, USA; London, UK, 1996. [Google Scholar]
- Alroy, J.; Goyal, V.; Skutelsky, E. Lectin histochemistry of mammalian endothelium. Histochemistry 1987, 86, 603–607. [Google Scholar] [CrossRef]
- Converse, A.K.; Larsen, E.C.; Engle, J.W.; Barnhart, T.E.; Nickles, R.J.; Duncan, I.D. 11C-(R)-pk11195 pet imaging of microglial activation and response to minocycline in zymosan-treated rats. J. Nucl. Med. 2011, 52, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Lartey, F.M.; Ahn, G.O.; Ali, R.; Rosenblum, S.; Miao, Z.; Arksey, N.; Shen, B.; Colomer, M.V.; Rafat, M.; Liu, H.; et al. The relationship between serial [18F] pbr06 pet imaging of microglial activation and motor function following stroke in mice. Mol. Imaging Biol. 2014, 16, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Boisgard, R.; Kassiou, M.; Dolle, F.; Tavitian, B. Reduced pbr/tspo expression after minocycline treatment in a rat model of focal cerebral ischemia: A pet study using [18F] dpa-714. Mol. Imaging Biol 2011, 13, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Szczupak, B.; Gomez-Vallejo, V.; Domercq, M.; Cano, A.; Padro, D.; Munoz, C.; Higuchi, M.; Matute, C.; Llop, J. In vivo pet imaging of the alpha4beta2 nicotinic acetylcholine receptor as a marker for brain inflammation after cerebral ischemia. J. Neurosci. 2015, 35, 5998–6009. [Google Scholar] [CrossRef]
- Wang, Y.; Yue, X.; Kiesewetter, D.O.; Wang, Z.; Lu, J.; Niu, G.; Teng, G.; Chen, X. [18F] dpa-714 pet imaging of amd3100 treatment in a mouse model of stroke. Mol. Pharm. 2014, 11, 3463–3470. [Google Scholar] [CrossRef]
- Santos-Galdiano, M.; Perez-Rodriguez, D.; Anuncibay-Soto, B.; Font-Belmonte, E.; Ugidos, I.F.; Perez-Garcia, C.C.; Fernandez-Lopez, A. Celecoxib treatment improves neurologic deficit and reduces selective neuronal loss and glial response in rats after transient middle cerebral artery occlusion. J. Pharmacol. Exp. Ther. 2018, 367, 528–542. [Google Scholar] [CrossRef]
- Brown, B.M.; Shim, H.; Christophersen, P.; Wulff, H. Pharmacology of small- and intermediate-conductance calcium-activated potassium channels. Annu. Rev. Pharmacol. Toxicol. 2019, 60. [Google Scholar] [CrossRef]
- Cremer, J.E.; Hume, S.P.; Cullen, B.M.; Myers, R.; Manjil, L.G.; Turton, D.R.; Luthra, S.K.; Bateman, D.M.; Pike, V.W. The distribution of radioactivity in brains of rats given [n-methyl-11C]pk 11195 in vivo after induction of a cortical ischaemic lesion. Int. J. Rad. Appl. Instrum. B 1992, 19, 159–166. [Google Scholar] [CrossRef]
- Lartey, F.M.; Ahn, G.O.; Shen, B.; Cord, K.T.; Smith, T.; Chua, J.Y.; Rosenblum, S.; Liu, H.; James, M.L.; Chernikova, S.; et al. Pet imaging of stroke-induced neuroinflammation in mice using [18F] pbr06. Mol. Imaging Biol. 2014, 16, 109–117. [Google Scholar] [CrossRef]
- Walberer, M.; Jantzen, S.U.; Backes, H.; Rueger, M.A.; Keuters, M.H.; Neumaier, B.; Hoehn, M.; Fink, G.R.; Graf, R.; Schroeter, M. In-vivo detection of inflammation and neurodegeneration in the chronic phase after permanent embolic stroke in rats. Brain Res. 2014, 1581, 80–88. [Google Scholar] [CrossRef]
