Striatal Neurodegeneration that Mimics Huntington’s Disease Modifies GABA-induced Currents


Abstract
:1. Introduction
2. Material and Methods
2.1. Animals
2.2. Reagents
2.3. Slice Preparation
2.4. Whole-Cell Patch-Clamp Technique
2.5. Drug Application
2.6. Electrophysiology Protocols
2.7. Statistical Analysis
3. Results
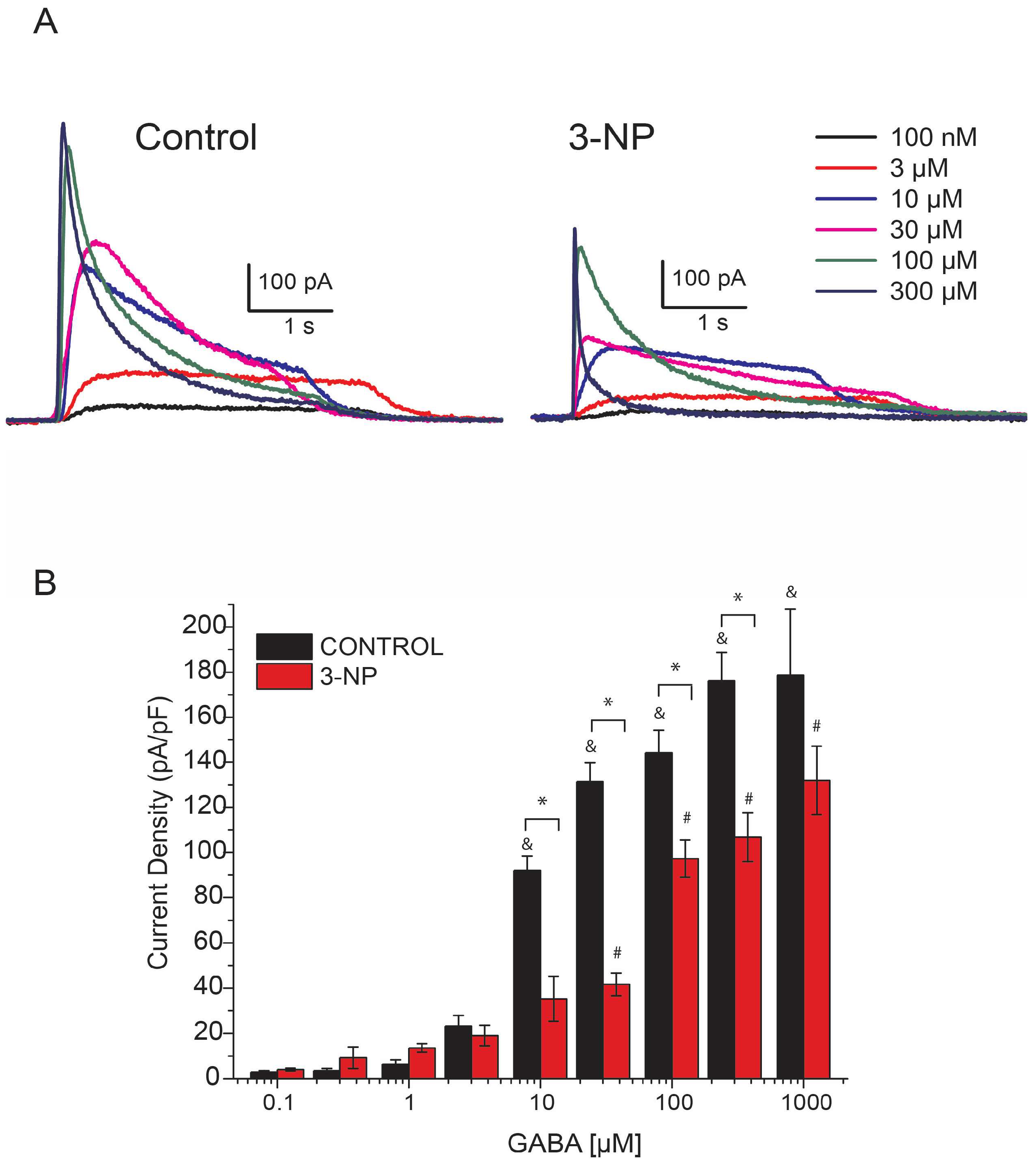
3.1. Recording IGABA
Equilibrium Potential for IGABA
3.2. GABA-Activated Currents (IGABA)
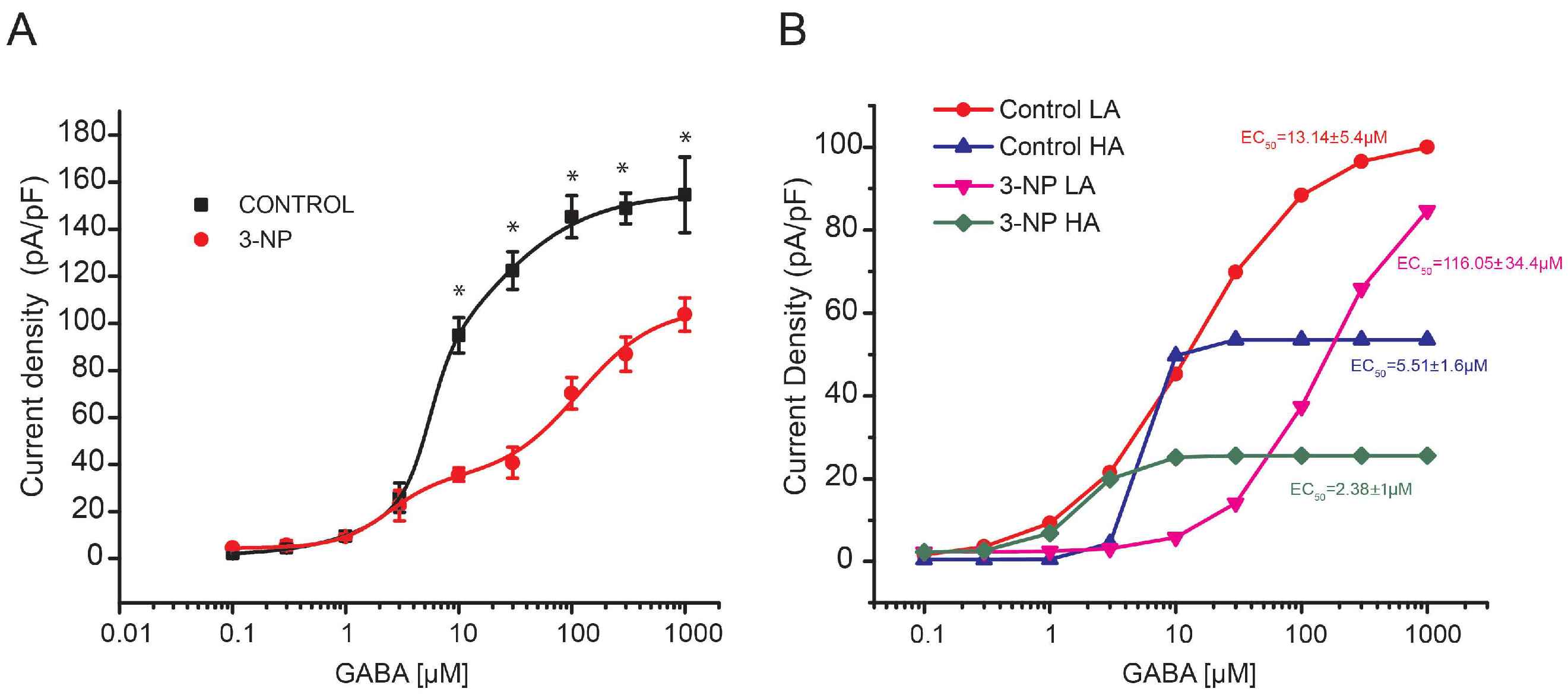
GABA Dose-Response Curve in Medium Spiny Neurons
3.3. GABA Dose-Response Curves in Giant Interneurons
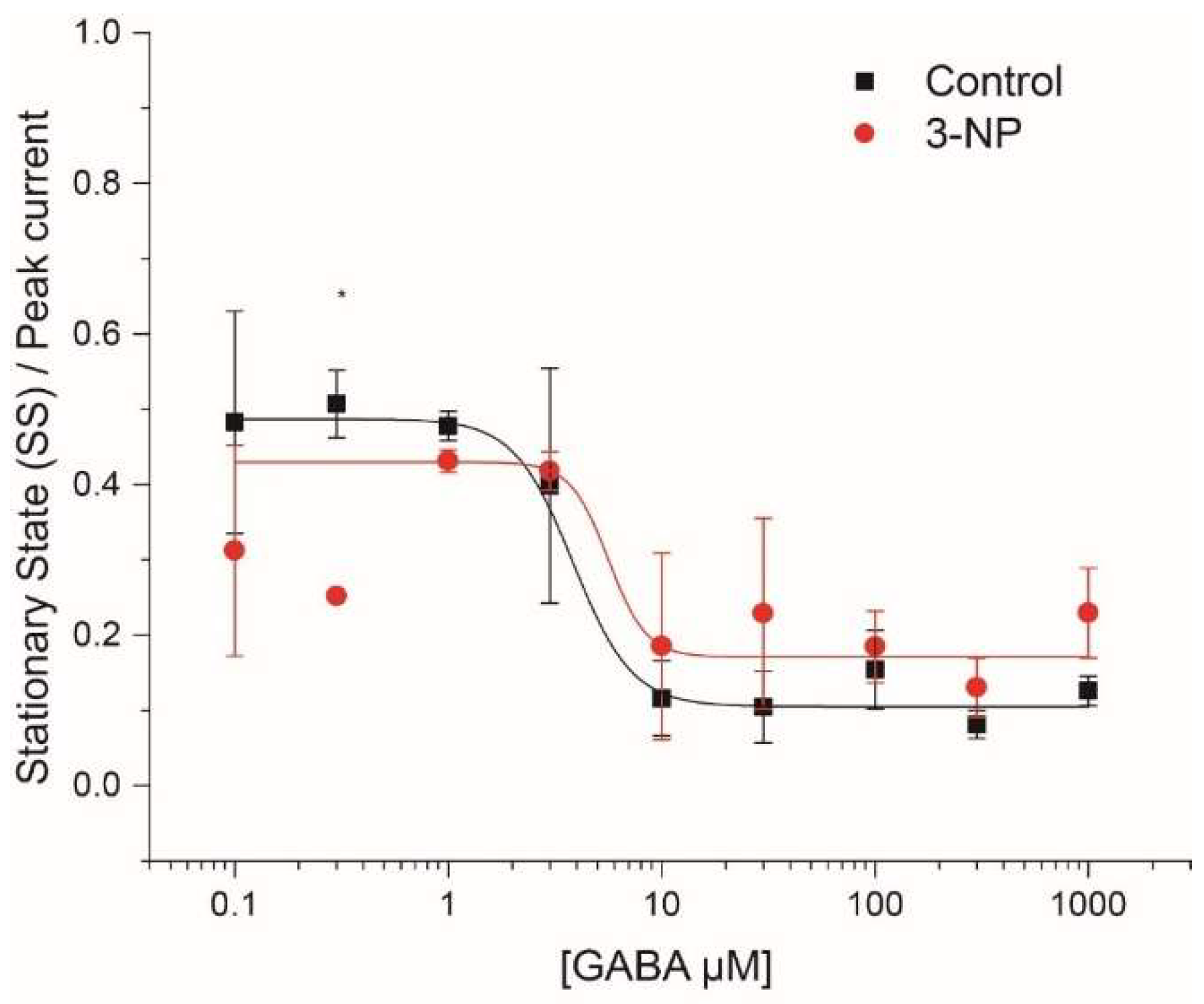
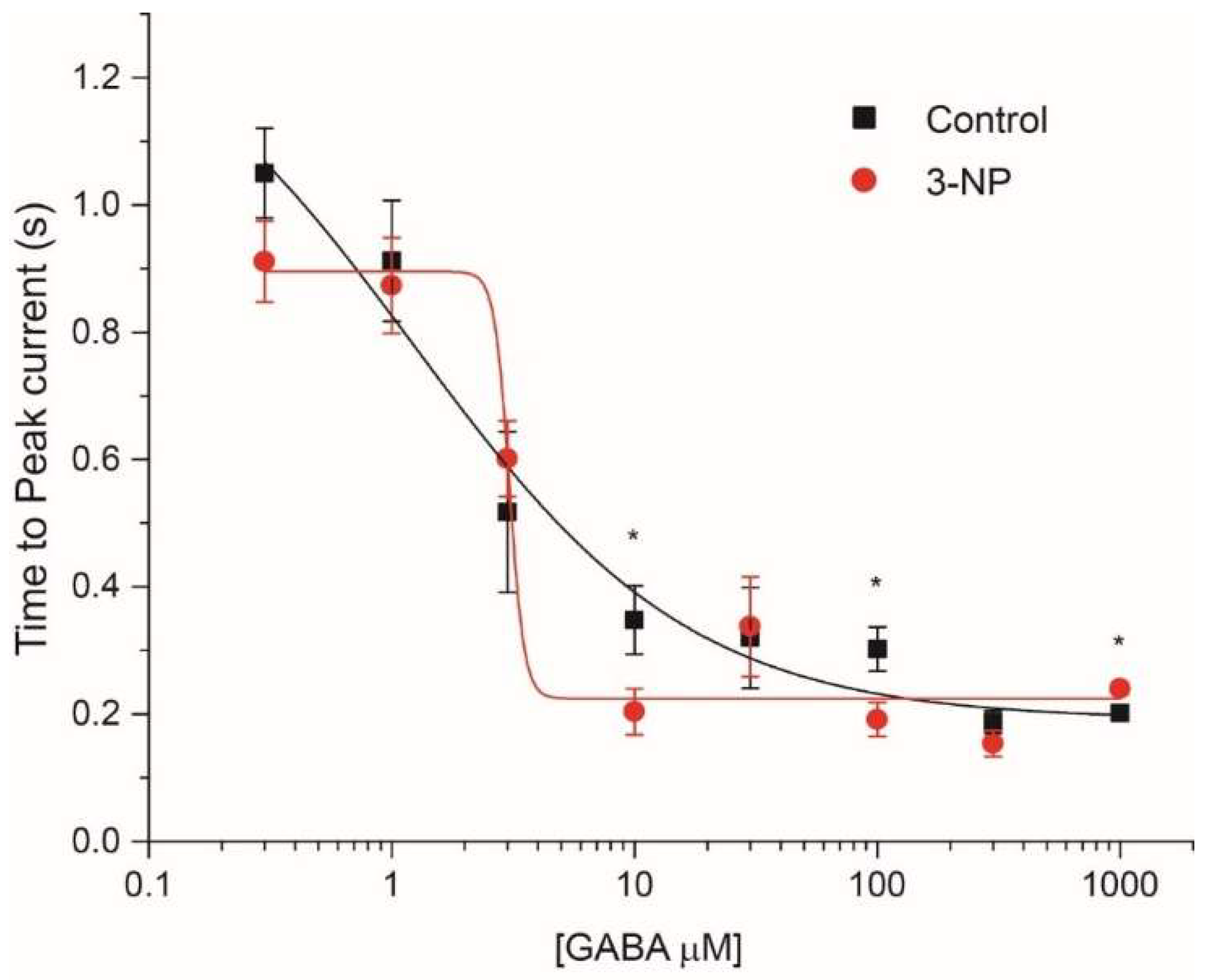
3.4. Kinetics of the IGABA
3.4.1. Steady-State (SS)/Peak Current
3.4.2. Time to Peak
3.4.3. Time Constant of Desensitization (tau,τ)
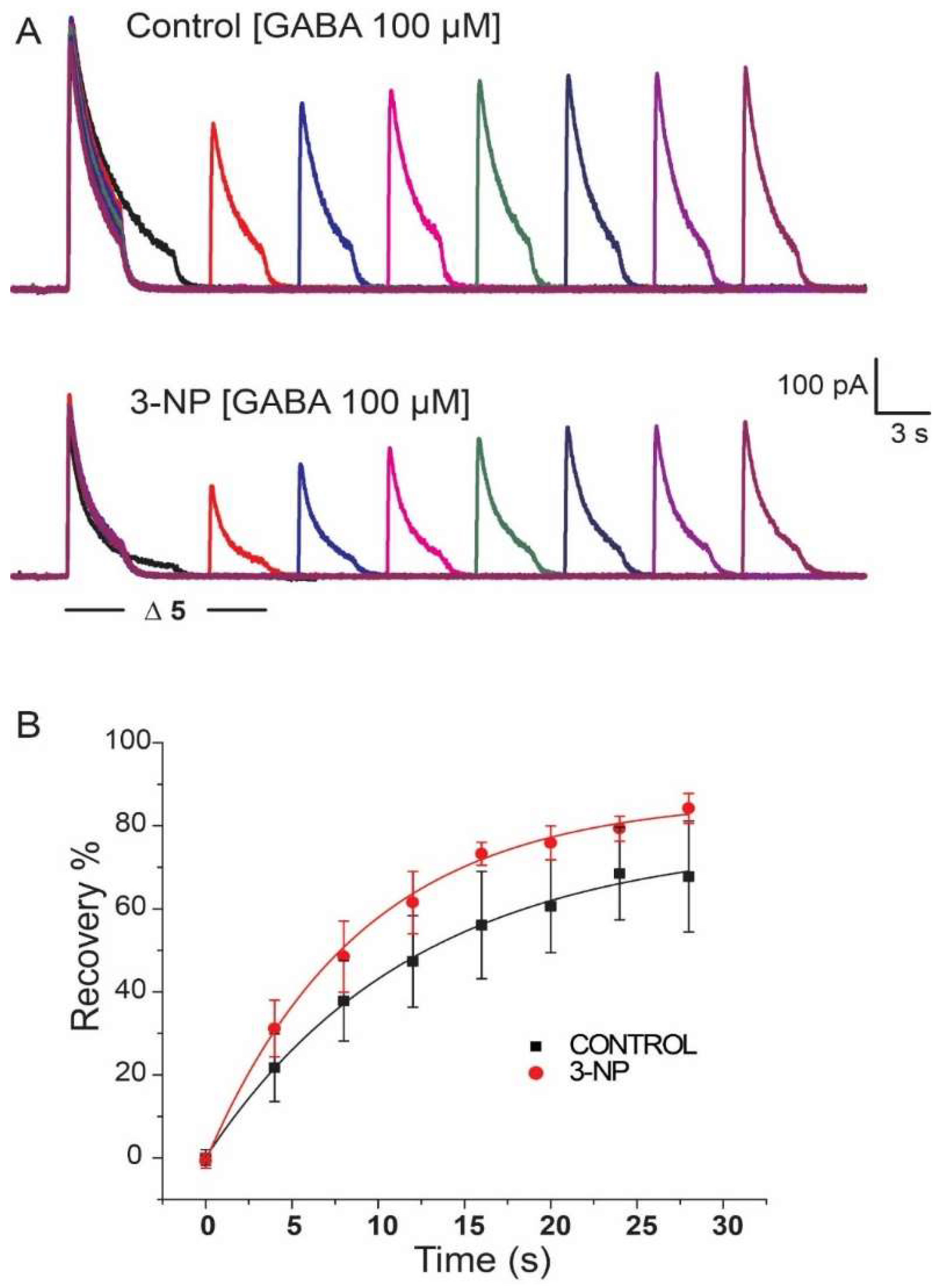
3.5. Recovery of IGABA
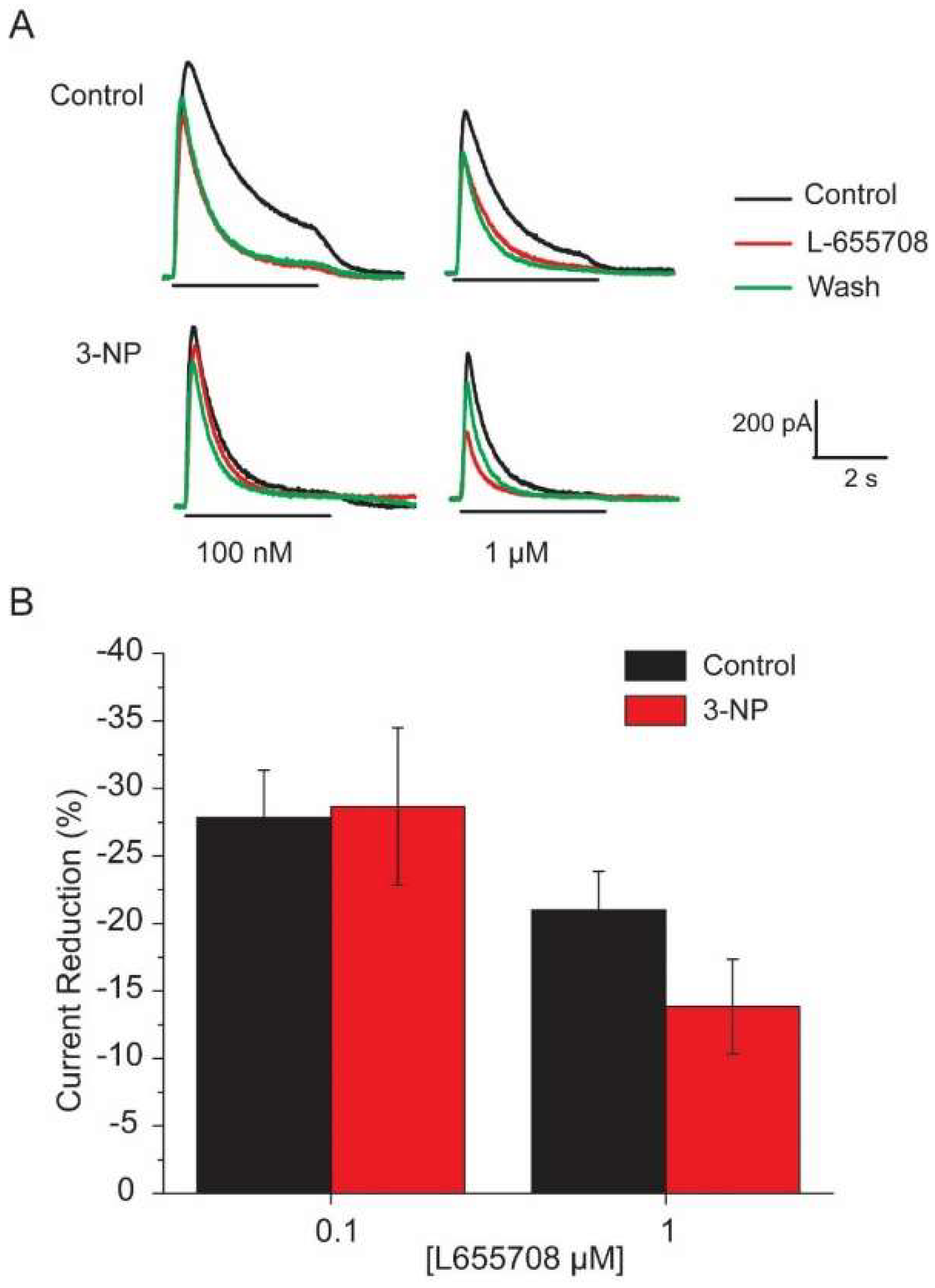
3.6. Effects of the α5 Subunit Inverse Agonist L-655708
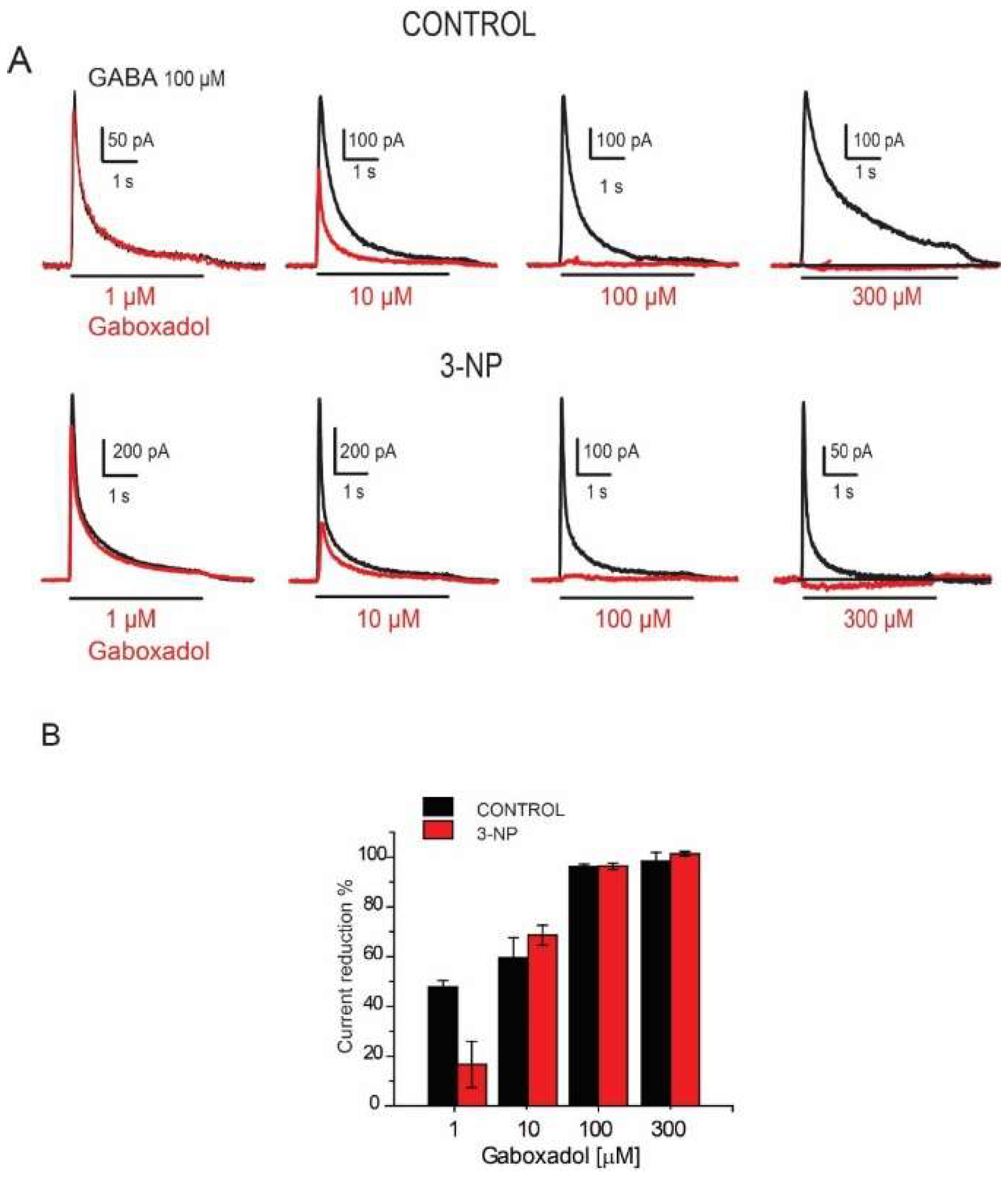
3.7. Effects of the Gaboxadol
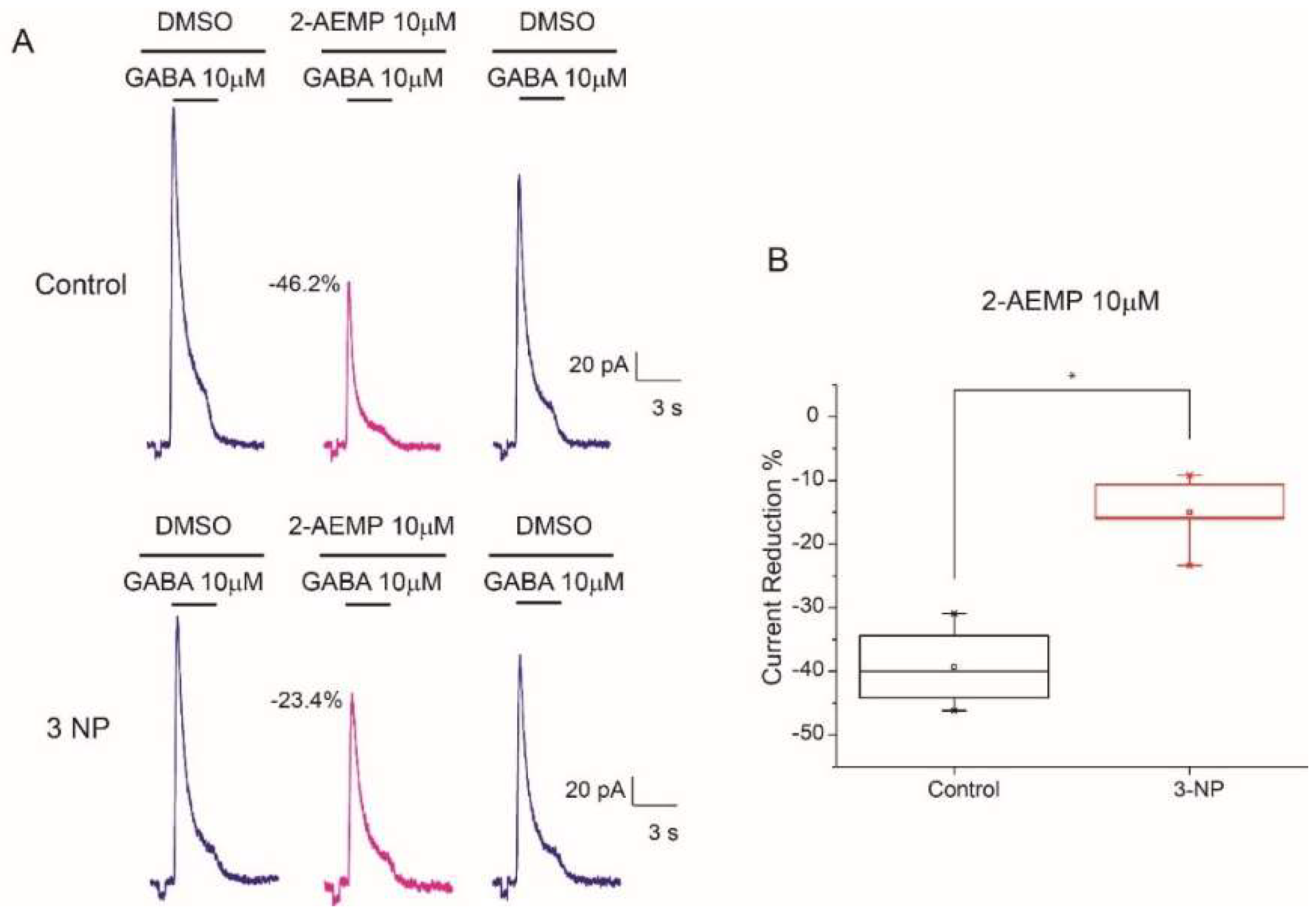
3.8. GABAC Receptors Reduction in 3-NP Group
4. Discussion
4.1. MSN Capacitance Decreases as a Result of 3-NP
4.2. Recording IGABA
4.3. Effect of L-655708
4.4. Effect of Gaboxadol
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Browne, S.E.; Howling, A.C.; MacGarvey, U.; Balk, M.; Bcrgvr, S.C.; Muqit, M.M.; Bird, E.D.; Beal, M.F. Oxidative damage and metabolic dysfunction in Huntington’s disease: Selective vulnerability of the basal ganglia. Ann. Neurol. 1997, 41, 646–653. [Google Scholar] [CrossRef]
