
Neuroimmune Support of Neuronal Regeneration and Neuroplasticity following Cerebral Ischemia in Juvenile Mice

 , ,
, ,
Abstract
:
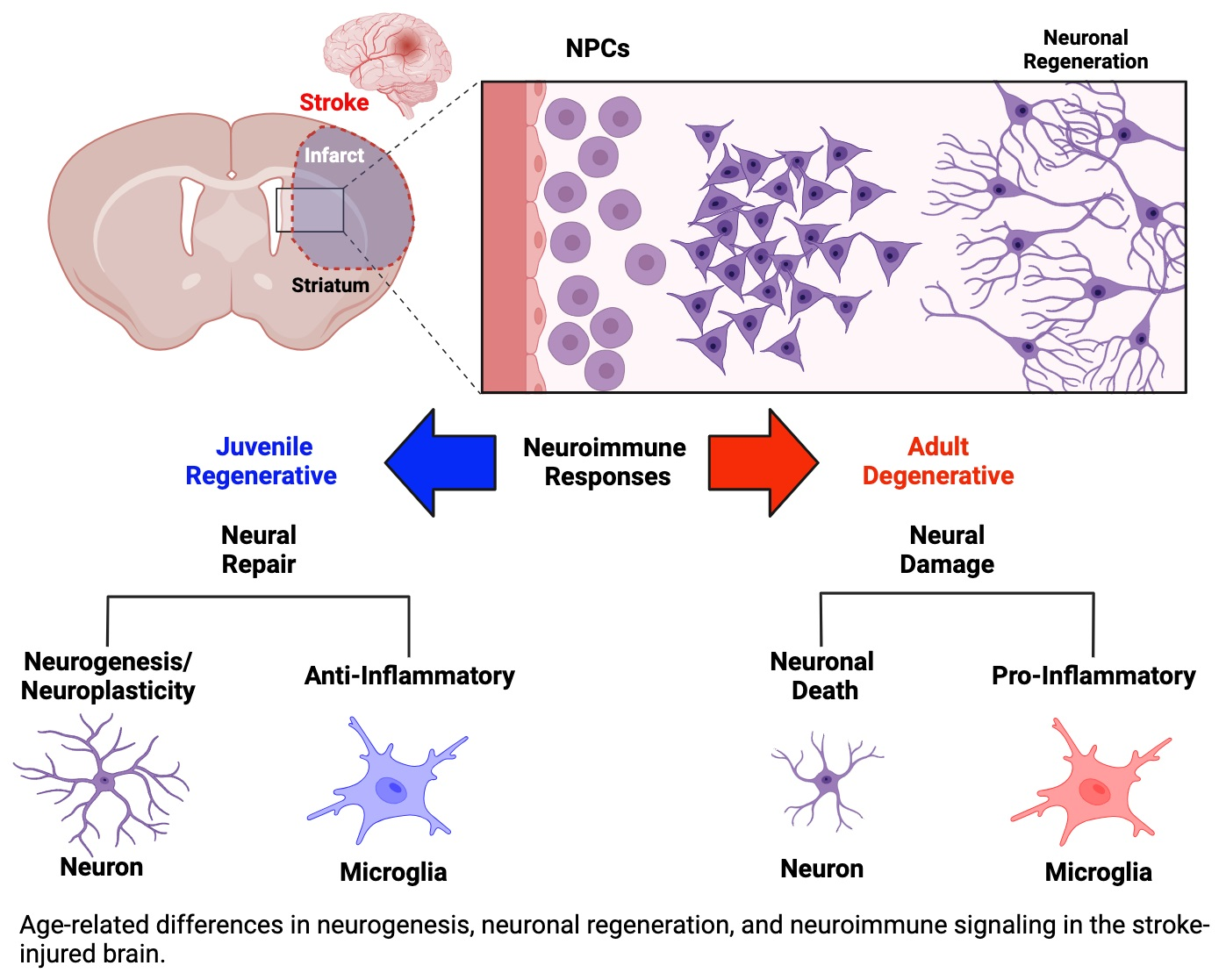{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
3.1. Equivalent Neurogenesis at Acute Time Points
3.2. Neuronal Regeneration in Juveniles at Chronic Time Points
3.3. Neuroimmune Support of Juvenile Neurogenesis and Neuroplasticity
3.4. Age-Associated Neuroimmune Responses Promote Neuroprotective Tissue Preservation after Juvenile Stroke
3.5. Proinflammatory Mediators following Stroke
3.6. Anti-Inflammatory Mediators following Stroke
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Disease, G.B.D.; Injury, I.; Prevalence, C. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1545–1602. [Google Scholar] [CrossRef]
- Shakir, R. The struggle for stroke reclassification. Nat. Rev. Neurol. 2018, 14, 447–448. [Google Scholar] [CrossRef]
- Collaborators, G.B.D.S. Global, regional, and national burden of stroke and its risk factors, 1990-2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol 2021, 20, 795–820. [Google Scholar] [CrossRef]
- Donkor, E.S. Stroke in the 21(st) Century: A Snapshot of the Burden, Epidemiology, and Quality of Life. Stroke Res. Treat. 2018, 2018, 3238165. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.T.; Han, J.; Kim, H. Pediatric stroke recovery: A descriptive analysis. Arch. Phys. Med. Rehabil. 2009, 90, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Anderson, V.; Spencer-Smith, M.; Wood, A. Do children really recover better? Neurobehavioural plasticity after early brain insult. Brain 2011, 134 Pt 8, 2197–2221. [Google Scholar] [CrossRef] [PubMed]
- Elbers, J.; deVeber, G.; Pontigon, A.M.; Moharir, M. Long-term outcomes of pediatric ischemic stroke in adulthood. J. Child. Neurol. 2014, 29, 782–788. [Google Scholar] [CrossRef]
- Hollist, M.; Au, K.; Morgan, L.; Shetty, P.A.; Rane, R.; Hollist, A.; Amaniampong, A.; Kirmani, B.F. Pediatric Stroke: Overview and Recent Updates. Aging Dis. 2021, 12, 1043–1055. [Google Scholar] [CrossRef]
- Bernard, T.J.; Manco-Johnson, M.J.; Lo, W.; MacKay, M.T.; Ganesan, V.; DeVeber, G.; Goldenberg, N.A.; Armstrong-Wells, J.; Dowling, M.M.; Roach, E.S.; et al. Towards a consensus-based classification of childhood arterial ischemic stroke. Stroke 2012, 43, 371–377. [Google Scholar] [CrossRef] [PubMed]
- deVeber, G.; Roach, E.S.; Riela, A.R.; Wiznitzer, M. Stroke in children: Recognition, treatment, and future directions. Semin. Pediatr. Neurol. 2000, 7, 309–317. [Google Scholar] [CrossRef]
- Beslow, L.A.; Dowling, M.M.; Hassanein, S.M.A.; Lynch, J.K.; Zafeiriou, D.; Sun, L.R.; Kopyta, I.; Titomanlio, L.; Kolk, A.; Chan, A.; et al. Mortality After Pediatric Arterial Ischemic Stroke. Pediatrics 2018, 141, e20174146. [Google Scholar] [CrossRef] [PubMed]
