Lipopolysaccharide Challenge Reveals Hypothalamic-Pituitary-Adrenal Axis Dysfunction in Murine Systemic Lupus Erythematosus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
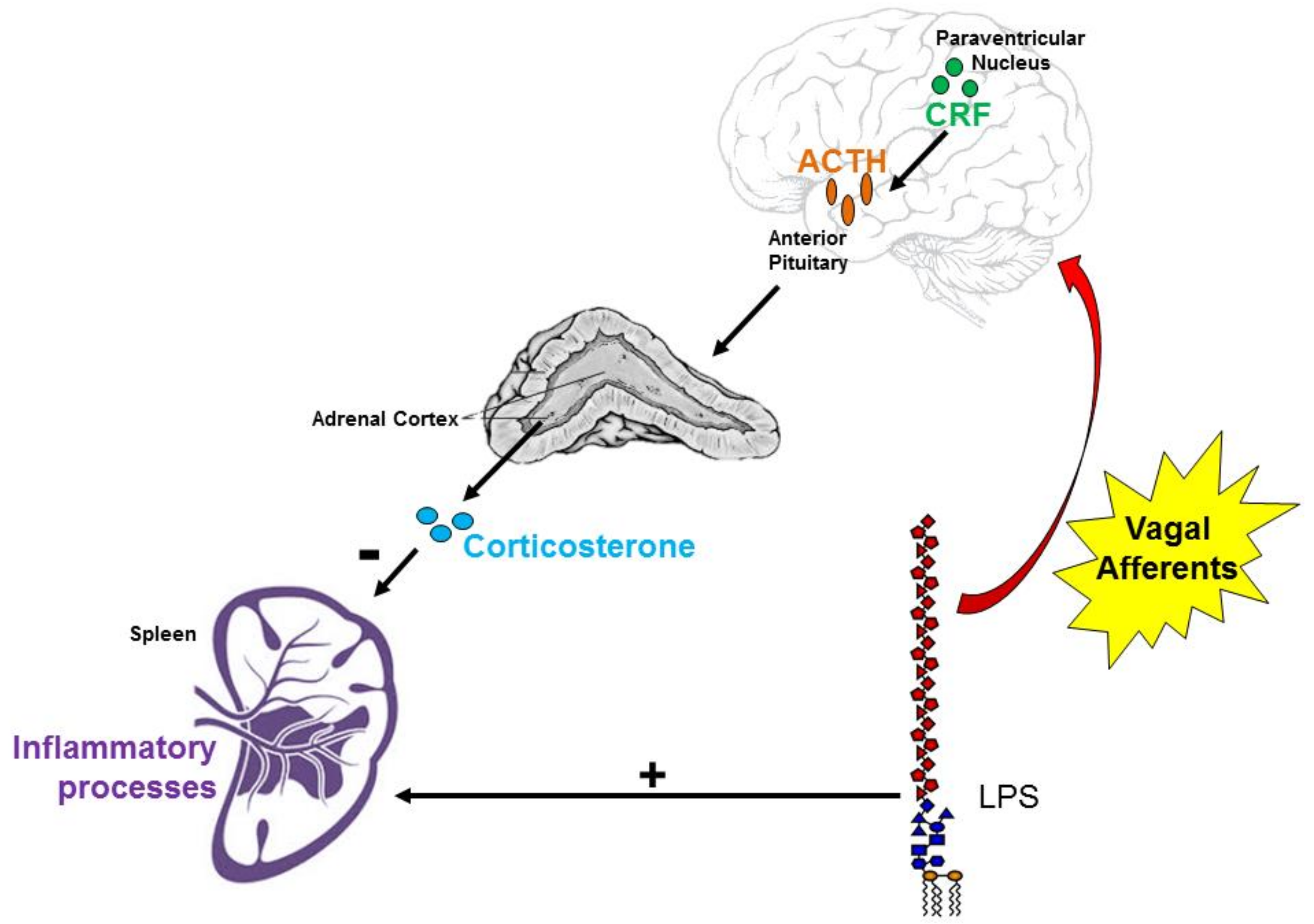
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Acute LPS Challenge
2.3. c-Fos and CRF Immunohistochemistry
2.4. Plasma Corticosterone and Adrenocorticotrophic Hormone (ACTH)
2.5. Brain and Spleen Cytokines
2.6. Statistical Analysis
3. Results
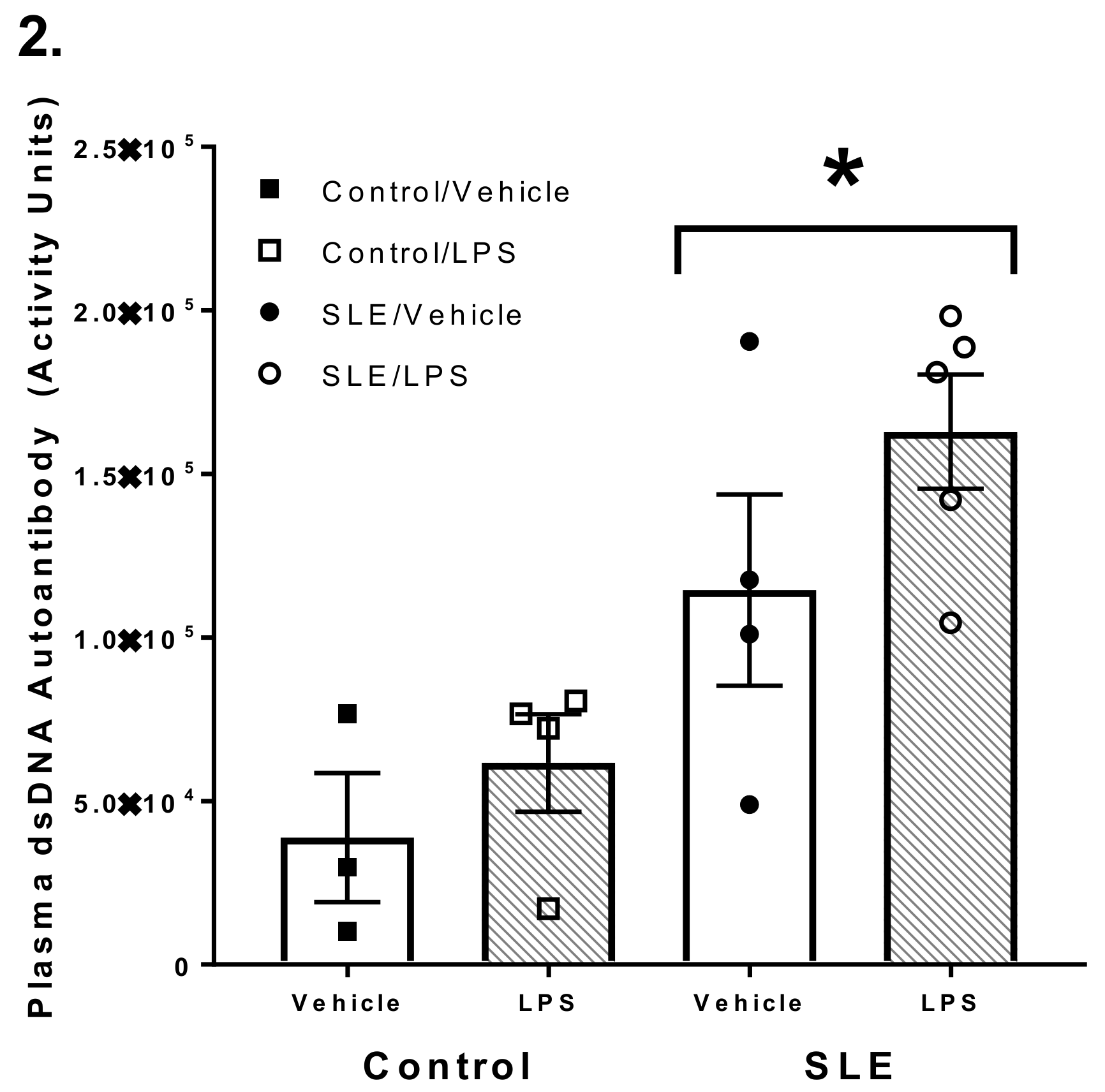
3.1. SLE Mice had Elevated Anti-dsDNA Autoantibody Titer Compared to NZW and C57/Bl6 Mice
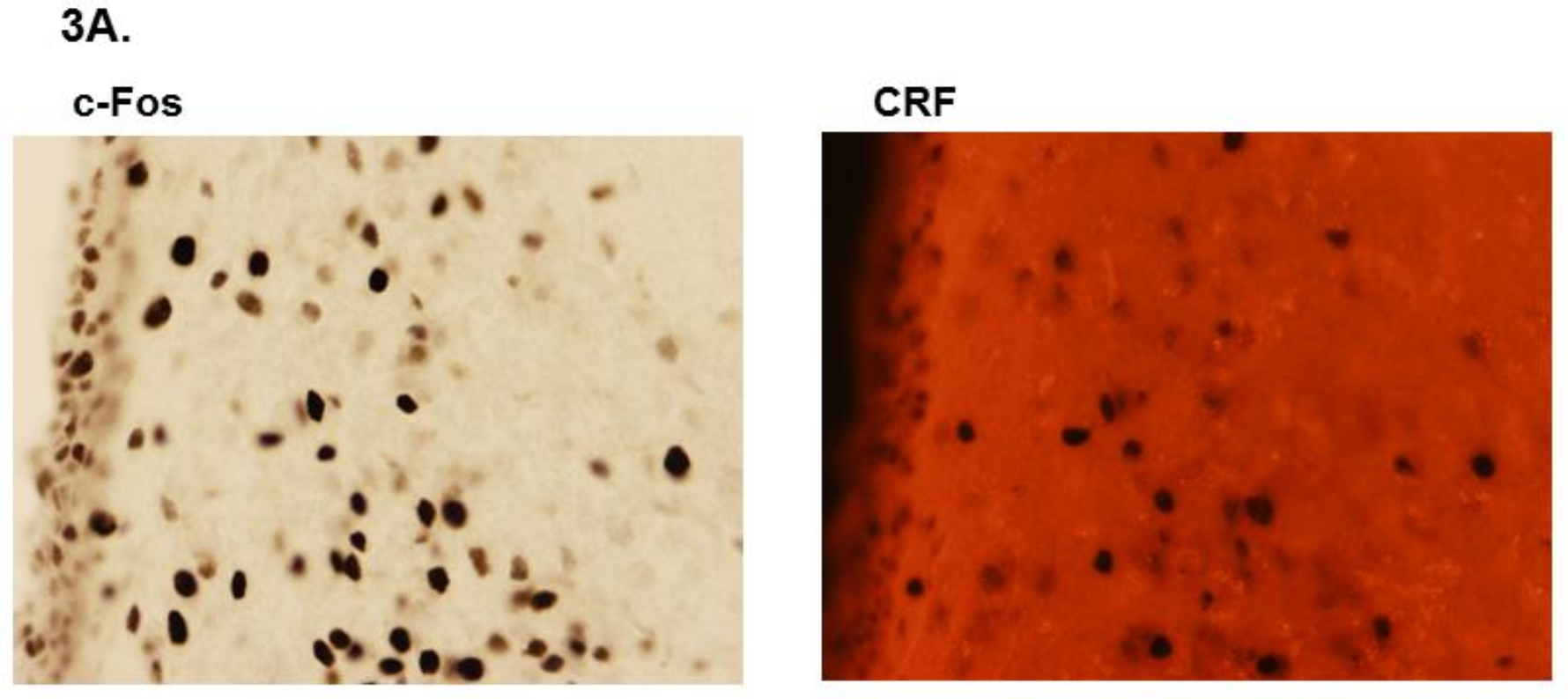
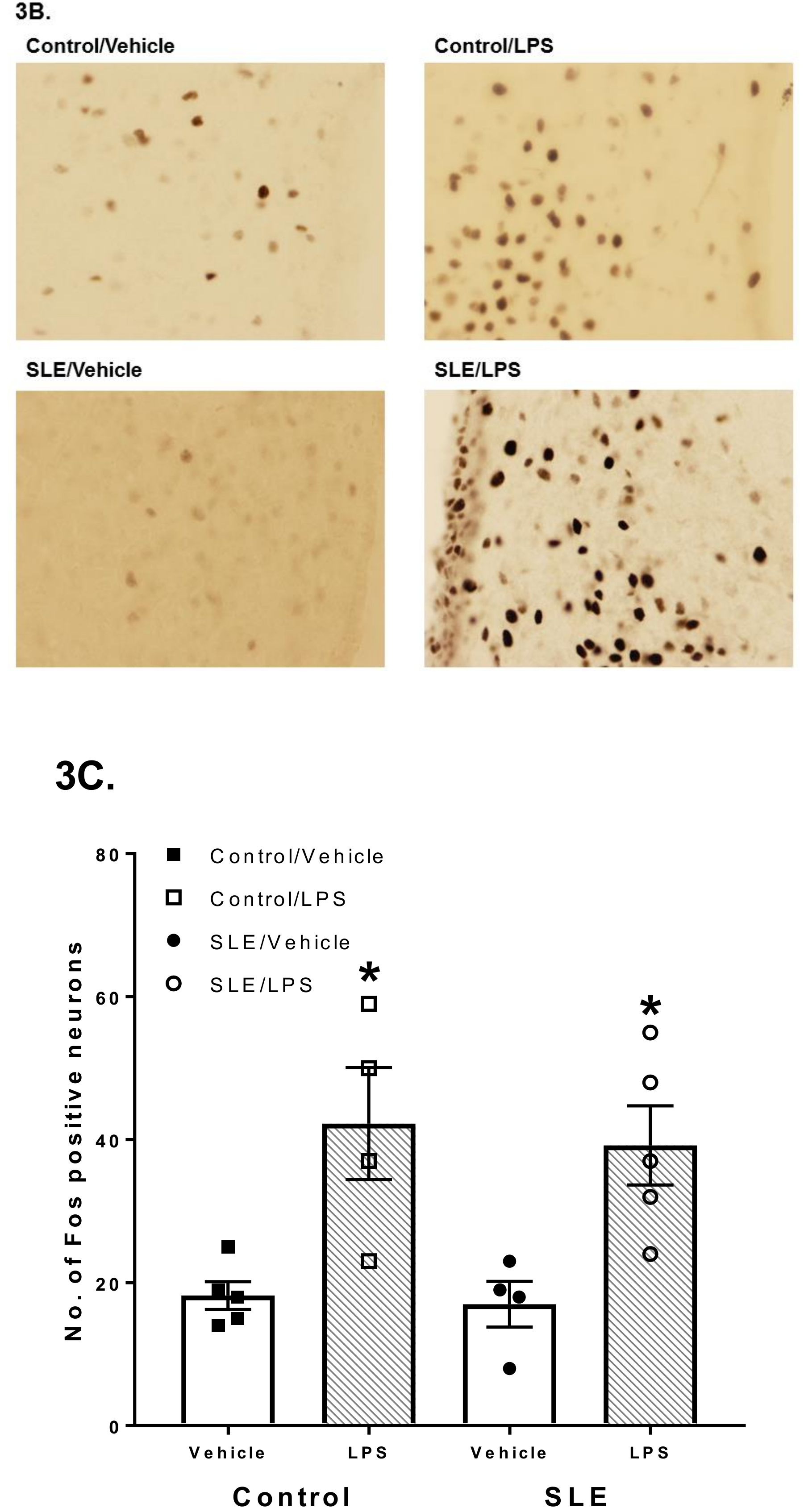
3.2. LPS Challenge Elicits Similar Parvocellular Paraventricular Nuclei Activation between SLE and Control Mice
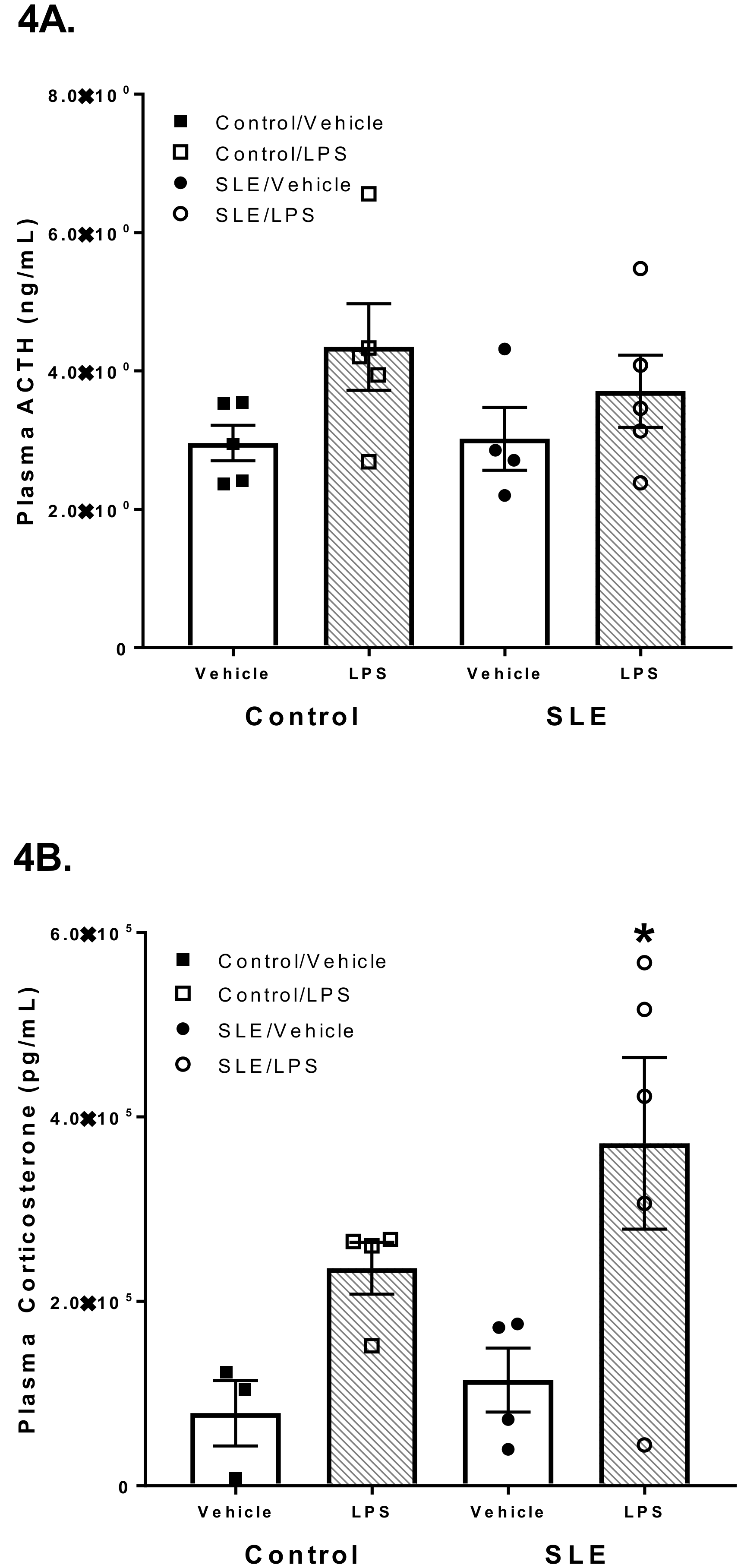
3.3. LPS has Differential Effects on HPA Axis Hormones in SLE Mice
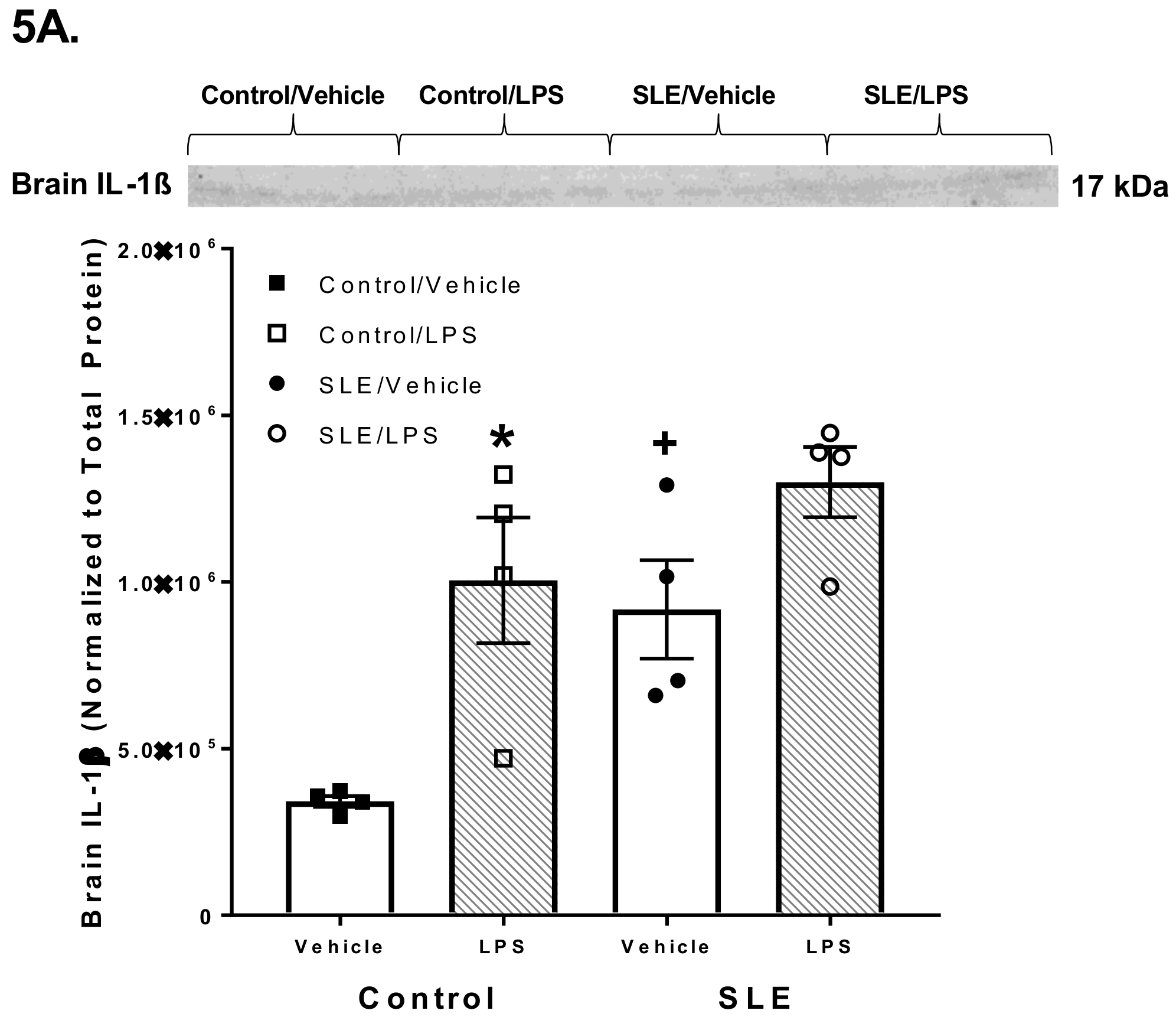
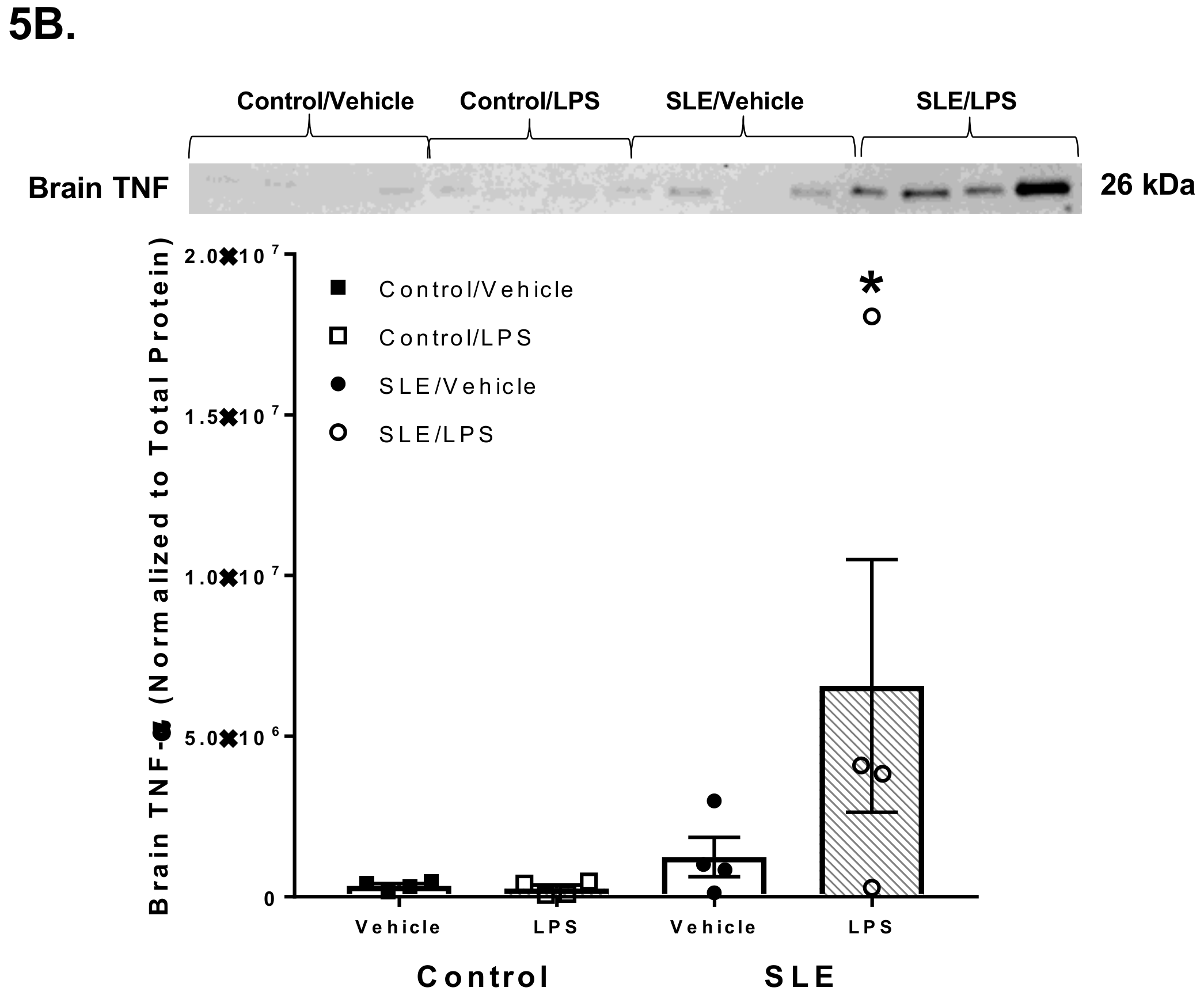
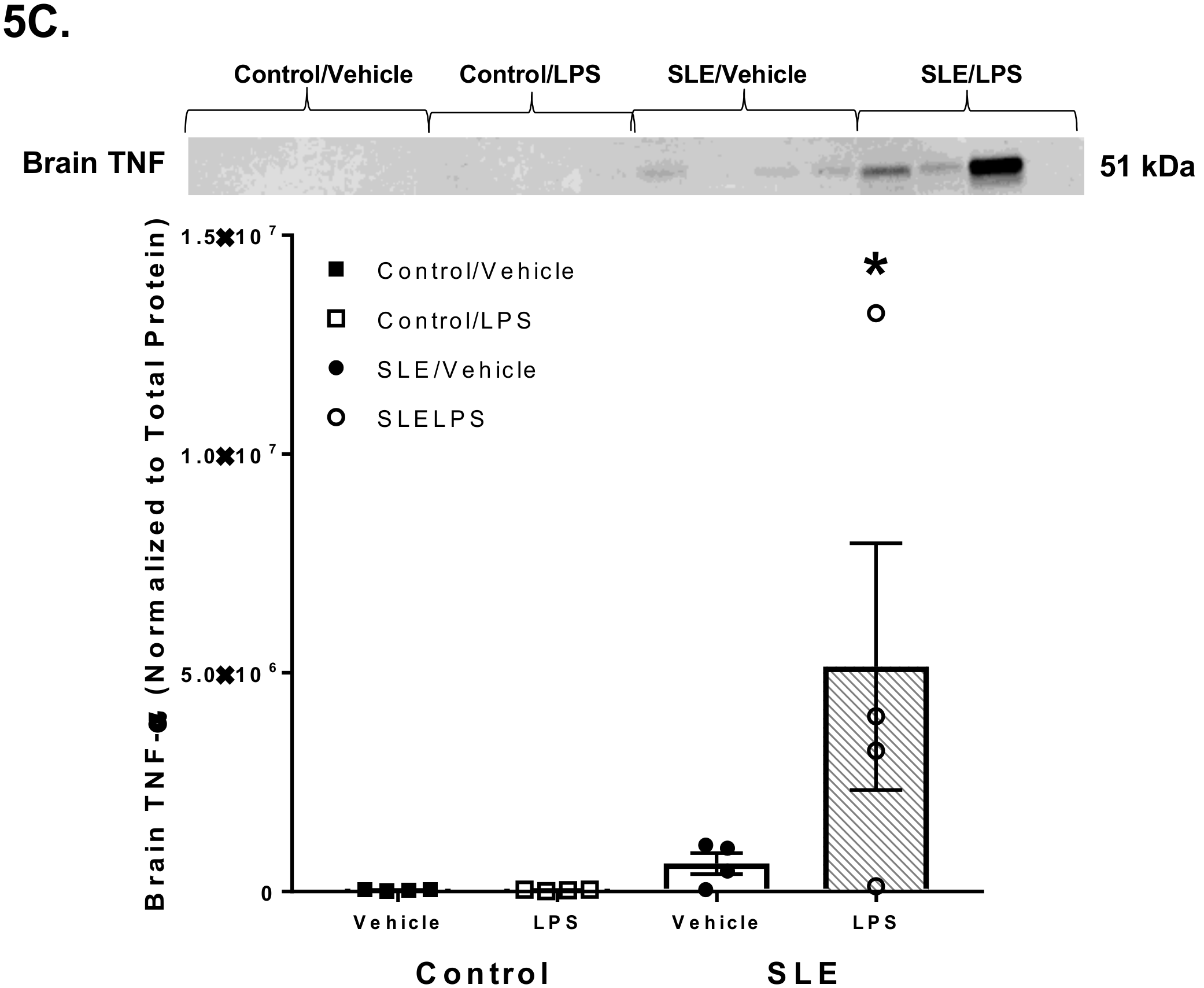
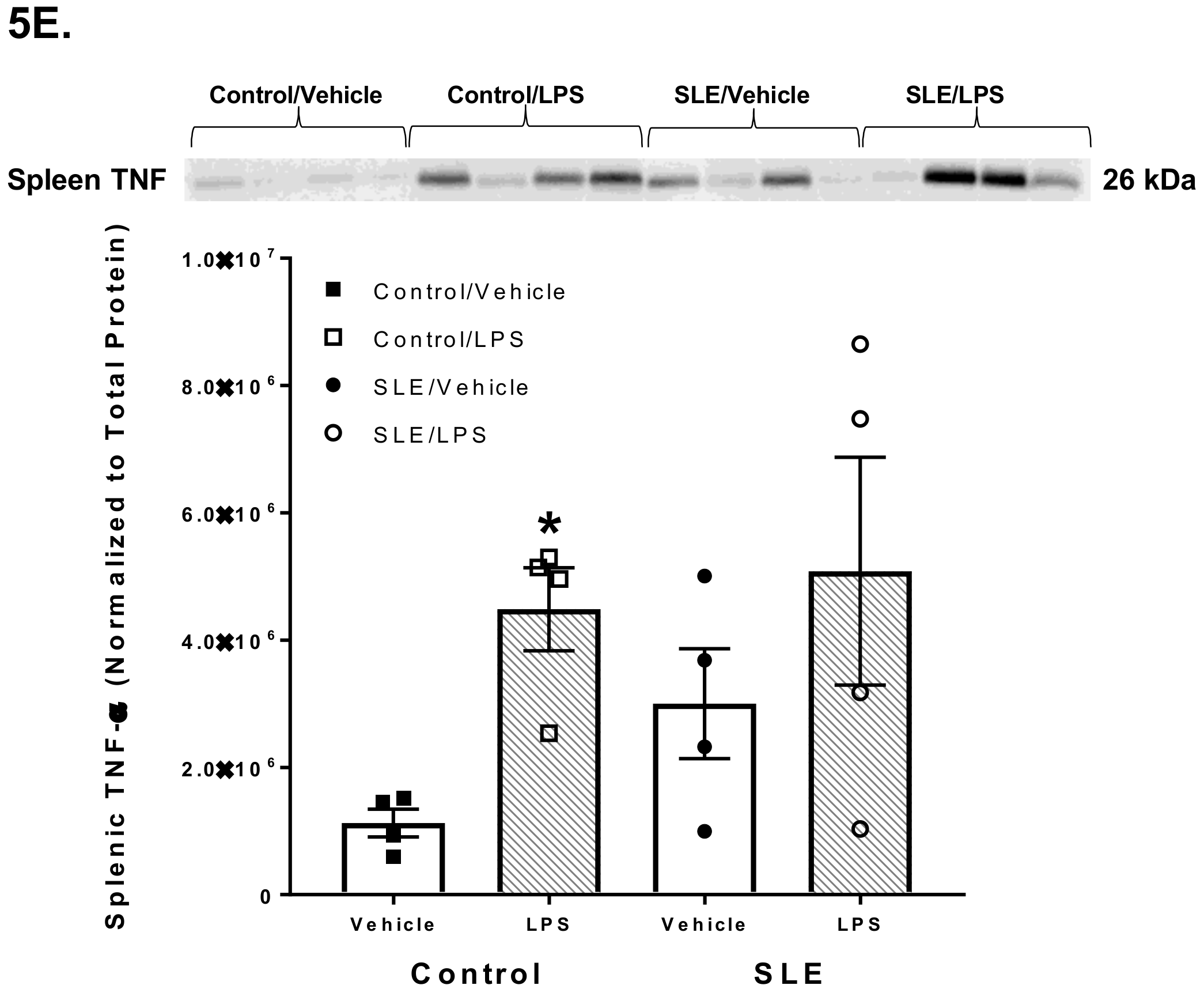
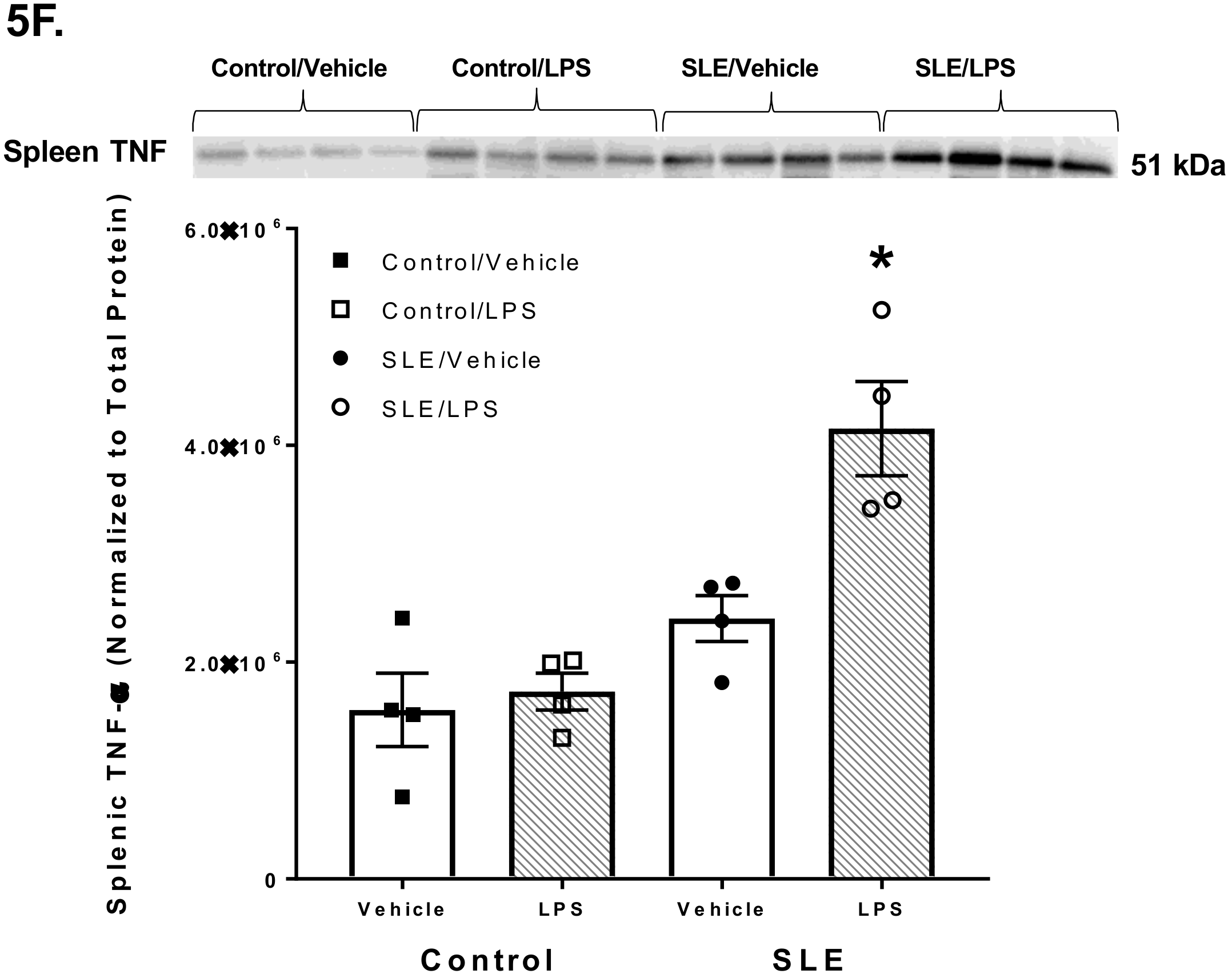
3.4. LPS Alters Brain and Splenic Pro-Inflammatory Cytokine Expression
4. Discussion
Reference
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maule, S.; Quadri, R.; Mirante, D.; Pellerito, R.A.; Marucco, E.; Marinone, C.; Vergani, D.; Chiandussi, L.; Zanone, M.M. Autonomic nervous dysfunction in systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA): Possible pathogenic role of autoantibodies to autonomic nervous structures. Clin. Exp. Immunol. 1997, 110, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Barrat, F.J.; Meeker, T.; Gregorio, J.; Chan, J.H.; Uematsu, S.; Akira, S.; Chang, B.; Duramad, O.; Coffman, R.L. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med. 2005, 202, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Ganguly, D.; Facchinetti, V.; Frasca, L.; Conrad, C.; Gregorio, J.; Meller, S.; Chamilos, G.; Sebasigari, R.; Riccieri, V.; et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra19. [Google Scholar] [CrossRef] [PubMed]
