The Genetic Diagnosis of Neurodegenerative Diseases and Therapeutic Perspectives


Abstract
:
1. Introduction
2. Obtaining Genetic Information
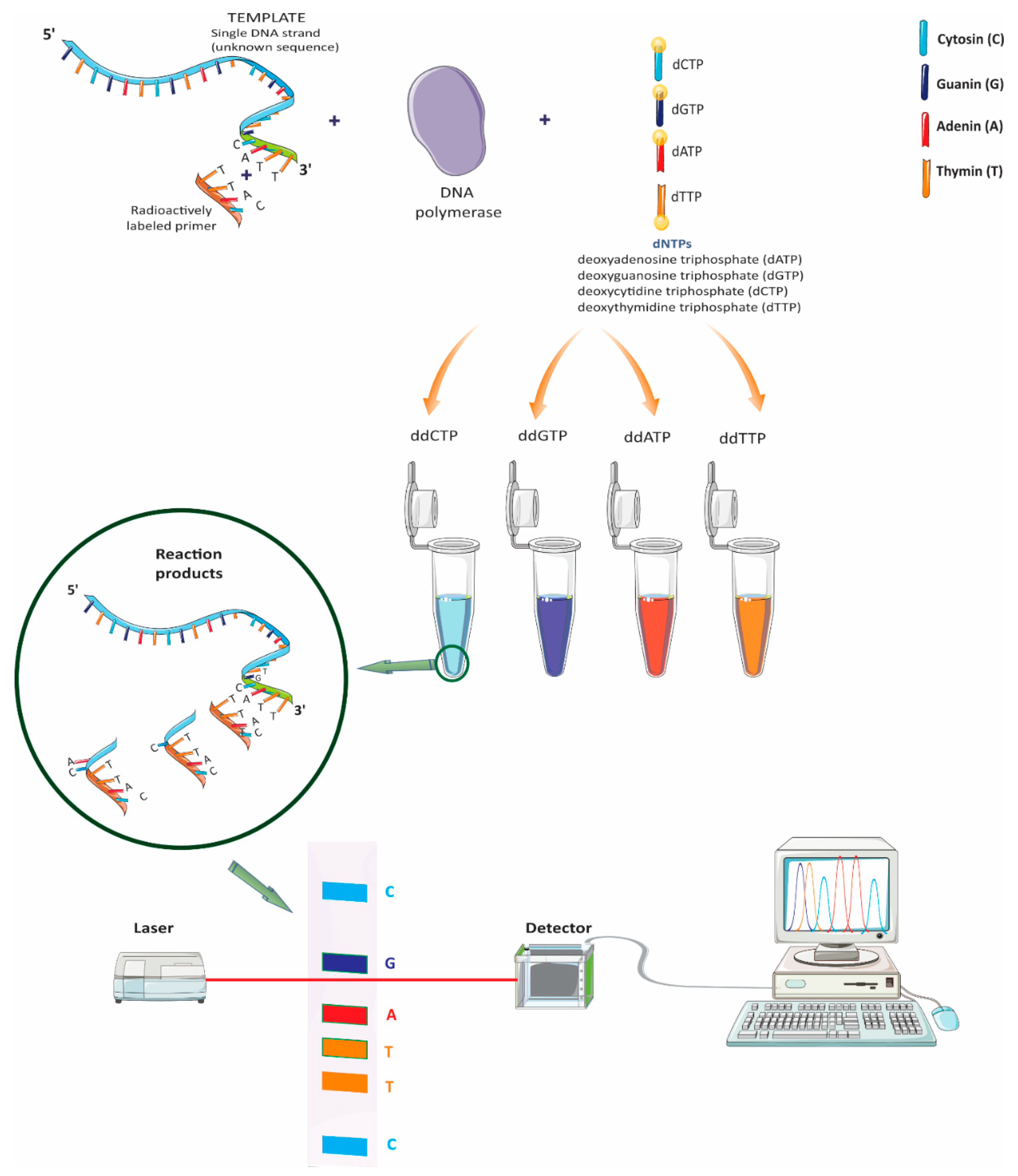
2.1. Sanger Sequencing
2.2. Next Generation Sequencing (NGS)
3. The Genetics of Neurodegenerative Diseases
3.1. Alzheimer’s Disease (AD)
3.2. Amyotrophic Lateral Sclerosis (ALS)
3.3. Parkinson’s Disease (PD)
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- University of Maryland Medical System. Neurological Diseases and Movement Rehabilitation. Available online: https://www.umms.org/health-services/rehabilitation/services/neuro/neurological-diseases-movement-rehabilitation (accessed on 29 November 2018).