- Felling, R.J.; Sun, L.R.; Maxwell, E.C.; Goldenberg, N.; Bernard, T. Pediatric arterial ischemic stroke: Epidemiology, risk factors, and management. Blood Cells Mol. Dis. 2017, 67, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Malone, L.A.; Felling, R.J. Pediatric Stroke: Unique Implications of the Immature Brain on Injury and Recovery. Pediatr. Neurol. 2020, 102, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Harrison, T.C.; Silasi, G.; Boyd, J.D.; Murphy, T.H. Displacement of sensory maps and disorganization of motor cortex after targeted stroke in mice. Stroke 2013, 44, 2300–2306. [Google Scholar] [CrossRef]
- Jones, T.A.; Adkins, D.L. Motor System Reorganization After Stroke: Stimulating and Training Toward Perfection. Physiology 2015, 30, 358–370. [Google Scholar] [CrossRef]
- Parent, J.M.; Vexler, Z.S.; Gong, C.; Derugin, N.; Ferriero, D.M. Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann. Neurol. 2002, 52, 802–813. [Google Scholar] [CrossRef]
- Jin, K.; Wang, X.; Xie, L.; Mao, X.O.; Zhu, W.; Wang, Y.; Shen, J.; Mao, Y.; Banwait, S.; Greenberg, D.A. Evidence for stroke-induced neurogenesis in the human brain. Proc. Natl. Acad. Sci. USA 2006, 103, 13198–13202. [Google Scholar] [CrossRef]
- Parent, J.M.; Silverstein, F.S. Replacing neocortical neurons after stroke. Ann. Neurol. 2007, 61, 185–186. [Google Scholar] [CrossRef]
- Arvidsson, A.; Collin, T.; Kirik, D.; Kokaia, Z.; Lindvall, O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 2002, 8, 963–970. [Google Scholar] [CrossRef]
- Lichtenwalner, R.J.; Parent, J.M. Adult neurogenesis and the ischemic forebrain. J. Cereb. Blood Flow. Metab. 2006, 26, 1–20. [Google Scholar] [CrossRef]
- Danzer, S.C. Postnatal and adult neurogenesis in the development of human disease. Neuroscientist 2008, 14, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, K.M.; Ahrendsen, J.T.; Patsos, O.P.; Strnad, F.A.; Yonchek, J.C.; Traystman, R.J.; Macklin, W.B.; Herson, P.S. Endogenous Neuronal Replacement in the Juvenile Brain Following Cerebral Ischemia. Neuroscience 2018, 380, 45. [Google Scholar] [CrossRef]
- Gemma, C.; Bachstetter, A.D. The role of microglia in adult hippocampal neurogenesis. Front. Cell Neurosci. 2013, 7, 229. [Google Scholar] [CrossRef]
- Rodriguez-Iglesias, N.; Sierra, A.; Valero, J. Rewiring of Memory Circuits: Connecting Adult Newborn Neurons With the Help of Microglia. Front. Cell Dev. Biol. 2019, 7, 24. [Google Scholar] [CrossRef]
- Chan, J.K.; Roth, J.; Oppenheim, J.J.; Tracey, K.J.; Vogl, T.; Feldmann, M.; Horwood, N.; Nanchahal, J. Alarmins: Awaiting a clinical response. J. Clin. Investig. 2012, 122, 2711–2719. [Google Scholar] [CrossRef]