- Johnston, G.R.; Webster, N.R. Cytokines and the immunomodulatory function of the vagus nerve. Br. J. Anaesth. 2009, 102, 453–452. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, K.; Takeuchi, O.; Kawai, T.; Sanjo, H.; Ogawa, T.; Takeda, Y.; Takeda, K.; Akira, S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: Evidence for TLR4 as the Lps gene product. J. Immunol. 1999, 162, 3749–3752. [Google Scholar] [PubMed]
- Hosoi, T.; Okuma, Y.; Matsuda, T.; Nomura, Y. Novel pathway for LPS-induced afferent vagus nerve activation: Possible role of nodose ganglion. Auton. Neurosci. 2005, 120, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Goehler, L.E.; Gaykema, R.P.; Hammack, S.E.; Maier, S.F.; Watkins, L.R. Interleukin-1 induces c-Fos immunoreactivity in primary afferent neurons of the vagus nerve. Brain Res. 1998, 804, 306–310. [Google Scholar] [CrossRef]
- Wieczorek, M.; Dunn, A.J. Effect of subdiaphragmatic vagotomy on the noradrenergic and HPA axis activation induced by intraperitoneal interleukin-1 administration in rats. Brain Research 2006, 1101, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinenov, Y.; Rogatsky, I. Glucocorticoids and the innate immune system Crosstalk with the Toll-like receptor signaling network. Mol. Cell. Endocrinol. 2007, 275, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Elenkov, I.J. Glucocorticoids and the Th1/Th2 balance. Ann. N. Y. Acad. Sci. 2004, 1024, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, J.B.; Martins, A.S.; Pinheiro, H.T.; Secchin, N.A.; Moura, R.L.; Bastos, A.C. Traditional ecological knowledge and the mapping of benthic marine habitats. J. Environ. Manage. 2013, 115, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Dujmovic, I.; Mangano, K.; Pekmezovic, T.; Quattrocchi, C.; Mesaros, S.; Stojsavljevic, N.; Nicoletti, F.; Drulovic, J.J. The analysis of IL-1 beta and its naturally occurring inhibitors in multiple sclerosis: The elevation of IL-1 receptor antagonist and IL-1 receptor type II after steroid therapy. J. Neuroimmunol. 