- Reddy, P.H.; Shirendeb, U.P. Mutant Huntingtin, Abnormal Mitochondrial Dynamics, Defective Axonal Transport of Mitochondria, and Selective Synaptic Degeneration in Huntington’s Disease. Biochim. Biophys. Acta 2012, 1822, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, A.; Sandhir, R. Mitochondrial cofactors in experimental Huntington’s disease: Behavioral, biochemical and histological evaluation. Behav. Brain Res. 2014, 261, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.K.; Hilsabeck, T.; Rea, S.L. The role of mitochondrial dysfunction in age-related diseases. Biochim. Biophys. Acta 2015, 1847, 1387–1400. [Google Scholar] [CrossRef] [PubMed]
- Ludolph, A.C.; He, F.; Spencer, P.S.; Hammerstad, J.; Sabri, M. 3-Nitropropinic acid-exogenous animal neurotoxin and possible human striatal toxin. Can. J. Neurol. Sci. 1991, 18, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Brouillet, E.; Jenkins, B.G.; Ferrante, R.J.; Kowall, N.W.; Miller, J.M.; Storey, E.; Srivastava, R.; Rosen, B.R.; Hyman, B.T. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J. Neurosci. 1993, 13, 4181–4192. [Google Scholar] [CrossRef] [Green Version]
- Borlongan, C.V.; Koutouzis, T.K.; Freeman, T.B.; Hauser, R.A.; Cahill, D.W.; Sanberg, R. Hyperactivity and hypo activity in a rat model of Huntington’s disease: The systemic 3- nitropropionic acid model. Brain Res. Protoc. 1997, 1, 253–257. [Google Scholar] [CrossRef]
- Brouillet, E.; Jacquard, C.; Bizat, N.; Blum, D. 3-Nitropropionic acid: A mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington’s disease. J. Neurochem. 2005, 95, 1521–1540. [Google Scholar] [CrossRef]
- Liu, H.-G.; Ma, Y.; Meng, D.-W.; Yang, A.-C.; Zhang, J.-G. A Rat Model of Hemidystonia Induced by 3-Nitropropionic Acid. PLoS ONE 2013, 8, e79199. [Google Scholar] [CrossRef]