- Rider, P.; Voronov, E.; Dinarello, C.A.; Apte, R.N.; Cohen, I. Alarmins: Feel the Stress. J. Immunol. 2017, 198, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Anrather, J.; Iadecola, C. Inflammation and Stroke: An Overview. Neurotherapeutics 2016, 13, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Casse, F.; Richetin, K.; Toni, N. Astrocytes’ Contribution to Adult Neurogenesis in Physiology and Alzheimer’s Disease. Front. Cell Neurosci. 2018, 12, 432. [Google Scholar] [CrossRef]
- Zhang, D.; Hu, X.; Qian, L.; O’Callaghan, J.P.; Hong, J.S. Astrogliosis in CNS pathologies: Is there a role for microglia? Mol. Neurobiol. 2010, 41, 232–241. [Google Scholar] [CrossRef]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and foe for ischemic stroke. J. Neuroinflamm. 2019, 16, 142. [Google Scholar] [CrossRef]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Ore, M.V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Savman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef]
- Zhao, S.C.; Ma, L.S.; Chu, Z.H.; Xu, H.; Wu, W.Q.; Liu, F. Regulation of microglial activation in stroke. Acta Pharmacol. Sin. 2017, 38, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Guruswamy, R.; ElAli, A. Complex Roles of Microglial Cells in Ischemic Stroke Pathobiology: New Insights and Future Directions. Int. J. Mol. Sci. 2017, 18, 496. [Google Scholar] [CrossRef]
- Zera, K.A.; Buckwalter, M.S. The Local and Peripheral Immune Responses to Stroke: Implications for Therapeutic Development. Neurotherapeutics 2020, 17, 414–435. [Google Scholar] [CrossRef] [PubMed]
- Ritzel, R.M.; Patel, A.R.; Grenier, J.M.; Crapser, J.; Verma, R.; Jellison, E.R.; McCullough, L.D. Functional differences between microglia and monocytes after ischemic stroke. J. Neuroinflamm. 2015, 12, 106. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Crichlow, G.V.; Vermeire, J.J.; Leng, L.; Du, X.; Hodsdon, M.E.; Bucala, R.; Cappello, M.; Gross, M.; Gaeta, F.; et al. Allosteric inhibition of macrophage migration inhibitory factor revealed by ibudilast. Proc. Natl. Acad. Sci. USA 2010, 107, 11313–11318. [Google Scholar] [CrossRef]
- Ha, W.; Sevim-Nalkiran, H.; Zaman, A.M.; Matsuda, K.; Khasraw, M.; Nowak, A.K.; Chung, L.; Baxter, R.C.; McDonald, K.L. Ibudilast sensitizes glioblastoma to temozolomide by targeting Macrophage Migration Inhibitory Factor (MIF). Sci. Rep. 2019, 9, 2905. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Qin, L.; Kim, E.; Bhosle, S.; Guo, H.; Febbraio, M.; Haskew-Layton, R.E.; Ratan, R.; Cho, S. CD36 is involved in astrocyte activation and astroglial scar formation. J. Cereb. Blood Flow. Metab. 2012, 32, 1567–1577. [Google Scholar] [CrossRef] [PubMed]
- Rolan, P.; Gibbons, J.A.; He, L.; Chang, E.; Jones, D.; Gross, M.I.; Davidson, J.B.; Sanftner, L.M.; Johnson, K.W. Ibudilast in healthy volunteers: Safety, tolerability and pharmacokinetics with single and multiple doses. Br. J. Clin. Pharmacol. 2008, 66, 792–801. [Google Scholar] [CrossRef]
