NRSF and Its Epigenetic Effectors: New Treatments for Neurological Disease

{kind=link}
{kind=link}
Abstract
:1. Introduction
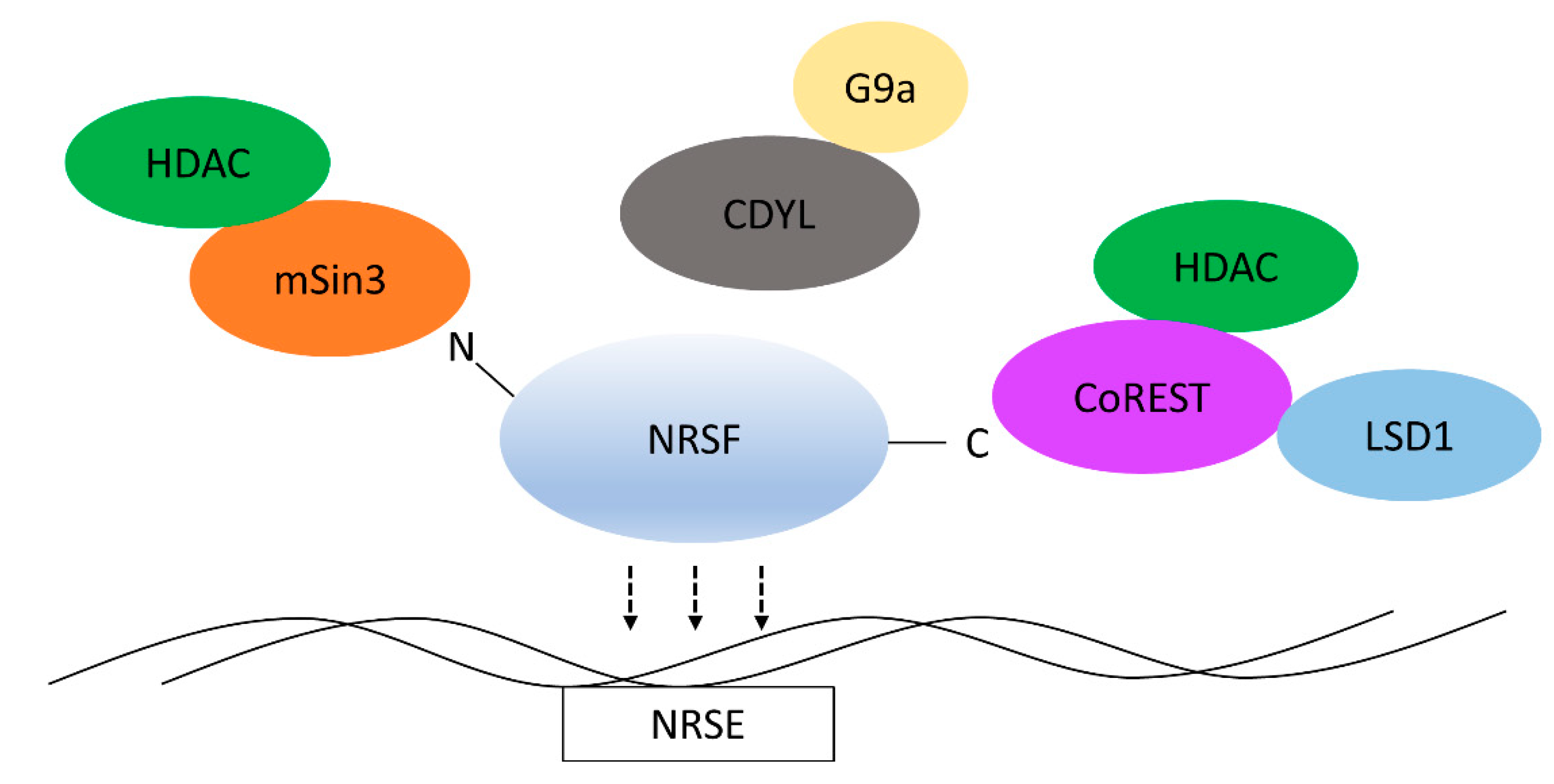
2. Structure and Function of NRSF
3. REST-Interacting LIM Domain Protein
4. REST4
5. CoREST
6. NRSF Recruits Chromatin Remodelers
7. NRSF-Related Diseases
7.1. Epilepsy
7.2. Neuropathic Pain
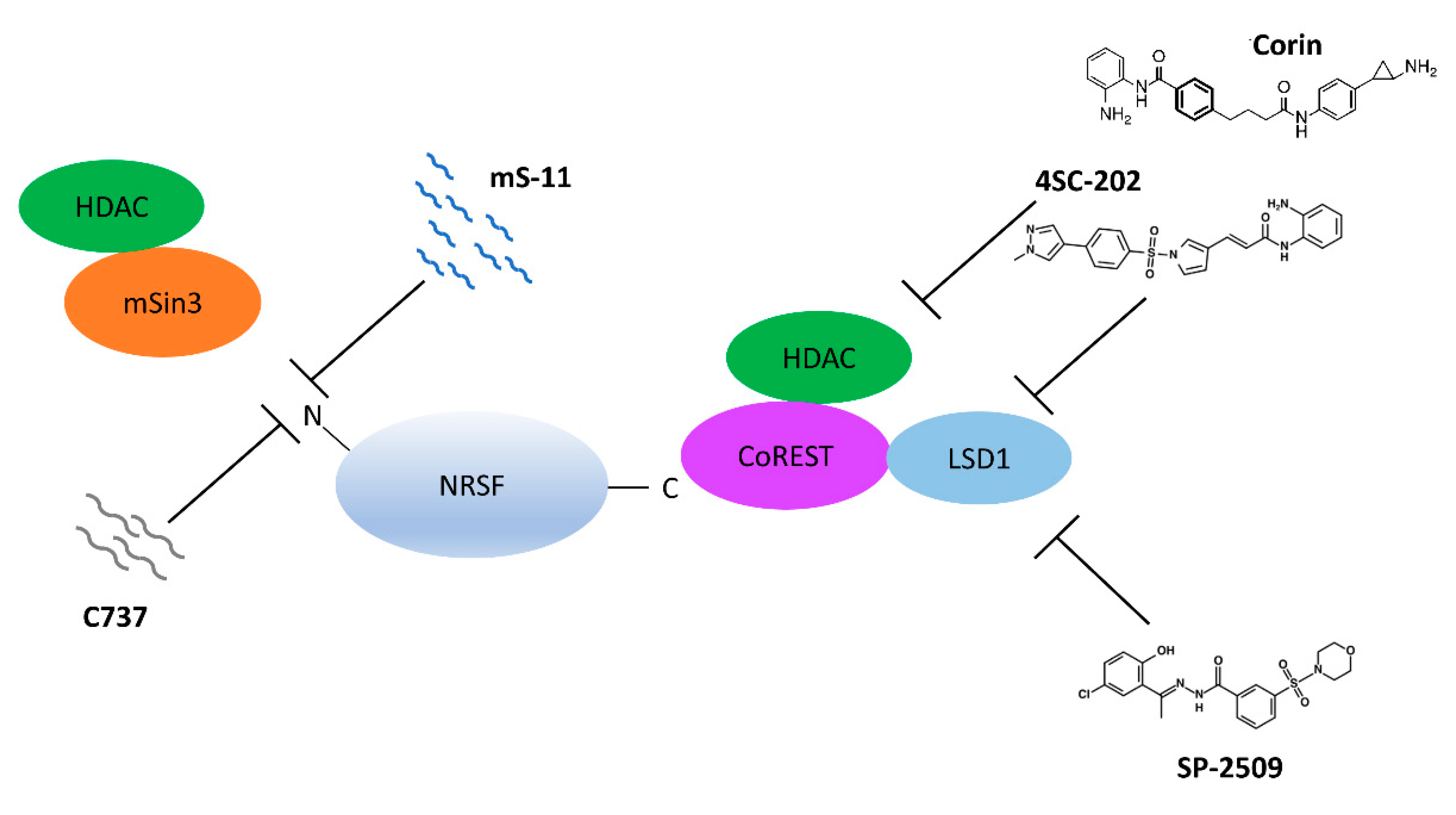
8. Epigenetic Inhibitors
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kraner, S.D.; Chong, J.A.; Tsay, H.J.; Mandel, G. Silencing the type II sodium channel gene: A model for neural-specific gene regulation. Neuron 1992, 9, 37–44. [Google Scholar] [CrossRef]
- Mori, N.; Schoenherr, C.; Vandenbergh, D.J.; Anderson, D.J. A common silencer element in the SCG10 and type II Na+ channel genes binds a factor present in nonneuronal cells but not in neuronal cells. Neuron 1992, 9, 45–54. [Google Scholar] [CrossRef]
- Chong, J.A.; Tapia-Ramirez, J.; Kim, S.; Toledo-Aral, J.J.; Zheng, Y.; Boutros, M.C.; Altshuller, Y.M.; Frohman, M.A.; Kraner, S.D.; Mandel, G. REST: A mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell 1995, 80, 949–957. [Google Scholar] [CrossRef]
- Schoenherr, C.J.; Anderson, D.J. The neuron-restrictive silencer factor (NRSF): A coordinate repressor of multiple neuron-specific genes. Science 1995, 267, 1360–1363. [Google Scholar] [CrossRef] [PubMed]
- Bruce, A.W.; Donaldson, I.J.; Wood, I.C.; Yerbury, S.A.; Sadowski, M.I.; Chapman, M.; Gottgens, B.; Buckley, N.J. Genome-wide analysis of repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) target genes. Proc. Natl. Acad. Sci. USA 2004, 101, 10458–10463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palm, K.; Belluardo, N.; Metsis, M.; Timmusk, T. Neuronal expression of zinc finger transcription factor REST/NRSF/XBR gene. J. Neurosci. 1998, 18, 1280–1296. [Google Scholar] [CrossRef]
- Mori, N.; Mizuno, T.; Murai, K.; Nakano, I.; Yamashita, H. Effect of age on the gene expression of neural-restrictive silencing factor NRSF/REST. Neurobiol. Aging 2002, 23, 255–262. [Google Scholar] [CrossRef]
- Singh, S.K.; Kagalwala, M.N.; Parker-Thornburg, J.; Adams, H.; Majumder, S. REST maintains self-renewal and pluripotency of embryonic stem cells. Nature 2008, 453, 223–227. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.F.; Paquette, A.J.; Anderson, D.J. NRSF/REST is required in vivo for repression of multiple neuronal target genes during embryogenesis. Nat. Genet. 1998, 20, 136–142. [Google Scholar] [CrossRef]