- VonsatteI, J.P.; Nyers, R.H.; Stevens, T.; Ferrunte, R.; Bird, E.D.; Richardson, E.P., Jr. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 1995, 44, 559–577. [Google Scholar] [CrossRef]
- Borlongan, C.V.; Nishino, H.; Sanberg, P.R. Systemic but not intraparenquimal administration of 3-nitropropionic acid mimics the neuropathology of Huntington’s disease: A speculative explanation. Neurosci. Res. 1997, 28, 185–189. [Google Scholar] [CrossRef]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Sieradzan, K.A.; Mann, D.M.A. The selective vulnerability of nerve cells in Huntington’s disease. Neuropathol. Appl. Neurobiol. 2001, 27, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.W.; Copin, J.C.; Kawase, M.; Chen, S.F.; Sato, S.; Gobbel, G.T.; Chan, P.H. Excitotoxicity is required for induction of oxidative stress and apoptosis in mouse striatum by the mitochondrial toxin, 3-nitropropionic acid. J. Cereb. Blood Flow Metab. 2000, 20, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Mirandola, S.R.; Melo, D.R.; Saito, A.; Castilho, R.F. 3-nitropropionic acid-induced mitochondrial permeability transition: Comparative study of mitochondria from different tissues and brain regions. J. Neurosci. Res. 2010, 88, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Ade, K.K.; Jannsse, M.J.; Ortinski, P.I.; Vicini, S. Differential tonic GABA conductances in striatal medium spiny neurons. J. Neurosci. 2008, 28, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Santhakumar, V.; Jones, R.T.; Mody, I. Developmental regulation and neuroprotective effects of striatal tonic GABAA currents. Neurosci. 