- Herson, P.S.; Bombardier, C.G.; Parker, S.M.; Shimizu, T.; Klawitter, J.; Quillinan, N.; Exo, J.L.; Goldenberg, N.A.; Traystman, R.J. Experimental pediatric arterial ischemic stroke model reveals sex-specific estrogen signaling. Stroke 2013, 44, 759–763. [Google Scholar] [CrossRef]
- Ming, G.L.; Song, H. Adult neurogenesis in the mammalian central nervous system. Annu. Rev. Neurosci. 2005, 28, 223–250. [Google Scholar] [CrossRef]
- Liu, F.; McCullough, L.D. The middle cerebral artery occlusion model of transient focal cerebral ischemia. Methods Mol. Biol. 2014, 1135, 81–93. [Google Scholar] [CrossRef]
- Ahrendsen, J.T.; Grewal, H.S.; Hickey, S.P.; Culp, C.M.; Gould, E.A.; Shimizu, T.; Strnad, F.A.; Traystman, R.J.; Herson, P.S.; Macklin, W.B. Juvenile striatal white matter is resistant to ischemia-induced damage. Glia 2016, 64, 1972–1986. [Google Scholar] [CrossRef] [PubMed]
- Bercum, F.M.; Rodgers, K.M.; Benison, A.M.; Smith, Z.Z.; Taylor, J.; Kornreich, E.; Grabenstatter, H.L.; Dudek, F.E.; Barth, D.S. Maternal Stress Combined with Terbutaline Leads to Comorbid Autistic-Like Behavior and Epilepsy in a Rat Model. J. Neurosci. 2015, 35, 15894–15902. [Google Scholar] [CrossRef]
- Rodgers, K.M.; Dudek, F.E.; Barth, D.S. Progressive, Seizure-Like, Spike-Wave Discharges Are Common in Both Injured and Uninjured Sprague-Dawley Rats: Implications for the Fluid Percussion Injury Model of Post-Traumatic Epilepsy. J. Neurosci. 2015, 35, 9194–9204. [Google Scholar] [CrossRef]
- Rolan, P.; Hutchinson, M.; Johnson, K. Ibudilast: A review of its pharmacology, efficacy and safety in respiratory and neurological disease. Expert Opin. Pharmacother. 2009, 10, 2897–2904. [Google Scholar] [CrossRef]
- Mizuno, T.; Kurotani, T.; Komatsu, Y.; Kawanokuchi, J.; Kato, H.; Mitsuma, N.; Suzumura, A. Neuroprotective role of phosphodiesterase inhibitor ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology 2004, 46, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Angelopoulou, E.; Pyrgelis, E.S.; Piperi, C. Emerging Potential of the Phosphodiesterase (PDE) Inhibitor Ibudilast for Neurodegenerative Diseases: An Update on Preclinical and Clinical Evidence. Molecules 2022, 27, 8448. [Google Scholar] [CrossRef]
- Litak, J.; Grochowski, C.; Litak, J.; Osuchowska, I.; Gosik, K.; Radzikowska, E.; Kamieniak, P.; Rolinski, J. TLR-4 Signaling vs. Immune Checkpoints, miRNAs Molecules, Cancer Stem Cells, and Wingless-Signaling Interplay in Glioblastoma Multiforme-Future Perspectives. Int. J. Mol. Sci. 2020, 21, 3114. [Google Scholar] [CrossRef]
- Ohashi, M.; Ohkubo, H.; Kito, J.; Nishino, K. A new vasodilator 3-isobutyryl-2-isopropylpyrazolo[1,5-a]pyridine (KC-404) has a dual mechanism of action on platelet aggregation. Arch. Int. Pharmacodyn. Ther. 1986, 283, 321–334. [Google Scholar] [PubMed]