2009, 207, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcellini, W.; Rizzardi, G.P.; Borghi, M.O.; Nicoletti, F.; Fain, C.; Del Papa, N.; Meroni, P.L. In vitro type-1 and type-2 cytokine production in systemic lupus erythematosus: Lack of relationship with clinical disease activity. Lupus 1996, 5, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Bedoya, S.K.; Lam, B.; Lau, K.; Larkin, J.I. Th17 cells in immunity and autoimmynity. Clin. Dev. Immunol. 2013, 2013, 986789. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.M.; Garcia, M.E.; Rodriguez, J.A.; Rivero, S.; Jacobelli, S. Hypothalamic-pituitary-adrenal axis function and prolactin secretion in systemic lupus erythematosus. Lupus 1998, 7, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Rovensky, J.; Blazícková, S.; Rauová, L.; Jezová, D.; Koska, J.; Lukác, J.; Vigas, M. The hypothalamic response in SLE. Regulation of prolactin, growth hormone and cortisol release. Lupus 1998, 7, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Lechner, O.; Hu, Y.; Jafarian-Tehrani, M.; Dietrich, H.; Schwarz, S.; Herold, M.; Haour, F.; Wick, G. Disturbed immunoendocrine communication via the hypothalamo-pituitary-adrenal axis in murine lupus. Brain Behav. Immun. 1996, 10, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.H.; Weidler, C.; Demmel, B.; Herrmann, M.; Kees, F.; Schmidt, M.; Schölmerich, J.; Schedel, J. Renal clearance and daily excretion of cortisol and adrenal androgens in patients with rheumatoid arthritis and systemic lupus erythematosus. Ann. Rheum. Dis. 2014, 63, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Van der Goes, M.C.; Bossema, E.R.; Hartkamp, A.; Godaert, G.L.; Jacobs, J.W.; Kruize, A.A.; Derksen, R.H.; Bijlsma, J.W.; Geenen, R. Cortisol during the day in patients with systemic lupus erythematosus or primary Sjogren’s syndrome. J. Rheumatol. 2010, 38, 285–258. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, T.; Okuma, Y.; Nomura, Y. Electrical stimulation of afferent vagus nerve induces IL-1beta expression in the brain and activates HPA axis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, 141–147. [Google Scholar] [CrossRef] [PubMed]