- Su, X.; Kameoka, S.; Lentz, S.; Majumder, S. Activation of REST/NRSF target genes in neural stem cells is sufficient to cause neuronal differentiation. Mol. Cell. Biol. 2004, 24, 8018–8025. [Google Scholar] [CrossRef]
- Gao, Z.; Ure, K.; Ding, P.; Nashaat, M.; Yuan, L.; Ma, J.; Hammer, R.E.; Hsieh, J. The master negative regulator REST/NRSF controls adult neurogenesis by restraining the neurogenic program in quiescent stem cells. J. Neurosci. 2011, 31, 9772–9786. [Google Scholar] [CrossRef] [PubMed]
- Conti, L.; Crisafulli, L.; Caldera, V.; Tortoreto, M.; Brilli, E.; Conforti, P.; Zunino, F.; Magrassi, L.; Schiffer, D.; Cattaneo, E. REST controls self-renewal and tumorigenic competence of human glioblastoma cells. PLoS ONE 2012, 7, e38486. [Google Scholar] [CrossRef] [PubMed]
- Palm, K.; Metsis, M.; Timmusk, T. Neuron-specific splicing of zinc finger transcription factor REST/NRSF/XBR is frequent in neuroblastomas and conserved in human, mouse and rat. Brain Res. Mol. Brain Res. 1999, 72, 30–39. [Google Scholar] [CrossRef]
- Su, X.; Gopalakrishnan, V.; Stearns, D.; Aldape, K.; Lang, F.F.; Fuller, G.; Snyder, E.; Eberhart, C.G.; Majumder, S. Abnormal expression of REST/NRSF and Myc in neural stem/progenitor cells causes cerebellar tumors by blocking neuronal differentiation. Mol. Cell. Biol. 2006, 26, 1666–1678. [Google Scholar] [CrossRef] [PubMed]
- Lawinger, P.; Venugopal, R.; Guo, Z.S.; Immaneni, A.; Sengupta, D.; Lu, W.; Rastelli, L.; Marin Dias Carneiro, A.; Levin, V.; Fuller, G.N.; et al. The neuronal repressor REST/NRSF is an essential regulator in medulloblastoma cells. Nat. Med. 2000, 6, 826–831. [Google Scholar] [PubMed]
- Hwang, J.Y.; Zukin, R.S. REST, a master transcriptional regulator in neurodegenerative disease. Curr. Opin. Neurobiol. 2018, 48, 193–200. [Google Scholar] [CrossRef]
- Landgrave-Gomez, J.; Mercado-Gomez, O.; Guevara-Guzman, R. Epigenetic mechanisms in neurological and neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 58. [Google Scholar] [PubMed]
- Noh, K.M.; Hwang, J.Y.; Follenzi, A.; Athanasiadou, R.; Miyawaki, T.; Greally, J.M.; Bennett, M.V.; Zukin, R.S. Repressor element-1 silencing transcription factor (REST)-dependent epigenetic remodeling is critical to ischemia-induced neuronal death. Proc. Natl. Acad. Sci. USA 2012, 109, 201121568. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhu, M.; Yu, Y.; Qiu, L.; Zhang, Y.; He, L.; Zhang, J. Brain REST/NRSF Is Not Only a Silent Repressor but Also an Active Protector. Mol. Neurobiol. 2017, 54, 541–550. [Google Scholar] [CrossRef]
- Shimojo, M. Characterization of the nuclear targeting signal of REST/NRSF. Neurosci. Lett. 2006, 398, 161–166. [Google Scholar] [CrossRef]