2010, 167, 644–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Z.; Tertrais, M.; Courtand, G.; Leste-Lasserre, T.; Cardoit, L.; Masmejean, F.; Halgan, C.; Cho, Y.H.; Garret, M. Differential alteration in expression of striatal GABAAR subunits in mouse models of Huntington’s disease. Front. Mol. Neurosci. 2017, 10, 198. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, E.; Rivera, I.; Astorga, S.; Mendoza, E.; García, F.; Hernández-Echeagaray, E. Uncoupling oxidative/energy metabolism with low sub chronic doses of 3-nitropropionic acid or iodoacetate in vivo produces striatal cell damage. Int. J. Biol. Sci. 2010, 6, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Mendoza, E.; Hernández-Echeagaray, E. Dopaminergic Modulation of Striatal Inhibitory Transmission and Long-Term Plasticity. Neural. Plast. 2015, 2015, 1–15. [Google Scholar] [CrossRef]
- Kumar, P.; Kalonia, H.; Kumar, A. Possible GABAergic mechanism in the neuroprotective effect of gabapentin and lamotrigine against 3-nitropropionic acid induced neurotoxicity. Eur. J. Pharmacol. 2012, 674, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Flores-Hernández, J.; Hernandez, J.; Snyder, G.L.; Yan, Z.; Fienberg, A.A.; Moss, S.J.; Greengard, P.; Surmeier, D.J. D1 dopamine receptor activation reduces GABA (A) receptor currents in neostriatal neurons through a PKA/DARPP-32/PPI signaling casacade. J. Neurophysiol. 2000, 83, 2996–3004. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Echeagaray, E.; Cepeda, C.; Ariano, M.A.; Lobo, M.K.; Sibley, D.R.; Levine, M.S. Dopamine reduction of GABA currents in striatal Medium-Sized spiny Neurons is mediated principally by the D1 receptor subtype. Neurochem. Res. 2007, 32, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Yan, J.; Yue, L.; Feng, F.; Mir, F.; Abdel-Halim, H.; Pepperberg, D.R. 2-Aminoethyl Methylphosphonate, a Potent and Rapidly Acting Antagonist of GABAA-ρ1 Receptors. Mol. Pharmacol. 