- Verkhratsky, A.; Butt, A. Neuroglia in Neurological Diseases. In Glial Physiology and Pathophysiology; Wiley-Blackwell: West Sussex, UK, 2013; pp. 453–504. [Google Scholar]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Henkel, J.S.; Xiao, Q.; Zhao, W.; Wang, J.; Yen, A.A.; Siklos, L.; McKercher, S.R.; Appel, S.H. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 16021–16026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.H.; Parsadanian, A.S.; Andreeva, A.; Snider, W.D.; Elliott, J.L. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J. Neurosci. 2000, 20, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.D.; Julien, J.P.; Rivest, S. Induction of proinflammatory molecules in mice with amyotrophic lateral sclerosis: No requirement for proapoptotic interleukin-1beta in neurodegeneration. Ann. Neurol. 2001, 50, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Gowing, G.; Dequen, F.; Soucy, G.; Julien, J.P. Absence of tumor necrosis factor-alpha does not affect motor neuron disease caused by superoxide dismutase 1 mutations. J. Neurosci. 2006, 26, 11397–11402. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, P.; Robberecht, W. Recent advances in motor neuron disease. Curr. Opin. Neurol. 2009, 22, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Biotechnology 1992, 24, 104–108. [Google Scholar] [CrossRef]
- Jiménez-Escrig, A.; Gobernado, I.; Sánchez-Herranz, A. Secuenciación de genoma completo: Un salto cualitativo en los estudios genéticos. Rev. Neurol. 2012, 54, 692–698. [Google Scholar]
- Sanger, F.; Coulson, A.R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 1975, 94, 441–448. [Google Scholar] [CrossRef]
- Unamba, C.I.; Nag, A.; Sharma, R.K. Next Generation Sequencing Technologies: The Doorway to the Unexplored Genomics of Non-Model Plants. Front. Plant Sci. 2015, 6, 1074. [Google Scholar] [CrossRef]
- Ansorge, W.J. Next-generation DNA sequencing techniques. New Biotechnol. 2009, 25, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Diamandis, E.P. Next-generation sequencing: A new revolution in molecular diagnostics? Clin. Chem. 2009, 55, 2088–2092. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chiodini, R.; Badr, A.; Zhang, G. The impact of next-generation sequencing on genomics. J. Genet. Genomics Yi Chuan Xue Bao 2011, 38, 95–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bick, D.; Dimmock, D. Whole exome and whole genome sequencing. Curr. Opin. Pediatr. 2011, 23, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Biesecker, L.G.; Shianna, K.V.; Mullikin, J.C. Exome sequencing: The expert view. Genome Biol. 2011, 12, 128. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Wang, J.; Liu, Y.; Zhou, Y. Mechanisms of Long Non-Coding RNAs in the Assembly and Plasticity of Neural Circuitry. Front. Neural Circuits 2017, 11, 76. [Google Scholar] [CrossRef]
- Gilissen, C.; Hoischen, A.; Brunner, H.G.; Veltman, J.A. Disease gene identification strategies for exome sequencing. Eur. J. Hum. Genet. 2012, 20, 490–497. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.X.; Buckingham, K.J.; Jhangiani, S.N.; Boehm, C.; Sobreira, N.; Smith, J.D.; Harrell, T.M.; McMillin, M.J.; Wiszniewski, W.; Gambin, T.; et al. The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am. J. Hum. Genet. 2015, 97, 199–215. [Google Scholar] [CrossRef] [Green Version]
- Zemojtel, T.; Kohler, S.; Mackenroth, L.; Jager, M.; Hecht, J.; Krawitz, P.; Graul-Neumann, L.; Doelken, S.; Ehmke, N.; Spielmann, M.; et al. Effective diagnosis of genetic disease by computational phenotype analysis of the disease-associated genome. Sci. Transl. Med. 2014, 6, 252ra123. [Google Scholar] [CrossRef]
- Köhler, S.; Doelken, S.C.; Mungall, C.J.; Bauer, S.; Firth, H.V.; Bailleul-Forestier, I.; Black, G.C.; Brown, D.L.; Brudno, M.; Campbell, J.; et al. The Human Phenotype Ontology project: Linking molecular biology and disease through phenotype data. Nucleic Acids Res. 2014, 42, D966–D974. [Google Scholar] [CrossRef]