- Shimojo, M.; Lee, J.H.; Hersh, L.B. Role of zinc finger domains of the transcription factor neuron-restrictive silencer factor/repressor element-1 silencing transcription factor in DNA binding and nuclear localization. J. Biol. Chem. 2001, 276, 13121–13126. [Google Scholar] [CrossRef] [PubMed]
- Shimojo, M.; Hersh, L.B. REST/NRSF-interacting LIM domain protein, a putative nuclear translocation receptor. Mol. Cell. Biol. 2003, 23, 9025–9031. [Google Scholar] [CrossRef]
- Shimojo, M.; Hersh, L.B. Characterization of the REST/NRSF-interacting LIM domain protein (RILP): Localization and interaction with REST/NRSF. J. Neurochem. 2006, 96, 1130–1138. [Google Scholar] [CrossRef]
- Nomura, M.; Uda-Tochio, H.; Murai, K.; Mori, N.; Nishimura, Y. The neural repressor NRSF/REST binds the PAH1 domain of the Sin3 corepressor by using its distinct short hydrophobic helix. J. Mol. Biol. 2005, 354, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Myers, S.J.; Dingledine, R. Transcriptional repression by REST: Recruitment of Sin3A and histone deacetylase to neuronal genes. Nat. Neurosci. 1999, 2, 867–872. [Google Scholar] [CrossRef]
- Andres, M.E.; Burger, C.; Peral-Rubio, M.J.; Battaglioli, E.; Anderson, M.E.; Grimes, J.; Dallman, J.; Ballas, N.; Mandel, G. CoREST: A functional corepressor required for regulation of neural-specific gene expression. Proc. Natl. Acad. Sci. USA 1999, 96, 9873–9878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Gocke, C.B.; Luo, X.; Borek, D.; Tomchick, D.R.; Machius, M.; Otwinowski, Z.; Yu, H. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol. Cell 2006, 23, 377–387. [Google Scholar] [CrossRef]
- Westbrook, T.F.; Hu, G.; Ang, X.L.; Mulligan, P.; Pavlova, N.N.; Liang, A.; Leng, Y.; Maehr, R.; Shi, Y.; Harper, J.W.; et al. SCFbeta-TRCP controls oncogenic transformation and neural differentiation through REST degradation. Nature 2008, 452, 370–374. [Google Scholar] [CrossRef]
- Chen, G.L.; Miller, G.M. Alternative REST Splicing Underappreciated. eNeuro 2018, 5, 20–25. [Google Scholar] [CrossRef]
- Koenigsberger, C.; Chicca, J.J.; Amoureux, M.C.; Edelman, G.M.; Jones, F.S. Differential regulation by multiple promoters of the gene encoding the neuron-restrictive silencer factor. Proc. Natl. Acad. Sci. USA 2000, 97, 2291–2296. [Google Scholar] [CrossRef] [Green Version]
