
Diagnosis and Management of Seronegative Myasthenia Gravis: Lights and Shadows

 , and
, and 
Abstract
:1. Introduction
2. Epidemiological and Clinical Aspects
3. The Role of Neurophysiological Testing
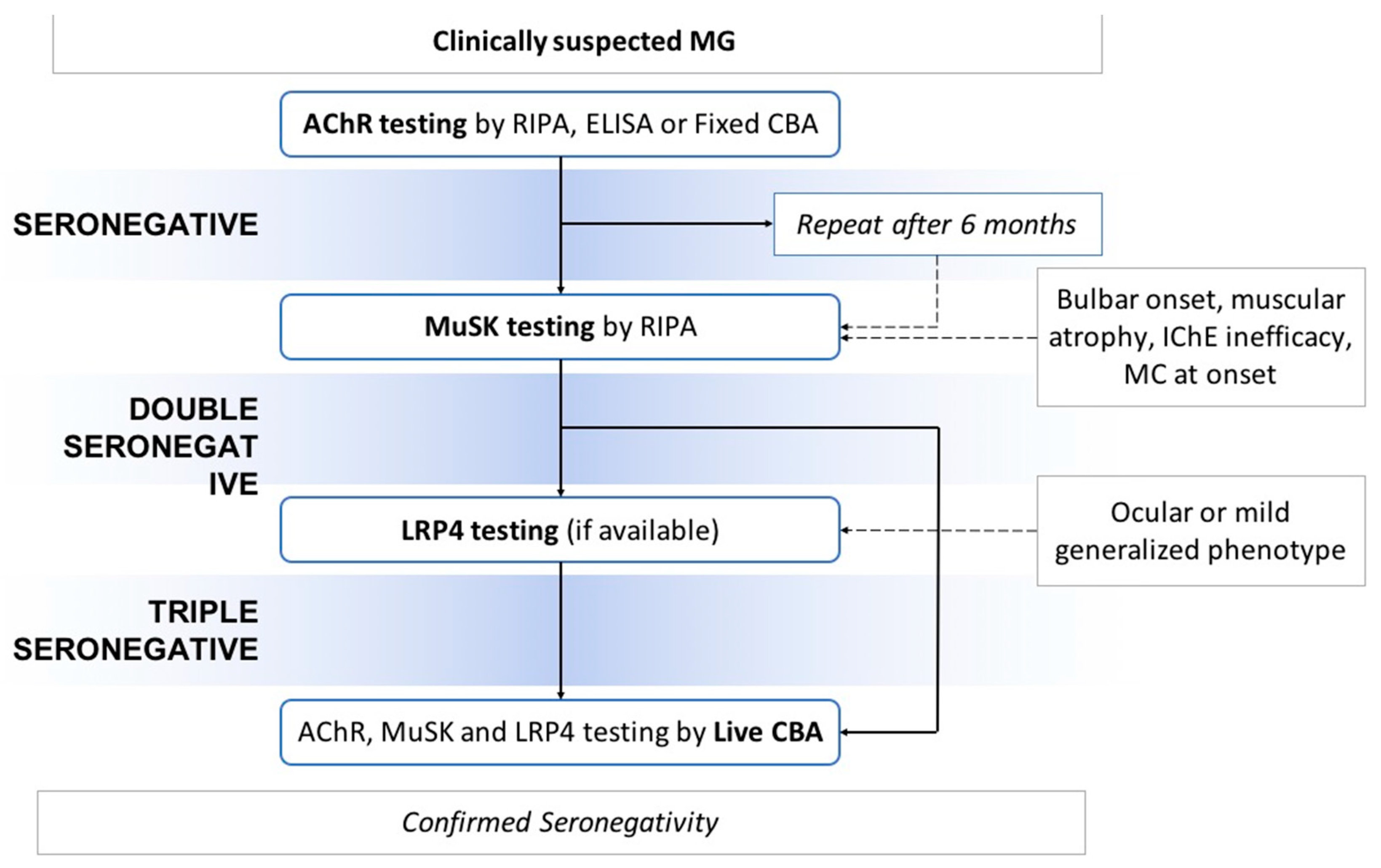
4. Serological Diagnosis: Different Methods of Antibodies’ Detection
5. Differential Diagnosis
6. Treatment
7. Conclusion Remarks and Future Directions
8. Study Limitations
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gilhus, N.E.; Tzartos, S.; Evoli, A.; Palace, J.; Burns, T.M.; Verschuuren, J.J.G.M. Myasthenia gravis. Nat. Rev. Dis. Primers 2019, 2, 30. [Google Scholar] [CrossRef]
- Gilhus, N.E.; Verschuuren, J.J. Myasthenia gravis: Subgroup classification and therapeutic strategies. Lancet Neurol. 2015, 14, 1023–1036. [Google Scholar] [CrossRef]
- Gilhus, N.E.; Skeie, G.O.; Romi, F.; Lazaridis, K.; Zisimopoulou, P.; Tzartos, S. Myasthenia gravis—autoantibody characteristics and their implications for therapy. Nat. Rev. Neurol. 2016, 12, 259–268. [Google Scholar] [CrossRef]
- Evoli, A.; Spagni, G.; Monte, G.; Damato, V. Heterogeneity in myasthenia gravis: Considerations for disease management. Expert. Rev. Clin. Immunol. 2021, 17, 761–771. [Google Scholar] [CrossRef]
- Verschuuren, J.J.; Huijbers, M.G.; Plomp, J.J.; Niks, E.H.; Molenaar, P.C.; Martinez-Martinez, P.; Gomez, A.M.; De Baets, M.H.; Losen, M. Pathophysiology of myasthenia gravis with antibodies to the acetylcholine receptor, muscle-specific kinase and low-density lipoprotein receptor-related protein 4. Autoimmun. Rev. 2013, 12, 918–923. [Google Scholar] [CrossRef]
- Hoch, W.; McConville, J.; Helms, S.; Newsom-Davis, J.; Melms, A.; Vincent, A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat. Med. 2001, 7, 365–368. [Google Scholar] [CrossRef]
- Zhang, B.; Tzartos, J.S.; Belimezi, M.; Ragheb, S.; Bealmear, B.; Lewis, R.A.; Xiong, W.-C.; Lisak, R.P.; Tzartos, S.J.; Mei, L. Autoantibodies to Lipoprotein-Related Protein 4 in Patients With Double-Seronegative Myasthenia Gravis. Arch. Neurol. 2012, 69, 445–451. [Google Scholar]
- Zisimopoulou, P.; Evangelakou, P.; Tzartos, J.; Lazaridis, K.; Zouvelou, V.; Mantegazza, R.; Antozzi, C.; Andreetta, F.; Evoli, A.; Deymeer, F.; et al. A comprehensive analysis of the epidemiology and clinical characteristics of anti-LRP4 in myasthenia gravis. J. Autoimmun. 2014, 52, 139–145. [Google Scholar] [CrossRef]
- Zhang, B.; Shen, C.; Bealmear, B.; Ragheb, S.; Xiong, W.-C.; Lewis, R.A.; Lisak, R.P.; Mei, L. Autoantibodies to agrin in myasthenia gravis patients. PLoS ONE 2014, 9, e91816. [Google Scholar] [CrossRef]
- Witzemann, V.; Chevessier, F.; Pacifici, P.G.; Yampolsky, P. The neuromuscular junction: Selective remodeling of synaptic regulators at the nerve/muscle interface. Mech. Dev. 2013, 130, 402–411. [Google Scholar] [CrossRef]