- Robinson, P.N.; Kohler, S.; Bauer, S.; Seelow, D.; Horn, D.; Mundlos, S. The Human Phenotype Ontology: A tool for annotating and analyzing human hereditary disease. Am. J. Hum. Genet. 2008, 83, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Köhler, S.; Schulz, M.H.; Krawitz, P.; Bauer, S.; Dolken, S.; Ott, C.E.; Mundlos, C.; Horn, D.; Mundlos, S.; Robinson, P.N. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am. J. Hum. Genet. 2009, 85, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Sifrim, A.; Popovic, D.; Tranchevent, L.C.; Ardeshirdavani, A.; Sakai, R.; Konings, P.; Vermeesch, J.R.; Aerts, J.; De Moor, B.; Moreau, Y. eXtasy: Variant prioritization by genomic data fusion. Nat. Methods 2013, 10, 1083–1084. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Kohler, S.; Schulz, M.H.; Robinson, P.N. Bayesian ontology querying for accurate and noise-tolerant semantic searches. Bioinformatics 2012, 28, 2502–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doelken, S.C.; Kohler, S.; Mungall, C.J.; Gkoutos, G.V.; Ruef, B.J.; Smith, C.; Smedley, D.; Bauer, S.; Klopocki, E.; Schofield, P.N.; et al. Phenotypic overlap in the contribution of individual genes to CNV pathogenicity revealed by cross-species computational analysis of single-gene mutations in humans, mice and zebrafish. Dis. Models Mech. 2013, 6, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Al-Qattan, M.M.; Al Abdulkareem, I.; Al Haidan, Y.; Al Balwi, M. A novel mutation in the SHH long-range regulator (ZRS) is associated with preaxial polydactyly, triphalangeal thumb, and severe radial ray deficiency. Am. J. Med. Genet. Part A 2012, 158A, 2610–2615. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Van Deerlin, V.M. The genetics and neuropathology of neurodegenerative disorders: Perspectives and implications for research and clinical practice. Acta Neuropathol. 2012, 124, 297–303. [Google Scholar] [CrossRef]
- Ferri, C.P.; Sousa, R.; Albanese, E.; Ribeiro, W.S.; Honyashiki, M. World Alzheimer Report 2009—Executive Summary; Alzheimer’s Disease International: London, UK, 2009; pp. 1–22. [Google Scholar]
- Perrin, R.J.; Fagan, A.M.; Holtzman, D.M. Multimodal techniques for diagnosis and prognosis of Alzheimer’s disease. Nature 2009, 461, 916–922. [Google Scholar] [CrossRef]
- Schellenberg, G.D.; Montine, T.J. The genetics and neuropathology of Alzheimer’s disease. Acta Neuropathol. 2012, 124, 305–323. [Google Scholar] [CrossRef] [Green Version]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Muller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Ryman, D.C.; Acosta-Baena, N.; Aisen, P.S.; Bird, T.; Danek, A.; Fox, N.C.; Goate, A.; Frommelt, P.; Ghetti, B.; Langbaum, J.B.; et al. Symptom onset in autosomal dominant Alzheimer disease: A systematic review and meta-analysis. Neurology 2014, 83, 253–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopera, F.; Ardilla, A.; Martinez, A.; Madrigal, L.; Arango-Viana, J.C.; Lemere, C.A.; Arango-Lasprilla, J.C.; Hincapie, L.; Arcos-Burgos, M.; Ossa, J.E.; et al. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA 1997, 277, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Wetzel-Smith, M.K.; Hunkapiller, J.; Bhangale, T.R.; Srinivasan, K.; Maloney, J.A.; Atwal, J.K.; Sa, S.M.; Yaylaoglu, M.B.; Foreman, O.; Ortmann, W.; et al. A rare mutation in UNC5C predisposes to late-onset Alzheimer’s disease and increases neuronal cell death. Nat. Med. 2014, 20, 1452–1457. [Google Scholar] [CrossRef]
- Logue, M.W.; Schu, M.; Vardarajan, B.N.; Farrell, J.; Bennett, D.A.; Buxbaum, J.D.; Byrd, G.S.; Ertekin-Taner, N.; Evans, D.; Foroud, T.; et al. Two rare AKAP9 variants are associated with Alzheimer’s disease in African Americans. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2014, 10, 609–618.e11. [Google Scholar] [CrossRef]
- Namba, Y.; Tomonaga, M.; Kawasaki, H.; Otomo, E.; Ikeda, K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991, 541, 163–166. [Google Scholar] [CrossRef]
- Holtzman, D.M. In vivo effects of ApoE and clusterin on amyloid-beta metabolism and neuropathology. J. Mol. Neurosci. 2004, 23, 247–254. [Google Scholar] [CrossRef]
- Ertekin-Taner, N. Genetics of Alzheimer’s disease: A centennial review. Neurol. Clin. 2007, 25, 611–667. [Google Scholar] [CrossRef] [PubMed]
- Sachidanandam, R.; Weissman, D.; Schmidt, S.C.; Kakol, J.M.; Stein, L.D.; Marth, G.; Sherry, S.; Mullikin, J.C.; Mortimore, B.J.; Willey, D.L.; et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 2001, 409, 928–933. [Google Scholar] [PubMed]
- Alzforum. AlzGene—Field Synopsis of Genetic Association Studies in AD. Available online: http://www.alzgene.org/ (accessed on 12 November 2018).