- Raj, B.; O’Hanlon, D.; Vessey, J.P.; Pan, Q.; Ray, D.; Buckley, N.J.; Miller, F.D.; Blencowe, B.J. Cross-regulation between an alternative splicing activator and a transcription repressor controls neurogenesis. Mol. Cell 2011, 43, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Hersh, L.B.; Shimojo, M. Regulation of cholinergic gene expression by the neuron restrictive silencer factor/repressor element-1 silencing transcription factor. Life Sci. 2003, 72, 2021–2028. [Google Scholar] [CrossRef]
- Coulson, J.M.; Edgson, J.L.; Woll, P.J.; Quinn, J.P. A splice variant of the neuron-restrictive silencer factor repressor is expressed in small cell lung cancer: A potential role in derepression of neuroendocrine genes and a useful clinical marker. Cancer Res. 2000, 60, 1840–1844. [Google Scholar] [PubMed]
- Shimojo, M.; Paquette, A.J.; Anderson, D.J.; Hersh, L.B. Protein kinase A regulates cholinergic gene expression in PC12 cells: REST4 silences the silencing activity of neuron-restrictive silencer factor/REST. Mol. Cell. Biol. 1999, 19, 6788–6795. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, A.; Yamada, T.; Sasagawa, S.; Naruse, Y.; Mori, N.; Tsuda, M. REST4-mediated modulation of REST/NRSF-silencing function during BDNF gene promoter activation. Biochem. Biophys. Res. Commun. 2002, 290, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Shudo, Y.; Shimojo, M.; Fukunaga, M.; Ito, S. Pituitary adenylate cyclase-activating polypeptide is regulated by alternative splicing of transcriptional repressor REST/NRSF in nerve injury. Life Sci. 2015, 143, 174–181. [Google Scholar] [CrossRef]
- Ren, H.; Gao, Z.; Wu, N.; Zeng, L.; Tang, X.; Chen, X.; Liu, Z.; Zhang, W.; Wang, L.; Li, Z. Expression of REST4 in human gliomas in vivo and influence of pioglitazone on REST in vitro. Biochem. Biophys. Res. Commun. 2015, 463, 504–509. [Google Scholar] [CrossRef]
- Yu, M.; Cai, L.; Liang, M.; Huang, Y.; Gao, H.; Lu, S.; Fei, J.; Huang, F. Alteration of NRSF expression exacerbating 1-methyl-4-phenyl-pyridinium ion-induced cell death of SH-SY5Y cells. Neurosci. Res. 2009, 65, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Spencer, E.M.; Chandler, K.E.; Haddley, K.; Howard, M.R.; Hughes, D.; Belyaev, N.D.; Coulson, J.M.; Stewart, J.P.; Buckley, N.J.A. Regulation and role of REST and REST4 variants in modulation of gene expression in in vivo and in vitro in epilepsy models. Neurobiol. Dis. 2006, 24, 41–52. [Google Scholar] [CrossRef]
- Abrajano, J.J.; Qureshi, I.A.; Gokhan, S.; Molero, A.E.; Zheng, D.; Bergman, A.; Mehler, M.F. Corepressor for element-1-silencing transcription factor preferentially mediates gene networks underlying neural stem cell fate decisions. Proc. Natl. Acad. Sci. USA 2010, 107, 16685–16690. [Google Scholar] [CrossRef] [PubMed]