- Gasperi, C.; Melms, A.; Schoser, B.; Zhang, Y.; Meltoranta, J.; Risson, V.; Schaeffer, L.; Schalke, B.; Kröger, S. Anti-agrin autoantibodies in myasthenia gravis. Neurology 2014, 82, 1976–1983. [Google Scholar] [CrossRef] [PubMed]
- Leite, M.I.; Jacob, S.; Viegas, S.; Cossins, J.; Clover, L.; Morgan, B.P.; Beeson, D.; Willcox, N.; Vincent, A. IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis. Brain 2008, 131 Pt 7, 1940–1952. [Google Scholar] [CrossRef] [PubMed]
- Tsonis, A.I.; Zisimopoulou, P.; Lazaridis, K.; Tzartos, J.; Matsigkou, E.; Zouvelou, V.; Mantegazza, R.; Antozzi, C.; Andreetta, F.; Evoli, A.; et al. MuSK autoantibodies in myasthenia gravis detected by cell based assay—A multinational study. J. Neuroimmunol. 2015, 284, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Jacob, S.; Viegas, S.; Leite, M.I.; Webster, R.; Cossins, J.; Kennett, R.; Hilton-Jones, D.; Morgan, B.P.; Vincent, A. Presence and pathogenic relevance of antibodies to clustered acetylcholine receptor in ocular and generalized myasthenia gravis. Arch. Neurol. 2012, 69, 994–1001. [Google Scholar] [CrossRef]
- Engel, A.G.; Shen, X.M.; Selcen, D.; Sine, S.M. Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015, 14, 420–434. [Google Scholar] [CrossRef]
- Kao, J.C.; Milone, M.; Selcen, D.; Shen, X.-M.; Engel, A.G.; Liewluck, T. Congenital Myasthenic Syndromes In Adult Neurology Clinic. A Long Road To Diagnosis And Therapy. Neurology 2018, 91, E1770–E1777. [Google Scholar] [CrossRef]
- Vincent, A.; Leite, M.I.; Farrugia, M.E.; Jacob, S.; Viegas, S.; Shiraishi, H.; Benveniste, O.; Morgan, B.P.; Hilton-Jones, D.; Newsom-Davis, J.; et al. Myasthenia Gravis Seronegative for Acetylcholine Receptor Antibodies. Ann. N. Y. Acad. Sci. 2008, 1132, 84–92. [Google Scholar] [CrossRef]
- Oh, S.J.; Morgan, M.B.; Lu, L.; Hatanaka, Y.; Hemmi, S.; Young, A.; Claussen, G.C. Racial differences in myasthenia gravis in Alabama. Muscle Nerve 2009, 39, 328–332. [Google Scholar] [CrossRef]
- Park, K.H.; Waters, P.; Woodhall, M.; Lang, B.; Smith, T.; Sung, J.-J.; Kim, K.-K.; Lim, Y.-M.; Kim, J.-E.; Kim, B.-J.; et al. Myasthenia gravis seronegative for acetylcholine receptor antibodies in South Korea: Autoantibody profiles and clinical features. PLoS ONE 2018, 13, e0193723. [Google Scholar]
- Romi, F.; Aarli, J.A.; Gilhus, N.E. Seronegative myasthenia gravis: Disease severity and prognosis. Eur. J. Neurol. 2005, 12, 413–418. [Google Scholar] [CrossRef]
- Li, W.; Liu, P.; Cui, W.; Wang, S.; Ji, Y.; Zhang, L.; He, X.; Zhou, S.; Shen, T.; Zhao, X.; et al. Clinical characteristics of anti-AChR-MuSK-LRP4 antibody-negative myasthenia gravis in China. Muscle Nerve 2023, 67, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Witoonpanich, R.; Dejthevaporn, C.; Sriphrapradang, A.; Pulkes, T. Electrophysiological and immunological study in myasthenia gravis: Diagnostic sensitivity and correlation. Clin. Neurophysiol. 2011, 122, 1873–1877. [Google Scholar] [CrossRef]