- Bertram, L.; McQueen, M.B.; Mullin, K.; Blacker, D.; Tanzi, R.E. Systematic meta-analyses of Alzheimer disease genetic association studies: The AlzGene database. Nat. Genet. 2007, 39, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Rogaeva, E.; Meng, Y.; Lee, J.H.; Gu, Y.; Kawarai, T.; Zou, F.; Katayama, T.; Baldwin, C.T.; Cheng, R.; Hasegawa, H.; et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet. 2007, 39, 168–177. [Google Scholar] [CrossRef] [Green Version]
- Dreses-Werringloer, U.; Lambert, J.C.; Vingtdeux, V.; Zhao, H.; Vais, H.; Siebert, A.; Jain, A.; Koppel, J.; Rovelet-Lecrux, A.; Hannequin, D.; et al. A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer’s disease risk. Cell 2008, 133, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Lohmueller, K.E.; Pearce, C.L.; Pike, M.; Lander, E.S.; Hirschhorn, J.N. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat. Genet. 2003, 33, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Newton-Cheh, C.; Hirschhorn, J.N. Genetic association studies of complex traits: Design and analysis issues. Mutat. Res. 2005, 573, 54–69. [Google Scholar] [CrossRef]
- Ertekin-Taner, N. Genetics of Alzheimer disease in the pre- and post-GWAS era. Alzheimer’s Res. Ther. 2010, 2, 3. [Google Scholar] [CrossRef]
- Grupe, A.; Abraham, R.; Li, Y.; Rowland, C.; Hollingworth, P.; Morgan, A.; Jehu, L.; Segurado, R.; Stone, D.; Schadt, E.; et al. Evidence for novel susceptibility genes for late-onset Alzheimer’s disease from a genome-wide association study of putative functional variants. Hum. Mol. Genet. 2007, 16, 865–873. [Google Scholar] [CrossRef]
- Feulner, T.M.; Laws, S.M.; Friedrich, P.; Wagenpfeil, S.; Wurst, S.H.; Riehle, C.; Kuhn, K.A.; Krawczak, M.; Schreiber, S.; Nikolaus, S.; et al. Examination of the current top candidate genes for AD in a genome-wide association study. Mol. Psychiatry 2010, 15, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Webster, J.A.; Myers, A.J.; Hardy, J.; Dunckley, T.; Zismann, V.L.; Joshipura, K.D.; Pearson, J.V.; Hu-Lince, D.; Huentelman, M.J.; et al. GAB2 alleles modify Alzheimer’s risk in APOE epsilon4 carriers. Neuron 2007, 54, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wetten, S.; Li, L.; St Jean, P.L.; Upmanyu, R.; Surh, L.; Hosford, D.; Barnes, M.R.; Briley, J.D.; Borrie, M.; et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch. Neurol. 2008, 65, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.; Moskvina, V.; Sims, R.; Hollingworth, P.; Morgan, A.; Georgieva, L.; Dowzell, K.; Cichon, S.; Hillmer, A.M.; O’Donovan, M.C.; et al. A genome-wide association study for late-onset Alzheimer’s disease using DNA pooling. BMC Med. Genomics 2008, 1, 44. [Google Scholar] [CrossRef] [PubMed]
- Carrasquillo, M.M.; Zou, F.; Pankratz, V.S.; Wilcox, S.L.; Ma, L.; Walker, L.P.; Younkin, S.G.; Younkin, C.S.; Younkin, L.H.; Bisceglio, G.D.; et al. Genetic variation in PCDH11X is associated with susceptibility to late-onset Alzheimer’s disease. Nat. Genet. 2009, 41, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Poduslo, S.E.; Huang, R.; Huang, J.; Smith, S. Genome screen of late-onset Alzheimer’s extended pedigrees identifies TRPC4AP by haplotype analysis. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2009, 150, 50–55. [Google Scholar] [CrossRef]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- Coon, K.D.; Myers, A.J.; Craig, D.W.; Webster, J.A.; Pearson, J.V.; Lince, D.H.; Zismann, V.L.; Beach, T.G.; Leung, D.; Bryden, L.; et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J. Clin. Psychiatry 2007, 68, 613–618. [Google Scholar] [CrossRef]
- Genin, E.; Hannequin, D.; Wallon, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Bullido, M.J.; Engelborghs, S.; De Deyn, P.; Berr, C.; et al. APOE and Alzheimer disease: A major gene with semi-dominant inheritance. Mol. Psychiatry 2011, 16, 903–907. [Google Scholar] [CrossRef]
- Chen, S.; Parmigiani, G. Meta-analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol. 2007, 25, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Hadano, S.; Hand, C.K.; Osuga, H.; Yanagisawa, Y.; Otomo, A.; Devon, R.S.; Miyamoto, N.; Showguchi-Miyata, J.; Okada, Y.; Singaraja, R.; et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 2001, 29, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Burgunder, J.M.; Schols, L.; Baets, J.; Andersen, P.; Gasser, T.; Szolnoki, Z.; Fontaine, B.; Van Broeckhoven, C.; Di Donato, S.; De Jonghe, P.; et al. EFNS guidelines for the molecular diagnosis of neurogenetic disorders: Motoneuron, peripheral nerve and muscle disorders. Eur. J. Neurol. 2011, 18, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.; Henders, A.K.; Nyholt, D.R.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W.; et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Manolio, T.A.; Pasquale, L.R.; Boerwinkle, E.; Caporaso, N.; Cunningham, J.M.; de Andrade, M.; Feenstra, B.; Feingold, E.; Hayes, M.G.; et al. Genome partitioning of genetic variation for complex traits using common SNPs. Nat. Genet. 2011, 43, 519–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. Genome-wide complex trait analysis (GCTA): Methods, data analyses, and interpretations. Methods Mol. Biol. 2013, 1019, 215–236. [Google Scholar] [PubMed]