- You, A.; Tong, J.K.; Grozinger, C.M.; Schreiber, S.L. CoREST is an integral component of the CoREST-human histone deacetylase complex. Proc. Natl. Acad. Sci. USA 2001, 98, 1454–1458. [Google Scholar] [CrossRef] [PubMed]
- Abrajano, J.J.; Qureshi, I.A.; Gokhan, S.; Zheng, D.; Bergman, A.; Mehler, M.F. REST and CoREST modulate neuronal subtype specification, maturation and maintenance. PLoS ONE 2009, 4, e7936. [Google Scholar] [CrossRef] [PubMed]
- Abrajano, J.J.; Qureshi, I.A.; Gokhan, S.; Zheng, D.; Bergman, A.; Mehler, M.F. Differential deployment of REST and CoREST promotes glial subtype specification and oligodendrocyte lineage maturation. PLoS ONE 2009, 4, e7665. [Google Scholar] [CrossRef] [PubMed]
- Ballas, N.; Grunseich, C.; Lu, D.D.; Speh, J.C.; Mandel, G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 2005, 121, 645–657. [Google Scholar] [CrossRef]
- Barrios, A.P.; Gomez, A.V.; Saez, J.E.; Ciossani, G.; Toffolo, E.; Battaglioli, E.; Mattevi, A.; Andres, M.E. Differential properties of transcriptional complexes formed by the CoREST family. Mol. Cell. Biol. 2014, 34, 2760–2770. [Google Scholar] [CrossRef] [PubMed]
- Laherty, C.D.; Yang, W.M.; Sun, J.M.; Davie, J.R.; Seto, E.; Eisenman, R.N. Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell 1997, 89, 349–356. [Google Scholar] [CrossRef]
- Roopra, A.; Qazi, R.; Schoenike, B.; Daley, T.J.; Morrison, J.F. Localized domains of G9a-mediated histone methylation are required for silencing of neuronal genes. Mol. Cell 2004, 14, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, P.; Westbrook, T.F.; Ottinger, M.; Pavlova, N.; Chang, B.; Macia, E.; Shi, Y.J.; Barretina, J.; Liu, J.; Howley, P.M.; et al. CDYL bridges REST and histone methyltransferases for gene repression and suppression of cellular transformation. Mol. Cell 2008, 32, 718–726. [Google Scholar] [CrossRef]
- Lunyak, V.V.; Burgess, R.; Prefontaine, G.G.; Nelson, C.; Sze, S.H.; Chenoweth, J.; Schwartz, P.; Pevzner, C.; Glass, P.A.; Mandel, G.; et al. Corepressor-dependent silencing of chromosomal regions encoding neuronal genes. Science 2002, 298, 1747–1752. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.N.; Filippi, D.; Hauser, W.A. The descriptive epidemiology of epilepsy-a review. Epilepsy Res. 2009, 85, 31–45. [Google Scholar] [CrossRef]
- Bassuk, A.G.; Wallace, R.H.; Buhr, A.; Buller, A.R.; Afawi, Z.; Shimojo, M.; Miyata, S.; Chen, S.; Gonzalez-Alegre, P.; Griesbach, H.L.; et al. A homozygous mutation in human PRICKLE1 causes an autosomal-recessive progressive myoclonus epilepsy-ataxia syndrome. Am. J. Hum. Genet. 2008, 83, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Gillies, S.; Haddley, K.; Vasiliou, S.; Bubb, V.J.; Quinn, J.P. The human neurokinin B gene, TAC3, and its promoter are regulated by Neuron Restrictive Silencing Factor (NRSF) transcription factor family. Neuropeptides 2009, 43, 333–340. [Google Scholar] [CrossRef] [PubMed]