- De Herdt, V.; Puimege, L.; De Waele, J.; Raedt, R.; Wyckhuys, T.; El Tahry, R.; Libert, C.; Wadman, W.; Boon, P.; Vonck, K. Increased rat serum corticosterone suggests immunomodulation by stimulation of the vagal nerve. J. Neuroimmunol. 2009, 212, 102–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathis, K.W.; Wallace, K.; Flynn, E.R.; Maric-Bilkan, C.; LaMarca, B.; Ryan, M.J. Preventing autoimmunity protects against the development of hypertension and renal injury. Hypertension 2014, 64, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Mathis, K.W.; Venegas-Pont, M.; Masterson, C.W.; Stewart, N.J.; Wasson, K.L.; Ryan, M.J. Oxidative stress promotes hypertension and albuminuria during the autoimmune disease systemic lupus erythematosus. Hypertension 2012, 59, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Mathis, K.W.; Venegas-Pont, M.; Flynn, E.R.; Williams, J.M.; Maric-Bilkan, C.; Dwyer, T.M.; Ryan, M.J. Hypertension in an experimental model of systemic lupus erythematosus occurs independently of the renal nerves. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R711–R719. [Google Scholar] [CrossRef] [PubMed]
- Mathis, K.W.; Venegas-Pont, M.; Masterson, C.W.; Wasson, K.L.; Ryan, M.J. Blood pressure in a hypertensive mouse model of SLE is not salt-sensitive. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, 1281–1285. [Google Scholar] [CrossRef] [PubMed]
- Venegas-Pont, M.; Mathis, K.W.; Iliescu, R.; Ray, W.H.; Glover, P.H.; Ryan, M.J. Blood pressure and renal hemodynamic responses to acute angiotensin II infusion are enhanced in a female mouse model of systemic lupus erythematosus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Fairley, A.S.; Mathis, K.W. Cholinergic agonists reduce blood pressure in a mouse model of systemic lupus erythematosus. Physiol. Rep. 2017, 5, e13213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haziot, A.; Ferrero, E.; Kontgen, F.; Hijiya, N.; Yamamoto, S.; Silver, J.; Stewart, C.L.; Goyert, S.M. Resistance to endotoxin shock and reduced dissemination of gram-negative bacteria in CD14-deficient mice. Immunity 1996, 4, 407–414. [Google Scholar] [CrossRef]