2011, 80, 965–978. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.; Kerby, J.; Bonnert, T.P.; Whiting, P.J.; Wafford, K.A. Pharmacological characterization of a novel cell line expressing human α4β3δGABAA receptors. Br. J. Pharmacol. 2002, 136, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.; Ebert, B.; Wafford, K.; Smart, T.G. Distinct activities of GABA agonists at synaptic-and extrasynaptic-type GABAA receptors. J. Physiol. 2010, 588, 1251–1268. [Google Scholar] [CrossRef] [PubMed]
- Krehan, D.; Frølund, B.; Ebert, B.; Nielsen, B.; Krogsgaard-Larsen, P.; Johnston, G.A.; Chebib, M. Aza-THIP and related analogues of THIP as GABA C. antagonists. Bioorg. Med. Chem. 2003, 11, 4891–4896. [Google Scholar] [CrossRef] [PubMed]
- Johnston, G.A.R.; Chebib, M.; Hanrahan, J.R.; Mewett, K.N. Neurochemicals for the investigation of GABAC Receptors. Neurochem. Res. 2010, 35, 1970–1977. [Google Scholar] [CrossRef]
- Mendoza, E.; Miranda-Barrientos, J.; Vázquez-Roque, R.A.; Morales-Herrera, E.; Ruelas, A.; De la Rosa, G.; Flores, G.; Hernández-Echeagaray, E. Mitochondrial inhibition in vivo alters corticostriatal function and the modulatory effects of neurotrophins. Neuroscience 2014, 280, 156–170. [Google Scholar] [CrossRef]
- Cepeda, C.; Starling, A.J.; Wu, N.; Nguyen, O.K.; Uzgil, B.; Soda, T.; Marjorie, V.M.A.; Levine, M.S. Increased GABAergic Function in Mouse Models of Huntington’s disease: Reversal by BDNF. J. Neurosci. Res. 2004, 78, 855–867. [Google Scholar] [CrossRef]