- McClelland, S.; Brennan, G.P.; Dube, C.; Rajpara, S.; Iyer, S.; Richichi, C.; Bernard, C.; Baram, T.Z. The transcription factor NRSF contributes to epileptogenesis by selective repression of a subset of target genes. eLife 2014, 3, e01267. [Google Scholar] [CrossRef] [PubMed]
- Mucha, M.; Ooi, L.; Linley, J.E.; Mordaka, P.; Dalle, C.; Robertson, B.; Gamper, N.; Wood, I.C. Transcriptional control of KCNQ channel genes and the regulation of neuronal excitability. J. Neurosci. 2010, 30, 13235–13245. [Google Scholar] [CrossRef] [PubMed]
- Escayg, A.; MacDonald, B.T.; Meisler, M.H.; Baulac, S.; Huberfeld, G.; An-Gourfinkel, I.; Brice, A.; LeGuern, E.; Moulard, B.; Chaigne, D.; et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat. Genet. 2000, 24, 343–345. [Google Scholar] [CrossRef]
- Laumet, G.; Garriga, J.; Chen, S.R.; Zhang, Y.; Li, D.P.; Smith, T.M.; Dong, Y.; Jelinek, J.; Cesaroni, M.; Issa, J.P.; et al. G9a is essential for epigenetic silencing of K(+) channel genes in acute-to-chronic pain transition. Nat. Neurosci. 2015, 18, 1746–1755. [Google Scholar] [CrossRef]
- McClelland, S.; Flynn, C.; Dube, C.; Richichi, C.; Zha, Q.; Ghestem, A.; Esclapez, M.; Bernard, C.; Baram, T.Z. Neuron-restrictive silencer factor-mediated hyperpolarization-activated cyclic nucleotide gated channelopathy in experimental temporal lobe epilepsy. Ann. Neurol. 2011, 70, 454–464. [Google Scholar] [CrossRef]
- Dibbens, L.M.; Reid, C.A.; Hodgson, B.; Thomas, E.A.; Phillips, A.M.; Gazina, E.; Cromer, B.A.; Clarke, A.L.; Baram, T.Z.; Scheffer, I.E.; et al. Augmented currents of an HCN2 variant in patients with febrile seizure syndromes. Ann. Neurol. 2010, 67, 542–546. [Google Scholar] [CrossRef]
- Garriga-Canut, M.; Schoenike, B.; Qazi, R.; Bergendahl, K.; Daley, T.J.; Pfender, R.M.; Morrison, J.F.; Ockuly, J.; Stafstrom, C.; Sutula, T.; et al. 2-Deoxy-D-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nat. Neurosci. 2006, 9, 1382–1387. [Google Scholar] [CrossRef]
- Costigan, M.; Scholz, J.; Woolf, C.J. Neuropathic pain: A maladaptive response of the nervous system to damage. Annu. Rev. Neurosci. 2009, 32, 1–32. [Google Scholar] [CrossRef]