- Fukuyama, H.; Kimura, J.; Yamaguchi, S.; Yamauchi, H.; Ogawa, M.; Doi, T.; Yonekura, Y.; Konishi, J. Pharmacological effects of ibudilast on cerebral circulation: A PET study. Neurol. Res. 1993, 15, 169–173. [Google Scholar] [CrossRef]
- Inoue, N.; Harada, M. Effect of ibudilast on non-specific symptoms in patients with chronic cerebral ischemia. Analysis of cerebral blood flow. Arzneimittelforschung 2008, 58, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Cho, E.; Ko, Y.E.; Kim, I.; Lee, K.J.; Kwon, S.U.; Kang, D.W.; Kim, J.S. Ibudilast, a phosphodiesterase inhibitor with anti-inflammatory activity, protects against ischemic brain injury in rats. Brain Res. 2012, 1431, 97–106. [Google Scholar] [CrossRef]
- Inoue, N.; Fukuda, S.; Inada, T.; Sameshima, E.; Tokushima, Y.; Harada, M. Effect of ibudilast on the reciprocal inhibitory visual-vestibular interaction closely related to dizziness after cerebral ischemia. J. Stroke Cerebrovasc. Dis. 2014, 23, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Kruger, C.; Steigleder, T.; Weber, D.; Pitzer, C.; Laage, R.; Aronowski, J.; Maurer, M.H.; Gassler, N.; Mier, W.; et al. The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. J. Clin. Investig. 2005, 115, 2083–2098. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, S.T. Cellular and molecular mechanisms of neural repair after stroke: Making waves. Ann. Neurol. 2006, 59, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Eminaga, S.; Teekakirikul, P.; Seidman, C.E.; Seidman, J.G. Detection of Cell Proliferation Markers by Immunofluorescence Staining and Microscopy Imaging in Paraffin-Embedded Tissue Sections. Curr. Protoc. Mol. Biol. 2016, 115, 142511–142514. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Kim, H.Y.; Rogowska, J.; Zhao, B.Q.; Bhide, P.; Parent, J.M.; Lo, E.H. Involvement of matrix metalloproteinase in neuroblast cell migration from the subventricular zone after stroke. J. Neurosci. 2006, 26, 3491–3495. [Google Scholar] [CrossRef]
- Liu, X.S.; Chopp, M.; Zhang, R.L.; Zhang, Z.G. MicroRNAs in cerebral ischemia-induced neurogenesis. J. Neuropathol. Exp. Neurol. 2013, 72, 718–722. [Google Scholar] [CrossRef]
- Tobin, M.K.; Bonds, J.A.; Minshall, R.D.; Pelligrino, D.A.; Testai, F.D.; Lazarov, O. Neurogenesis and inflammation after ischemic stroke: What is known and where we go from here. J. Cereb. Blood Flow. Metab. 2014, 34, 1573–1584. [Google Scholar] [CrossRef]
- Thored, P.; Arvidsson, A.; Cacci, E.; Ahlenius, H.; Kallur, T.; Darsalia, V.; Ekdahl, C.T.; Kokaia, Z.; Lindvall, O. Persistent production of neurons from adult brain stem cells during recovery after stroke. Stem Cells 2006, 24, 739–747. [Google Scholar] [CrossRef]
- Zhao, C.; Deng, W.; Gage, F.H. Mechanisms and functional implications of adult neurogenesis. Cell 2008, 132, 645–660. [Google Scholar] [CrossRef] [PubMed]