- Rocher, A.B.; Gubellini, P.; Merienne, N.; Boussicault, L.; Petit, F.; Gipchtein, P.; Jan, C.; Hantraye, P.; Brouillet, E.; Bonvento, G. Synaptic scaling up in medium spiny neurons of aged BACHD mice: A slow-progression model of Huntington’s disease. Neurobiol. Dis. 2016, 86, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Hines, R.M.; Davies, P.A.; Moss, S.J.; Maguire, J. Functional regulation of GABAA receptors in nervous system pathologies. Curr. Opin. Neurobiol. 2011, 22, 1–7. [Google Scholar]
- Bianchi, M.T.; Macdonald, R.L. Slow phases of GABA(A) receptor desensitization: Structural determinants and possible relevance for synaptic function. J. Physiol. 2002, 544, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Haas, K.F.; Macdonald, R.L. GABAA receptor subunit γ2 and δ subtypes confer unique kinetic properties on recombinant GABAA receptor currents in mouse fibroblasts. J. Physiol. 1999, 514, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Saxena, N.C.; Macdonald, R.L. Assembly of GABA(A) receptor subunits: Role of the delta subunit. J. Neurosci. 1994, 14, 7077–7086. [Google Scholar] [CrossRef] [PubMed]
- Graveland, G.A.; Williams, R.S.; DiFiglia, M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science 1985, 227, 770–773. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, R.; Kowall, N.W.; Beal, M.F.; Martin, J.B.; Bird, E.D.; Richardson, E.P., Jr. Morphologic and histochemical characteristics of a spared subset of striatal neurons in Huntington’s disease. J. Neuropathol. Exp. Neurol. 1987, 46, 12–27. [Google Scholar]
- Quirk, K.; Blurton, P.; Fletcher, S.; Leeson, P.; Tang, F.; Mellilo, D.; Ragan, C.I.; McKernan, R.M. [3H] L-655,708, a novel ligand selective for the benzodiazepine site of GABAA receptors which contain the α5 subunit. Neuropharmacology 1996, 35, 1331–1335. [Google Scholar] [CrossRef]