- Cunningham, J.T.; Mifflin, S.W.; Gould, G.G.; Frazer, A. Induction of c-Fos and DeltaFosB immunoreactivity in rat brain by Vagal nerve stimulation. Neuropsychopharmacology 2008, 33, 1884–1895. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.T.; Grindstaff, R.J.; Grindstaff, R.R.; Sullivan, M.J. Fos immunoreactivity in the diagonal band and the perinuclear zone of the supraoptic nucleus after hypertension and hypervolaemia in unanaesthetized rats. J. Neuroendocrinol. 2002, 14, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Baglioni, C. The active form of tumor necrosis factor is a trimer. J. Biol. Chem. 1987, 262, 6951–6954. [Google Scholar] [PubMed]
- Pons-Estel, G.J.; Alarcon, G.S.; Scofield, L.; Reinlib, L.; Cooper, G.S. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin. Arthritis Rheum. 2010, 39, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.S.; Dooley, M.A.; Treadwell, E.L.; St Clair, E.W.; Parks, C.G.; Gilkeson, G.S. Hormonal, environmental, and infectious risk factors for developing systemic lupus erythematosus. Arthritis Rheum. 1998, 41, 1714–1724. [Google Scholar] [CrossRef] [Green Version]
- Ippolito, A.; Wallace, D.J.; Gladman, D.; Fortin, P.R.; Urowitz, M.; Werth, V.; Costner, M.; Gordon, C.; Alarcón, G.S.; Ramsey-Goldman, R.; et al. Autoantibodies in systemic lupus erythematosus: comparison of historical and current assessment of seropositivity. Lupus 2011, 20, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Wajed, J.; Ahmad, Y.; Durrington, P.N.; Bruce, I.N. Prevention of cardiovascular disease in systemic lupus erythematosus-proposed guidelines for risk factor management. Rheumatology 2004, 43, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Amissah-Arthur, M.B.; Gordon, C. Contemporary treatment of systemic lupus erythematosus: An update for clinicians. Ther. Adv. Chronic Dis. 2010, 1, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Crofford, L.J. The hypothalamic-pituitary-adrenal axis in the pathogenesis of rheumatic diseases. Endocrinol. Metab. Clin. North Am. 2002, 31, 1–13. [Google Scholar] [CrossRef]