- Bang, O.Y.; Kim, E.H.; Cha, J.M.; Moon, G.J. Adult Stem Cell Therapy for Stroke: Challenges and Progress. J. Stroke 2016, 18, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Chrostek, M.R.; Fellows, E.G.; Crane, A.T.; Grande, A.W.; Low, W.C. Efficacy of stem cell-based therapies for stroke. Brain Res. 2019, 1722, 146362. [Google Scholar] [CrossRef]
- Shan, X.Q.; Luo, Y.Y.; Chang, J.; Song, J.J.; Hao, N.; Zhao, L. Immunomodulation: The next target of mesenchymal stem cell-derived exosomes in the context of ischemic stroke. World J. Stem Cells 2023, 15, 52–70. [Google Scholar] [CrossRef]
- Rawanduzy, C.A.; Earl, E.; Mayer, G.; Lucke-Wold, B. Pediatric Stroke: A Review of Common Etiologies and Management Strategies. Biomedicines 2022, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Veltkamp, R.; Gill, D. Clinical Trials of Immunomodulation in Ischemic Stroke. Neurotherapeutics 2016, 13, 791–800. [Google Scholar] [CrossRef]
- Santos Samary, C.; Pelosi, P.; Leme Silva, P.; Rieken Macedo Rocco, P. Immunomodulation after ischemic stroke: Potential mechanisms and implications for therapy. Crit. Care 2016, 20, 391. [Google Scholar] [CrossRef]
- Rabiller, G.; He, J.W.; Nishijima, Y.; Wong, A.; Liu, J. Perturbation of Brain Oscillations after Ischemic Stroke: A Potential Biomarker for Post-Stroke Function and Therapy. Int. J. Mol. Sci. 2015, 16, 25605–25640. [Google Scholar] [CrossRef]
- Foreman, B.; Claassen, J. Quantitative EEG for the detection of brain ischemia. Crit. Care 2012, 16, 216. [Google Scholar] [CrossRef] [PubMed]
- Glezer, I.; Simard, A.R.; Rivest, S. Neuroprotective role of the innate immune system by microglia. Neuroscience 2007, 147, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Han, X.; Li, Q.; Yang, Q.W.; Wang, J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat. Rev. Neurol. 2017, 13, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Ikegaya, Y.; Koyama, R. The effects of microglia- and astrocyte-derived factors on neurogenesis in health and disease. Eur. J. Neurosci. 2021, 54, 5880–5901. [Google Scholar] [CrossRef] [PubMed]
- Wellman, S.M.; Cambi, F.; Kozai, T.D. The role of oligodendrocytes and their progenitors on neural interface technology: A novel perspective on tissue regeneration and repair. Biomaterials 2018, 183, 200–217. [Google Scholar] [CrossRef]
- Bai, M.; Sun, R.; Cao, B.; Feng, J.; Wang, J. Monocyte-related cytokines/chemokines in cerebral ischemic stroke. CNS Neurosci. Ther. 2023. [Google Scholar] [CrossRef]
- Allan, S.M.; Rothwell, N.J. Cytokines and acute neurodegeneration. Nat. Rev. Neurosci. 2001, 2, 734–744. [Google Scholar] [CrossRef]
- Allan, S.M.; Tyrrell, P.J.; Rothwell, N.J. Interleukin-1 and neuronal injury. Nat. Rev. Immunol. 2005, 5, 629–640. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, H.; Sun, G.; Zhang, J.; Edwards, N.J.; Aronowski, J. Neuronal Interleukin-4 as a Modulator of Microglial Pathways and Ischemic Brain Damage. J. Neurosci. 2015, 35, 11281–11291. [Google Scholar] [CrossRef]