- Al-Chalabi, A.; Fang, F.; Hanby, M.F.; Leigh, P.N.; Shaw, C.E.; Ye, W.; Rijsdijk, F. An estimate of amyotrophic lateral sclerosis heritability using twin data. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1324–1326. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef]
- Byrne, S.; Hardiman, O. Familial aggregation in amyotrophic lateral sclerosis. Ann. Neurol. 2010, 67, 554. [Google Scholar] [CrossRef]
- Huisman, M.H.; de Jong, S.W.; Verwijs, M.C.; Schelhaas, H.J.; van der Kooi, A.J.; de Visser, M.; Veldink, J.H.; van den Berg, L.H. Family history of neurodegenerative and vascular diseases in ALS: A population-based study. Neurology 2011, 77, 1363–1369. [Google Scholar] [CrossRef]
- Johnston, C.A.; Stanton, B.R.; Turner, M.R.; Gray, R.; Blunt, A.H.; Butt, D.; Ampong, M.A.; Shaw, C.E.; Leigh, P.N.; Al-Chalabi, A. Amyotrophic lateral sclerosis in an urban setting: A population based study of inner city London. J. Neurol. 2006, 253, 1642–1643. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Jones, A.; Troakes, C.; King, A.; Al-Sarraj, S.; van den Berg, L.H. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 2012, 124, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.F.; Ferrucci, L.; Singleton, A.B.; Tienari, P.J.; Laaksovirta, H.; Restagno, G.; Chio, A.; Traynor, B.J.; Nalls, M.A. Genome-wide analysis of the heritability of amyotrophic lateral sclerosis. JAMA Neurol. 2014, 71, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Carson, M.; Smith, C.D.; Koppenol, W.H. ALS, SOD and peroxynitrite. Nature 1993, 364, 584. [Google Scholar] [CrossRef]
- Bosco, D.A.; Morfini, G.; Karabacak, N.M.; Song, Y.; Gros-Louis, F.; Pasinelli, P.; Goolsby, H.; Fontaine, B.A.; Lemay, N.; McKenna-Yasek, D.; et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 2010, 13, 1396–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crow, J.P.; Sampson, J.B.; Zhuang, Y.; Thompson, J.A.; Beckman, J.S. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J. Neurochem. 1997, 69, 1936–1944. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.M.; Jung, C.; Ding, H.; Xu, Z. Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J. Neurosci. 2002, 22, RC215. [Google Scholar] [CrossRef] [PubMed]
- Ligon, L.A.; LaMonte, B.H.; Wallace, K.E.; Weber, N.; Kalb, R.G.; Holzbaur, E.L. Mutant superoxide dismutase disrupts cytoplasmic dynein in motor neurons. Neuroreport 2005, 16, 533–536. [Google Scholar] [CrossRef]
- Spreux-Varoquaux, O.; Bensimon, G.; Lacomblez, L.; Salachas, F.; Pradat, P.F.; Le Forestier, N.; Marouan, A.; Dib, M.; Meininger, V. Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis: A reappraisal using a new HPLC method with coulometric detection in a large cohort of patients. J. Neurol. Sci. 2002, 193, 73–78. [Google Scholar] [CrossRef]
- Wiedau-Pazos, M.; Goto, J.J.; Rabizadeh, S.; Gralla, E.B.; Roe, J.A.; Lee, M.K.; Valentine, J.S.; Bredesen, D.E. Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis. Science 1996, 271, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Van Deerlin, V.M.; Leverenz, J.B.; Bekris, L.M.; Bird, T.D.; Yuan, W.; Elman, L.B.; Clay, D.; Wood, E.M.; Chen-Plotkin, A.S.; Martinez-Lage, M.; et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: A genetic and histopathological analysis. Lancet Neurol. 2008, 7, 409–416. [Google Scholar] [CrossRef]
- Yokoseki, A.; Shiga, A.; Tan, C.F.; Tagawa, A.; Kaneko, H.; Koyama, A.; Eguchi, H.; Tsujino, A.; Ikeuchi, T.; Kakita, A.; et al. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann. Neurol. 2008, 63, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lansbury, P.T.; Lashuel, H.A. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature 2006, 443, 774–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leigh, P.N.; Whitwell, H.; Garofalo, O.; Buller, J.; Swash, M.; Martin, J.E.; Gallo, J.M.; Weller, R.O.; Anderton, B.H. Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain A J. Neurol. 1991, 114, 775–788. [Google Scholar] [CrossRef]
- Ticozzi, N.; Vance, C.; Leclerc, A.L.; Keagle, P.; Glass, J.D.; McKenna-Yasek, D.; Sapp, P.C.; Silani, V.; Bosco, D.A.; Shaw, C.E.; et al. Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet. 2011, 156B, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Lill, C.M.; Abel, O.; Bertram, L.; Al-Chalabi, A. Keeping up with genetic discoveries in amyotrophic lateral sclerosis: The ALSoD and ALSGene databases. Amyotroph. Lateral Scler. Off. Publ. World Fed. Neurol. Res. Group Motor Neuron Dis. 2011, 12, 238–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodall, E.F.; Greenway, M.J.; van Marion, I.; Carroll, C.B.; Hardiman, O.; Morrison, K.E. Association of the H63D polymorphism in the hemochromatosis gene with sporadic ALS. Neurology 2005, 65, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Sutedja, N.A.; Sinke, R.J.; Van Vught, P.W.; Van der Linden, M.W.; Wokke, J.H.; Van Duijn, C.M.; Njajou, O.T.; Van der Schouw, Y.T.; Veldink, J.H.; Van den Berg, L.H. The association between H63D mutations in HFE and amyotrophic lateral sclerosis in a Dutch population. Arch. Neurol. 