- Mondal, T.K.; Saha, S.K.; Miller, V.M.; Seegal, R.F.; Lawrence, D.A. Autoantibody-mediated neuroinflammation: pathogenesis of neuropsychiatric systemic lupus erythematosus in the NZM88 murine model. Brain Behav. Immun. 2008, 22, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Catallani, B.; Palma, B.D.; Gil, F.Z.; Suchecki, D. Brief and long maternal separations decrease corticosterone secretion in a lupus-prone strain: dissociation from disease-related parameters. Brain Behav. Immun. 2008, 22, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Beishuizen, A.; Thijs, L.G. Endotoxin and the hypothalamo-pituitary-adrenal (HPA) axis. J. Endotoxin Res. 2003, 9, 3–24. [Google Scholar] [PubMed]
- Ryan, M.J.; McLemore, G.R., Jr.; Hendrix, S.T. Insulin resistance and obesity in a mouse model of systemic lupus erythematosus. Hypertension 2006, 48, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.; Buskila, D.; Gladman, D.; Bruser, B.; Malkin, A. Cortisol catabolism by lymphocytes of patients with systemic lupus erythematosus and rheumatoid arthritis. J. Rheumatol. 1990, 17, 30–33. [Google Scholar] [PubMed]
- Givalois, L.; Dornand, J.; Mekaouche, M.; Solier, M.D.; Bristow, A.F.; Ixart, G.; Siaud, P.; Assenmacher, I.; Barbanel, G. Temporal cascade of plasma level surges in ACTH, corticosterone, and cytokines in endotoxin-challenged rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1994, 267, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.F.; Xu, J.H.; Wang, F.; Liu, S.; Tao, J.H.; Cai, J.; Lian, L.; Xiao, H.; Chen, P.L.; Tian, G.; et al. Association study of glucocorticoid receptor genetic polymorphisms with efficacy of glucocorticoids in systemic lupus erythematosus: A prospective cohort study. Autoimmunity 2013, 46, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Xu, J.H.; Zou, Y.F.; Lian, L.; Wang, F.; Chen, S.Y.; Cai, J.; Li, M. Association of glucocorticoid receptor gene polymorphisms with systemic lupus erythematosus in a Chinese population. Int. J. Rheum. Dis. 2017, 20, 2053–2061. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Wang, Q.; Yu, X.; Liu, J.; Bai, S.; Feng, J.; Wu, B. Molecular mechanisms of glucocorticoid resistance in systemic lupus erythematosus: A review. Life Sci. 2018, 209, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Fagone, P.; Mazzon, E.; Cavalli, E.; Bramanti, A.; Petralia, M.C.; Mangano, K.; Al-Abed, Y.; Bramati, P.; Nicoletti, F. Contribution of the macrophage migration inhibitory factor superfamily of cytokines in the pathogenesis of preclinical and human multiple sclerosis: In silico and in vivo evidences. J. Neuroimmunol. 2018, 322, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, F.; Créange, A.; Orlikowski, D.; Bolgert, F.; Mangano, K.; Metz, C.; Di Marco, R.; Al Abed, Y. Macrophage migration inhibitory factor (MIF) seems crucially involved in Guillain-Barré syndrome and experimental allergic neuritis. J. Neuroimmunol. 2005, 168, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Lapter, S.; Ben-David, H.; Sharabi, A.; Zinger, H.; Telerman, A.; Gordin, M.; Leng, L.; Bucala, R.; Shachar, I.; Mozes, E. A role for the B-cell CD74/macrophage migration inhibitory factor pathway in the immunomodulation of systemic lupus erythematosus by a therapeutic tolerogenic peptide. Immunology 2011, 132, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Bollet, A.J.; Segal, S.; Bunim, J.J. Treatment of systemic lupus erythematosus with prednisone and prednisolone. J. Am. Med. Assoc. 1995, 159, 1501–1507. [Google Scholar] [CrossRef]
- Kasturi, S.; Sammaritano, L.R. Corticosteroids in lupus. Rheum. Dis. Clin. North Am. 2016, 42, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Harris-Jones, J.N. The role of ACTH and cortisone in the treatment of systemic lupus erythematosus. Postgrad. Med. J. 1956, 32, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Golubovsky, J.; Hui-Yuen, J.; Shah, U.; Olech, E.; Lomeo, R.; Singh, V.; Busch, H.; Strandberg, M.J.; Strandberg, K.; et al. Adrenocorticotropic hormone gel in the treatment of systemic lupus erythematosus: A retrospective study of patients. F1000Res. 2015, 4, 1–11. [Google Scholar] [CrossRef]