- Fukumoto, D.; Hosoya, T.; Nishiyama, S.; Harada, N.; Iwata, H.; Yamamoto, S.; Tsukada, H. Multiparametric assessment of acute and subacute ischemic neuronal damage: A small animal positron emission tomography study with rat photochemically induced thrombosis model. Synapse 2010, 65, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Benavides, J.; Capdeville, C.; Dauphin, F.; Dubois, A.; Duverger, D.; Fage, D.; Gotti, B.; MacKenzie, E.T.; Scatton, B. The quantification of brain lesions with an omega 3 site ligand: A critical analysis of animal models of cerebral ischaemia and neurodegeneration. Brain Res. 1990, 522, 275–289. [Google Scholar] [CrossRef]
- Yemisci, M.; Gursoy-Ozdemir, Y.; Vural, A.; Can, A.; Topalkara, K.; Dalkara, T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat. Med. 2009, 15, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, M.; Taoka, T.; Matsuo, Y.; Hiramatsu, K.I.; Sakaki, T. Novel brain ischemic change on mri. Delayed ischemic hyperintensity on t1-weighted images and selective neuronal death in the caudoputamen of rats after brief focal ischemia. Stroke 1999, 30, 1043–1046. [Google Scholar] [CrossRef]
- Fujioka, M.; Taoka, T.; Matsuo, Y.; Mishima, K.; Ogoshi, K.; Kondo, Y.; Tsuda, M.; Fujiwara, M.; Asano, T.; Sakaki, T.; et al. Magnetic resonance imaging shows delayed ischemic striatal neurodegeneration. Ann. Neurol. 2003, 54, 732–747. [Google Scholar] [CrossRef]
- Freret, T.; Chazalviel, L.; Roussel, S.; Bernaudin, M.; Schumann-Bard, P.; Boulouard, M. Long-term functional outcome following transient middle cerebral artery occlusion in the rat: Correlation between brain damage and behavioral impairment. Behav. Neurosci. 2006, 120, 1285–1298. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rat # | PK11195 PET | NeuN | IB4 |
|---|---|---|---|
| Controls | |||
| 1 | <0.001 | 0.052 | 0.57 |
| 2 | <0.001 | <0.001 | 0.44 |
| 3 | <0.001 | 0.41 | 0.09 |
| 4 | <0.001 | 0.53 | 0.08 |
| 5 | <0.001 | 0.006 | 0.007 |
| 6 | 0.266 | 0.036 | 0.014 |
| TRAM-34 treated | |||
| 7 | 0.099 | 0.012 | <0.001 |
| 8 | <0.001 | 0.25 | 0.29 |
| 9 | <0.001 | 0.88 | 0.031 |
| 10 | <0.001 | 0.20 | 0.02 |
| 11 | 0.029 | 0.11 | 0.002 |
| 12 | 0.009 | 0.08 | 0.003 |
| 11C-PK11195 PET | NeuN | IB4 | |
|---|---|---|---|
| Controls (mean ± SD) | +0.052 ± 0.015 | −2.32 ± 1.4% | +2.52 ± 4.9% |
| TRAM-34 treated (mean ± SD) | +0.066 ± 0.071 | +1.68 ± 2.36% | +0.08 ± 3.2% |
| Comparison | p = 1.0 | p = 0.009 | p = 0.337 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emmrich, J.V.; Ejaz, S.; Williamson, D.J.; Hong, Y.T.; Sitnikov, S.; Fryer, T.D.; Aigbirhio, F.I.; Wulff, H.; Baron, J.-C. Assessing the Effects of Cytoprotectants on Selective Neuronal Loss, Sensorimotor Deficit and Microglial Activation after Temporary Middle Cerebral Occlusion. Brain Sci. 2019, 9, 287. https://doi.org/10.3390/brainsci9100287
Emmrich JV, Ejaz S, Williamson DJ, Hong YT, Sitnikov S, Fryer TD, Aigbirhio FI, Wulff H, Baron J-C. Assessing the Effects of Cytoprotectants on Selective Neuronal Loss, Sensorimotor Deficit and Microglial Activation after Temporary Middle Cerebral Occlusion. Brain Sciences. 2019; 9(10):287. https://doi.org/10.3390/brainsci9100287
Chicago/Turabian StyleEmmrich, Julius V., Sohail Ejaz, David J. Williamson, Young T. Hong, Sergey Sitnikov, Tim D. Fryer, Franklin I. Aigbirhio, Heike Wulff, and Jean-Claude Baron. 2019. "Assessing the Effects of Cytoprotectants on Selective Neuronal Loss, Sensorimotor Deficit and Microglial Activation after Temporary Middle Cerebral Occlusion" Brain Sciences 9, no. 10: 287. https://doi.org/10.3390/brainsci9100287