2007, 64, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Praline, J.; Blasco, H.; Vourc’h, P.; Rat, V.; Gendrot, C.; Camu, W.; Andres, C.R. Study of the HFE gene common polymorphisms in French patients with sporadic amyotrophic lateral sclerosis. J. Neurol. Sci. 2012, 317, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Greenway, M.J.; Andersen, P.M.; Russ, C.; Ennis, S.; Cashman, S.; Donaghy, C.; Patterson, V.; Swingler, R.; Kieran, D.; Prehn, J.; et al. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat. Genet. 2006, 38, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Rooke, K.; Figlewicz, D.A.; Han, F.Y.; Rouleau, G.A. Analysis of the KSP repeat of the neurofilament heavy subunit in familiar amyotrophic lateral sclerosis. Neurology 1996, 46, 789–790. [Google Scholar] [CrossRef] [PubMed]
- Couthouis, J.; Hart, M.P.; Erion, R.; King, O.D.; Diaz, Z.; Nakaya, T.; Ibrahim, F.; Kim, H.J.; Mojsilovic-Petrovic, J.; Panossian, S.; et al. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2012, 21, 2899–2911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couthouis, J.; Hart, M.P.; Shorter, J.; DeJesus-Hernandez, M.; Erion, R.; Oristano, R.; Liu, A.X.; Ramos, D.; Jethava, N.; Hosangadi, D.; et al. A yeast functional screen predicts new candidate ALS disease genes. Proc. Natl. Acad. Sci. USA 2011, 108, 20881–20890. [Google Scholar] [CrossRef] [Green Version]
- Shatunov, A.; Mok, K.; Newhouse, S.; Weale, M.E.; Smith, B.; Vance, C.; Johnson, L.; Veldink, J.H.; van Es, M.A.; van den Berg, L.H.; et al. Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: A genome-wide association study. Lancet Neurol. 2010, 9, 986–994. [Google Scholar] [CrossRef]
- van Rheenen, W.; Shatunov, A.; Dekker, A.M.; McLaughlin, R.L.; Diekstra, F.P.; Pulit, S.L.; van der Spek, R.A.; Vosa, U.; de Jong, S.; Robinson, M.R.; et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1043–1048. [Google Scholar] [CrossRef] [Green Version]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef] [Green Version]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef]
- Vucic, S.; Kiernan, M.C. Cortical excitability testing distinguishes Kennedy’s disease from amyotrophic lateral sclerosis. Clin. Neurophysiol. 2008, 119, 1088–1096. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Martin, L.J.; Kuncl, R.W. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 1992, 326, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Sengillo, J.D.; Sagare, A.P.; Zhao, Z.; Ma, Q.; Zuniga, E.; Wang, Y.; Zhong, Z.; Sullivan, J.S.; Griffin, J.H.; et al. Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc. Natl. Acad. Sci. USA 2014, 111, E1035–E1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Lillo, C.; Jonsson, P.A.; Vande Velde, C.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M.; et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Vande Velde, C.; Miller, T.M.; Cashman, N.R.; Cleveland, D.W. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 4022–4027. [Google Scholar] [CrossRef]
- Sathasivam, S.; Grierson, A.J.; Shaw, P.J. Characterization of the caspase cascade in a cell culture model of SOD1-related familial amyotrophic lateral sclerosis: Expression, activation and therapeutic effects of inhibition. Neuropathol. Appl. Neurobiol. 2005, 31, 467–485. [Google Scholar] [CrossRef]
- Sathasivam, S.; Shaw, P.J. Apoptosis in amyotrophic lateral sclerosis--what is the evidence? Lancet Neurol. 2005, 4, 500–509. [Google Scholar] [CrossRef]
- Wiedemann, F.R.; Manfredi, G.; Mawrin, C.; Beal, M.F.; Schon, E.A. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem. 2002, 80, 616–625. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, D.; Sargsyan, S.; Monk, P.N.; Shaw, P.J. Astrocyte function and role in motor neuron disease: A future therapeutic target? Glia 2009, 57, 1251–1264. [Google Scholar] [CrossRef]
- Duffy, L.M.; Chapman, A.L.; Shaw, P.J.; Grierson, A.J. Review: The role of mitochondria in the pathogenesis of amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 2011, 37, 336–352. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.D.; Weiss, J.H. Excitotoxic and oxidative cross-talk between motor neurons and glia in ALS pathogenesis. Trends Neurosci. 2004, 27, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, S.A.; Monk, P.N.; Shaw, P.J. Microglia as potential contributors to motor neuron injury in amyotrophic lateral sclerosis. Glia 2005, 51, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef]