- Trimmer, J.S. Ion channels and pain: Important steps towards validating a new therapeutic target for neuropathic pain. Exp. Neurol. 2014, 254, 190–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devor, M. Sodium channels and mechanisms of neuropathic pain. J. Pain 2006, 7 (Suppl. 1), S3–S12. [Google Scholar] [CrossRef]
- Liu, M.; Wood, J.N. The roles of sodium channels in nociception: Implications for mechanisms of neuropathic pain. Pain Med. 2011, 12 (Suppl. 3), S93–S99. [Google Scholar] [CrossRef] [PubMed]
- Busserolles, J.; Tsantoulas, C.; Eschalier, A.; García, J.A. Potassium channels in neuropathic pain: Advances, challenges, and emerging ideas. Pain 2016, 157 (Suppl. 1), S7–S14. [Google Scholar] [CrossRef]
- Zhang, X.; Bao, L.; Shi, T.J.; Ju, G.; Elde, R.; Hokfelt, T. Down-regulation of mu-opioid receptors in rat and monkey dorsal root ganglion neurons and spinal cord after peripheral axotomy. Neuroscience 1998, 82, 223–240. [Google Scholar] [CrossRef]
- Formisano, L.; Noh, K.M.; Miyawaki, T.; Mashiko, T.; Bennett, M.V.; Zukin, R.S. Ischemic insults promote epigenetic reprogramming of mu opioid receptor expression in hippocampal neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 4170–4175. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.M.; Geng, L.J.; Shi, X.Y.; Zhang, C.G.; Wang, S.Y.; Zhang, G.M. By up-regulating mu- and delta-opioid receptors, neuron-restrictive silencer factor knockdown promotes neurological recovery after ischemia. Oncotarget 2017, 8, 101012–101025. [Google Scholar]
- Uchida, H.; Sasaki, K.; Ma, L.; Ueda, H. Neuron-restrictive silencer factor causes epigenetic silencing of Kv4.3 gene after peripheral nerve injury. Neuroscience 2010, 166, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.S.; Hwang, C.K.; Choi, H.S.; Song, K.Y.; Law, P.Y.; Wei, L.N.; Loh, H.H. Neuron-restrictive silencer factor (NRSF) functions as a repressor in neuronal cells to regulate the mu opioid receptor gene. J. Biol. Chem. 2004, 279, 46464–46473. [Google Scholar] [CrossRef]
- Kim, C.S.; Choi, H.S.; Hwang, C.K.; Song, K.Y.; Lee, B.K.; Law, P.Y.; Wei, L.N.; Loh, H.H. Evidence of the neuron-restrictive silencer factor (NRSF) interaction with Sp3 and its synergic repression to the mu opioid receptor (MOR) gene. Nucl. Acids Res. 2006, 34, 6392–6403. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, S.R.; Chen, H.; Pan, H.L. RE1-silencing transcription factor controls the acute-to-chronic neuropathic pain transition and Chrm2 receptor gene expression in primary sensory neurons. J. Biol. Chem. 2018, 293, 19078–19091. [Google Scholar] [CrossRef] [PubMed]