- Nusser, Z.; Sieghart, W.; Somogyi, P. Segregation of different GABA(A) receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J. Neurosci. 1998, 18, 1693–1703. [Google Scholar] [CrossRef]
- Drasbek, K.R.; Jensen, K. THIP, a Hypnotic and Antinociceptive Drug, Enhances an Extrasynaptic GABAA Receptor-mediated Conductance in Mouse Neocortex. Cer. Cortex 2006, 16, 1134–1141. [Google Scholar] [CrossRef]
- Rosas-Arellano, A.; Machuca-Parra, A.I.; Reyes-Haro, D.; Miledi, R.; Martinez-Torres, A. Expression of GABA rho receptors in the neostriatum: Localization in aspiny, medium spiny neurons and GFAP-positive cells. J. Neurochem. 2012, 122, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.H.; Zhang, D.; Zhang, X.; Lipton, S.A. Evidence for coassembly of mutant GABAC rho1 with GABAA gamma2S, glycine alpha1 and glycine alpha2 receptor subunits in vitro. Eur. J. Neurosci. 2000, 12, 3137–3145. [Google Scholar] [CrossRef] [PubMed]
- Milligan, C.J.; Buckley, N.J.; Garret, M.; Deuchars, J.; Deuchars, S.A. Evidence for inhibition mediated by coassembly of GABAA and GABA-C receptor subunits in native central neurons. J. Neurosci. 2004, 24, 7241–7250. [Google Scholar] [CrossRef] [PubMed]
- Kumar, U.; Heer, M.; Somvanshi, R.K. Regional and subcellular distribution of GABAC rho3 receptor in the brain of R6/2 mouse model of Huntington´s Disease. Neurosci. Lett. 2017, 640, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Hasbani, M.J.; Underhill, S.M.; De Erausquin, G.A.; Goldberg, M.P. Synapse loss and regeneration: A mechanism for functional decline and recovery after cerebral ischemia? Neuroscientist 2000, 6, 110–119. [Google Scholar] [CrossRef]