- Piao, Y.S.; Wakabayashi, K.; Kakita, A.; Yamada, M.; Hayashi, S.; Morita, T.; Ikuta, F.; Oyanagi, K.; Takahashi, H. Neuropathology with clinical correlations of sporadic amyotrophic lateral sclerosis: 102 autopsy cases examined between 1962 and 2000. Brain Pathol. 2003, 13, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Hooten, K.G.; Beers, D.R.; Zhao, W.; Appel, S.H. Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis. Neurother. J. Am. Soc. Exp. Neurother. 2015, 12, 364–375. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, H.; Elgueta, D.; Montoya, A.; Pacheco, R. Neuroimmune regulation of microglial activity involved in neuroinflammation and neurodegenerative diseases. J. Neuroimmunol. 2014, 274, 1–13. [Google Scholar] [CrossRef]
- More, S.V.; Kumar, H.; Kim, I.S.; Song, S.Y.; Choi, D.K. Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson’s disease. Mediat. Inflamm. 2013, 2013, 952375. [Google Scholar] [CrossRef]
- Gendelman, H.E.; Appel, S.H. Neuroprotective activities of regulatory T cells. Trends Mol. Med. 2011, 17, 687–688. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, A.; Kenna, K.P.; Renton, A.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.; Kenna, B.; Nalls, M.A.; Keagle, P.; et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1268–1283. [Google Scholar] [CrossRef]
- Diekstra, F.P.; van Vught, P.W.; van Rheenen, W.; Koppers, M.; Pasterkamp, R.J.; van Es, M.A.; Schelhaas, H.J.; de Visser, M.; Robberecht, W.; Van Damme, P.; et al. UNC13A is a modifier of survival in amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 630. [Google Scholar] [CrossRef] [PubMed]
- Fogh, I.; Lin, K.; Tiloca, C.; Rooney, J.; Gellera, C.; Diekstra, F.P.; Ratti, A.; Shatunov, A.; van Es, M.A.; Proitsi, P.; et al. Association of a Locus in the CAMTA1 Gene with Survival in Patients with Sporadic Amyotrophic Lateral Sclerosis. JAMA Neurol. 2016, 73, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Lill, C.M.; Rengmark, A.; Pihlstrom, L.; Fogh, I.; Shatunov, A.; Sleiman, P.M.; Wang, L.S.; Liu, T.; Lassen, C.F.; Meissner, E.; et al. The role of TREM2 R47H as a risk factor for Alzheimer’s disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2015, 11, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Holm, I.E.; Alstrup, A.K.; Luo, Y. Genetically modified pig models for neurodegenerative disorders. J. Pathol. 2016, 238, 267–287. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Tieu, K. A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harbor Perspect. Med. 2011, 1, a009316. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, K.; Holm, I.E.; Zimmer, J. Tyrosine hydroxylase and acetylcholinesterase in the domestic pig mesencephalon: An immunocytochemical and histochemical study. J. Comp. Neurol. 1992, 322, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.H.; Kim, J.H.; Im, H.J.; Lee, D.S.; Park, E.J.; Song, K.; Oh, H.J.; Hyun, S.B.; Kang, S.C.; Kim, H.; et al. Proposed Motor Scoring System in a Porcine Model of Parkinson’s Disease induced by Chronic Subcutaneous Injection of MPTP. Exp. Neurobiol. 2014, 23, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Larsen, K.; Bendixen, C. Characterization of the porcine FBX07 gene: The first step towards generation of a pig model for Parkinsonian pyramidal syndrome. Mol. Biol. Rep. 2012, 39, 1517–1526. [Google Scholar] [CrossRef]
- Larsen, K.; Madsen, L.B.; Farajzadeh, L.; Bendixen, C. Splicing variants of porcine synphilin-1. Meta Gene 2015, 5, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Lucking, C.B.; Durr, A.; Bonifati, V.; Vaughan, J.; De Michele, G.; Gasser, T.; Harhangi, B.S.; Meco, G.; Denefle, P.; Wood, N.W.; et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N. Engl. J. Med. 2000, 342, 1560–1567. [Google Scholar] [CrossRef] [PubMed]
- Pankratz, N.; Pauciulo, M.W.; Elsaesser, V.E.; Marek, D.K.; Halter, C.A.; Wojcieszek, J.; Rudolph, A.; Shults, C.W.; Foroud, T.; Nichols, W.C. Mutations in DJ-1 are rare in familial Parkinson disease. Neurosci. Lett. 2006, 408, 209–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chartier-Harlin, M.C.; Dachsel, J.C.; Vilarino-Guell, C.; Lincoln, S.J.; Lepretre, F.; Hulihan, M.M.; Kachergus, J.; Milnerwood, A.J.; Tapia, L.; Song, M.S.; et al. Translation initiator EIF4G1 mutations in familial Parkinson disease. Am. J. Hum. Genet. 2011, 89, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Benet-Pages, A.; Struhal, W.; Graf, E.; Eck, S.H.; Offman, M.N.; Haubenberger, D.; Spielberger, S.; Schulte, E.C.; Lichtner, P.; et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 2011, 89, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Vilarino-Guell, C.; Wider, C.; Ross, O.A.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 2011, 89, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Foo, J.N.; Liu, J.; Tan, E.K. Next-generation sequencing diagnostics for neurological diseases/disorders: From a clinical perspective. Hum. Genet. 2013, 132, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Houlden, H.; Singleton, A.B. The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol. 2012, 124, 325–338. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Singleton, A.; Gwinn-Hardy, K. Parkinson’s disease and dementia with Lewy bodies: A difference in dose? Lancet 2004, 364, 1105–1107. [Google Scholar] [CrossRef]