- Rossi, C.; Cusimano, M.; Zambito, M.; Finardi, A.; Capotondo, A.; Garcia-Manteiga, J.M.; Comi, G.; Furlan, R.; Martino, G.; Muzio, L. Interleukin 4 modulates microglia homeostasis and attenuates the early slowly progressive phase of amyotrophic lateral sclerosis. Cell Death Dis. 2018, 9, 250. [Google Scholar] [CrossRef]
- Yuan, J.; Ge, H.; Liu, W.; Zhu, H.; Chen, Y.; Zhang, X.; Yang, Y.; Yin, Y.; Chen, W.; Wu, W.; et al. M2 microglia promotes neurogenesis and oligodendrogenesis from neural stem/progenitor cells via the PPARgamma signaling pathway. Oncotarget 2017, 8, 19855–19865. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Ziv, Y.; Schwartz, A.; Landa, G.; Talpalar, A.E.; Pluchino, S.; Martino, G.; Schwartz, M. Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol. Cell Neurosci. 2006, 31, 149–160. [Google Scholar] [CrossRef]
- Ooboshi, H.; Ibayashi, S.; Shichita, T.; Kumai, Y.; Takada, J.; Ago, T.; Arakawa, S.; Sugimori, H.; Kamouchi, M.; Kitazono, T.; et al. Postischemic gene transfer of interleukin-10 protects against both focal and global brain ischemia. Circulation 2005, 111, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Lambertsen, K.L.; Finsen, B.; Clausen, B.H. Post-stroke inflammation-target or tool for therapy? Acta Neuropathol. 2019, 137, 693–714. [Google Scholar] [CrossRef] [PubMed]
- Biscaro, B.; Lindvall, O.; Tesco, G.; Ekdahl, C.T.; Nitsch, R.M. Inhibition of microglial activation protects hippocampal neurogenesis and improves cognitive deficits in a transgenic mouse model for Alzheimer’s disease. Neurodegener. Dis. 2012, 9, 187–198. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marquez-Ortiz, R.A.; Tesic, V.; Hernandez, D.R.; Akhter, B.; Aich, N.; Boudreaux, P.M.; Clemons, G.A.; Wu, C.Y.-C.; Lin, H.W.; Rodgers, K.M. Neuroimmune Support of Neuronal Regeneration and Neuroplasticity following Cerebral Ischemia in Juvenile Mice. Brain Sci. 2023, 13, 1337. https://doi.org/10.3390/brainsci13091337
Marquez-Ortiz RA, Tesic V, Hernandez DR, Akhter B, Aich N, Boudreaux PM, Clemons GA, Wu CY-C, Lin HW, Rodgers KM. Neuroimmune Support of Neuronal Regeneration and Neuroplasticity following Cerebral Ischemia in Juvenile Mice. Brain Sciences. 2023; 13(9):1337. https://doi.org/10.3390/brainsci13091337
Chicago/Turabian StyleMarquez-Ortiz, Ricaurte A., Vesna Tesic, Daniel R. Hernandez, Bilkis Akhter, Nibedita Aich, Porter M. Boudreaux, Garrett A. Clemons, Celeste Yin-Chieh Wu, Hung Wen Lin, and Krista M. Rodgers. 2023. "Neuroimmune Support of Neuronal Regeneration and Neuroplasticity following Cerebral Ischemia in Juvenile Mice" Brain Sciences 13, no. 9: 1337. https://doi.org/10.3390/brainsci13091337