- Fiechtner, J.J.; Montroy, T. Treatment of moderately to severely active systemic lupus erythematosus with adrenocorticotropic hormone: A single-site, open-label trial. Lupus 2014, 23, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Lightstone, L.; Doria, A.; Wilson, H.; Ward, F.L.; Larosa, M.; Bargman, J.M. Can we manage lupus nephritis without chronic corticosteroids administration? Autoimmun. Rev. 2018, 17, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Abe, C.; Sung, S.S.; Moscalu, S.; Jankowski, J.; Huang, L.; Ye, H.; Rosin, D.L.; Guyenet, P.G.; Okusa, M.D. Vagus nerve stimulation mediates protection from kidney ischemia-reperfusion injury through α7nAChR+ splenocytes. J. Clin. Invest. 2016, 126, 1939–1952. [Google Scholar] [CrossRef] [PubMed]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Koopman, F.A.; Schuurman, P.R.; Vervoordeldonk, M.J.; Tak, P.P. Vagus nerve stimulation: A new bioelectronics approach to treat rheumatoid arthritis? Best Pract. Res. Clin. Rheumatol. 2014, 28, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Mathis, K.W.; Stauss, H.; Pham, G.; Kim, S.; Kulp, D. Effects of vagus nerve stimulation in a murine model of systemic lupus erythematosus. In Proceedings of the APS Annual Meeting, San Diego, CA, USA, 21–25 April 2018. [Google Scholar]
- Bonaz, B.; Sinniger, V.; Pellissier, S. Anti-inflammatory properties of the vagus nerve: Potential therapeutic implications of vagus nerve stimulation. J. Physiol. 2016, 594, 5781–5790. [Google Scholar] [CrossRef] [PubMed]











© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pham, G.S.; Mathis, K.W. Lipopolysaccharide Challenge Reveals Hypothalamic-Pituitary-Adrenal Axis Dysfunction in Murine Systemic Lupus Erythematosus. Brain Sci. 2018, 8, 184. https://doi.org/10.3390/brainsci8100184
Pham GS, Mathis KW. Lipopolysaccharide Challenge Reveals Hypothalamic-Pituitary-Adrenal Axis Dysfunction in Murine Systemic Lupus Erythematosus. Brain Sciences. 2018; 8(10):184. https://doi.org/10.3390/brainsci8100184
Chicago/Turabian StylePham, Grace S., and Keisa W. Mathis. 2018. "Lipopolysaccharide Challenge Reveals Hypothalamic-Pituitary-Adrenal Axis Dysfunction in Murine Systemic Lupus Erythematosus" Brain Sciences 8, no. 10: 184. https://doi.org/10.3390/brainsci8100184