- Shimura, H.; Hattori, N.; Kubo, S.; Yoshikawa, M.; Kitada, T.; Matsumine, H.; Asakawa, S.; Minoshima, S.; Yamamura, Y.; Shimizu, N.; et al. Immunohistochemical and subcellular localization of Parkin protein: Absence of protein in autosomal recessive juvenile parkinsonism patients. Ann. Neurol. 1999, 45, 668–672. [Google Scholar] [CrossRef]
- Nagakubo, D.; Taira, T.; Kitaura, H.; Ikeda, M.; Tamai, K.; Iguchi-Ariga, S.M.; Ariga, H. DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem. Biophys. Res. Commun. 1997, 231, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Unoki, M.; Nakamura, Y. Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene 2001, 20, 4457–4465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, M.; Corey, D.R. Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 2017, 16, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Davidovich, C.; Cech, T.R. The recruitment of chromatin modifiers by long noncoding RNAs: Lessons from PRC2. RNA 2015, 21, 2007–2022. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Sun, B.K.; Erwin, J.A.; Song, J.J.; Lee, J.T. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 2008, 322, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Leucci, E.; Vendramin, R.; Spinazzi, M.; Laurette, P.; Fiers, M.; Wouters, J.; Radaelli, E.; Eyckerman, S.; Leonelli, C.; Vanderheyden, K.; et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature 2016, 531, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Person, R.E.; Beaudet, A.L. Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum. Mol. Genet. 2012, 21, 3001–3012. [Google Scholar] [CrossRef]
- Eissmann, M.; Gutschner, T.; Hammerle, M.; Gunther, S.; Caudron-Herger, M.; Gross, M.; Schirmacher, P.; Rippe, K.; Braun, T.; Zornig, M.; et al. Loss of the abundant nuclear non-coding RNA MALAT1 is compatible with life and development. RNA Biol. 2012, 9, 1076–1087. [Google Scholar] [CrossRef]
- Wang, D.Q.; Fu, P.; Yao, C.; Zhu, L.S.; Hou, T.Y.; Chen, J.G.; Lu, Y.; Liu, D.; Zhu, L.Q. Long Non-coding RNAs, Novel Culprits, or Bodyguards in Neurodegenerative Diseases. Mol. Ther. Nucleic Acids 2018, 10, 269–276. [Google Scholar] [CrossRef]
- Yohe, S.; Thyagarajan, B. Review of Clinical Next-Generation Sequencing. Arch. Pathol. Lab. Med. 2017, 141, 1544–1557. [Google Scholar] [CrossRef]
- Zhu, W.; Zhang, X.Y.; Marjani, S.L.; Zhang, J.; Zhang, W.; Wu, S.; Pan, X. Next-generation molecular diagnosis: Single-cell sequencing from bench to bedside. Cell. Mol. Life Sci. 2017, 74, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.C.; Liu, X. Toward Best Practice in Using Molecular Diagnosis to Guide Medical Management, Are We There Yet? N. Am. J. Med. Sci. 2014, 7, 199–200. [Google Scholar]
- Andersen, P.M.; Abrahams, S.; Borasio, G.D.; de Carvalho, M.; Chio, A.; Van Damme, P.; Hardiman, O.; Kollewe, K.; Morrison, K.E.; Petri, S.; et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)—revised report of an EFNS task force. Eur. J. Neurol. 2012, 19, 360–375. [Google Scholar] [PubMed]


{kind=link}
{kind=link}
| WES | WGS |
|---|---|
| Only done in encoding regions (exons) | Complete sequence: exons and introns |
| Cheap and fast | Currently expensive |
| Incomplete analysis of the target region | Technology-related challenges: an enormous amount of data is produced |
| Inclined towards known biology (medically-relevant genes) | Increased precision providing information about position and orientation |
| Gene | Symbol | Inheritance | Location | Risk (%) |
|---|---|---|---|---|
| Amyloid precursor protein | APP | Autosomal dominant | 21q21.3 | 38–69 |
| Presenilin 1 | PSEN1 | Autosomal dominant | 14q24.2 | 25–65 |
| Presenilin 2 | PSEN2 | Autosomal dominant, reduced penetrance | 1q42.13 | 41–88 |
| Mechanism | Mutated Genes |
|---|---|
| Dynamics of the cytoskeleton | PFN1, TUBA4A, DCTN1, and KIF5A |
| RNA processing | C9orf72, TDP-43, FUS, and MATR3 |
| Protein homeostasis | UBQLN2, VCP, OPTN, and VAPB |
| Protein | Gene | Function | Location | Risk (%) |
|---|---|---|---|---|
| Parkin [144] | PARK2 | Ubiquitin proteasome system | Cortex, hippocampus, basal ganglions and cerebellum | 10% of early onset PD |
| Deglycase protein (DJ1) [145] | PARK7 | Controlling cell cycle and oncogenesis | Basal nucleus neurons and astrocytes | Rare, early onset |
| PTEN 1-induced kinase [146] | PARK6 | Neuroprotector function: mitochondria-dependent cell apoptosis | Distributed throughout different tissue | Rare, familial, appearing during the 40s and 50s |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García, J.-C.; Bustos, R.-H. The Genetic Diagnosis of Neurodegenerative Diseases and Therapeutic Perspectives. Brain Sci. 2018, 8, 222. https://doi.org/10.3390/brainsci8120222
García J-C, Bustos R-H. The Genetic Diagnosis of Neurodegenerative Diseases and Therapeutic Perspectives. Brain Sciences. 2018; 8(12):222. https://doi.org/10.3390/brainsci8120222
Chicago/Turabian StyleGarcía, Julio-César, and Rosa-Helena Bustos. 2018. "The Genetic Diagnosis of Neurodegenerative Diseases and Therapeutic Perspectives" Brain Sciences 8, no. 12: 222. https://doi.org/10.3390/brainsci8120222