- Belelli, D.; Harrison, N.L.; Maguire, J.; Macdonald, R.L.; Walker, M.C.; Cope, D.W. Extrasynaptic GABAA receptors: Form, Pharmacology and Function. J. Neurosci. 2009, 29, 12757–12763. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | 3-NP | |||
|---|---|---|---|---|
| GABA µM | τ ms | SE | τ ms | SE |
| 0.1 | 975.73 | ± 254.90 | 391.70 | * ± 47.44 |
| 0.3 | 674.24 | ± 133.83 | 444.63 | ± 52.48 |
| 1 | 539.43 | ± 133.35 | 565.41 | ± 72.35 |
| 3 | 1056.66 | ± 503.34 | 1035.37 | ± 366.46 |
| 10 | 874.02 | ± 321.01 | 713.63 | ± 236.11 |
| 30 | 851.91 | ± 206.72 | 601.76 | ± 170.57 |
| 100 | 957.68 | ± 115.54 | 903.12 | ± 104.11 |
| 300 | 468.44 | ± 63.53 | 766.92 | ± 144.60 |
| 1000 | 761.81 | ± 330.15 | 1133.05 | ± 148.23 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flores-Hernández, J.; Garzón-Vázquez, J.A.; Hernández-Carballo, G.; Nieto-Mendoza, E.; Ruíz-Luna, E.A.; Hernández-Echeagaray, E. Striatal Neurodegeneration that Mimics Huntington’s Disease Modifies GABA-induced Currents. Brain Sci. 2018, 8, 217. https://doi.org/10.3390/brainsci8120217
Flores-Hernández J, Garzón-Vázquez JA, Hernández-Carballo G, Nieto-Mendoza E, Ruíz-Luna EA, Hernández-Echeagaray E. Striatal Neurodegeneration that Mimics Huntington’s Disease Modifies GABA-induced Currents. Brain Sciences. 2018; 8(12):217. https://doi.org/10.3390/brainsci8120217
Chicago/Turabian StyleFlores-Hernández, Jorge, Jeanette A. Garzón-Vázquez, Gustavo Hernández-Carballo, Elizabeth Nieto-Mendoza, Evelyn A. Ruíz-Luna, and Elizabeth Hernández-Echeagaray. 2018. "Striatal Neurodegeneration that Mimics Huntington’s Disease Modifies GABA-induced Currents" Brain Sciences 8, no. 12: 217. https://doi.org/10.3390/brainsci8120217