- Mattson, R.H.; Cramer, J.A.; Williamson, P.D.; Novelly, R.A. Valproic acid in epilepsy: Clinical and pharmacological effects. Ann. Neurol. 1978, 3, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Emrich, H.M.; von Zerssen, D.; Kissling, W.; Moller, H.J. Therapeutic effect of valproate in mania. Am. J. Psychiatry 1981, 138, 256. [Google Scholar] [PubMed]
- Uchida, H.; Matsushita, Y.; Araki, K.; Mukae, T.; Ueda, H. Histone deacetylase inhibitors relieve morphine resistance in neuropathic pain after peripheral nerve injury. J. Pharmacol. Sci. 2015, 128, 208–211. [Google Scholar] [CrossRef]
- Ueda, H.; Kurita, J.I.; Neyama, H.; Hirao, Y.; Kouji, H.; Mishina, T.; Kasai, M.; Nakano, H.; Yoshimori, A.; Nishimura, Y. A mimetic of the mSin3-binding helix of NRSF/REST ameliorates abnormal pain behavior in chronic pain models. Bioorg. Med. Chem. Lett. 2017, 27, 4705–4709. [Google Scholar] [CrossRef] [PubMed]
- Hai-Ying, C.; Nagano, K.; Ezzikouri, S.; Yamaguchi, C.; Kayesh, M.E.; Rebbani, K.; Kitab, B.; Nakano, H.; Kouji, H.; Kohara, M.; et al. Establishment of an intermittent cold stress model using Tupaia belangeri and evaluation of compound C737 targeting neuron-restrictive silencer factor. Exp. Anim. 2016, 65, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Conforti, P.; Zuccato, C.; Gaudenzi, G.; Ieraci, A.; Camnasio, S.; Buckley, N.J.; Mutti, C.; Cotelli, F.; Contini, A.; Cattaneo, E. Binding of the repressor complex REST-mSIN3b by small molecules restores neuronal gene transcription in Huntington’s disease models. J. Neurochem. 2013, 127, 22–35. [Google Scholar] [CrossRef]
- Inui, K.; Zhao, Z.; Yuan, J.; Jayaprakash, S.; Le, L.T.M.; Drakulic, S.; Sander, B.; Golas, M.M. Stepwise assembly of functional C-terminal REST/NRSF transcriptional repressor complexes as a drug target. Protein Sci. 2017, 26, 997–1011. [Google Scholar] [CrossRef] [Green Version]
- Kalin, J.H.; Wu, M.; Gomez, A.V.; Song, Y.; Das, J.; Hayward, D.; Adejola, N.; Wu, M.; Panova, I.; Chung, H.J.; et al. Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nat. Commun. 2018, 9, 53. [Google Scholar] [CrossRef]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thompson, R.; Chan, C. NRSF and Its Epigenetic Effectors: New Treatments for Neurological Disease. Brain Sci. 2018, 8, 226. https://doi.org/10.3390/brainsci8120226
Thompson R, Chan C. NRSF and Its Epigenetic Effectors: New Treatments for Neurological Disease. Brain Sciences. 2018; 8(12):226. https://doi.org/10.3390/brainsci8120226
Chicago/Turabian StyleThompson, Ryan, and Christina Chan. 2018. "NRSF and Its Epigenetic Effectors: New Treatments for Neurological Disease" Brain Sciences 8, no. 12: 226. https://doi.org/10.3390/brainsci8120226