- Harrison, P.; Barton, J.; Winkel, A. Chronic mimics of myasthenia gravis: A retrospective case series. Neuromuscul. Disord. 2023, 33, 250–256. [Google Scholar] [CrossRef]
- Padua, L.; Caliandro, P.; Di Iasi, G.; Pazzaglia, C.; Ciaraffa, F.; Evoli, A. Reliability of SFEMG in diagnosing myasthenia gravis: Sensitivity and specificity calculated on 100 prospective cases. Clin. Neurophysiol. 2014, 125, 1270–1273. [Google Scholar] [CrossRef] [PubMed]
- AAEM Quality Assurance Committee; American Association of Electrodiagnostic Medicine. Literature review of the usefulness of repetitive nerve stimulation and single fiber EMG in the electrodiagnostic evaluation of patients with suspected myasthenia gravis or Lambert-Eaton myasthenic syndrome. Muscle Nerve 2001, 24, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.L.; Najjar, R.P.; Teo, K.Y.; Tow, S.L.; Loo, J.L.; Milea, D. A reappraisal of diagnostic tests for myasthenia gravis in a large Asian cohort. J. Neurol. Sci. 2017, 376, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Andrapalliyal, N.; Claytor, B.; Li, Y. Incidence and causes of overdiagnosis of myasthenia gravis. Muscle Nerve 2023, 67, 464–468. [Google Scholar] [CrossRef]
- Gastaldi, M.; Scaranzin, S.; Businaro, P.; Mobilia, E.; Benedetti, L.; Pesce, G.; Franciotta, D. Improving laboratory diagnostics in myasthenia gravis. Expert. Rev. Mol. Diagn. 2021, 21, 579–590. [Google Scholar] [CrossRef]
- Vernino, S. Unraveling the Enigma of Seronegative Myasthenia Gravis. JAMA Neurol. 2015, 72, 630–631. [Google Scholar] [CrossRef]
- Hong, Y.; Zisimopoulou, P.; Trakas, N.; Karagiorgou, K.; Stergiou, C.; Skeie, G.O.; Hao, H.-J.; Gao, X.; Owe, J.F.; Zhang, X.; et al. Multiple antibody detection in ‘seronegative’ myasthenia gravis patients. Eur. J. Neurol. 2017, 24, 844–850. [Google Scholar] [CrossRef]
- Maddison, P.; Sadalage, G.; Ambrose, P.A.; Jacob, S.; Vincent, A. False-positive acetylcholine receptor antibody results in patients without myasthenia gravis. J. Neuroimmunol. 2019, 332, 69–72. [Google Scholar] [CrossRef]
- Hewer, R.; Matthews, I.; Chen, S.; McGrath, V.; Evans, M.; Roberts, E.; Nute, S.; Sanders, J.; Furmaniak, J.; Smith, B.R. A sensitive non-isotopic assay for acetylcholine receptor autoantibodies. Clin. Chim. Acta 2006, 364, 159–166. [Google Scholar] [CrossRef]
- Oger, J.; Frykman, H. An update on laboratory diagnosis in myasthenia gravis. Clin. Chim. Acta 2015, 449, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Cruz, P.M.R.; Al-Hajjar, M.; Huda, S.; Jacobson, L.; Woodhall, M.; Jayawant, S.; Buckley, C.; Hilton-Jones, D.; Beeson, D.; Vincent, A.; et al. Clinical Features and Diagnostic Usefulness of Antibodies to Clustered Acetylcholine Receptors in the Diagnosis of Seronegative Myasthenia Gravis. JAMA Neurol. 2015, 72, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Spagni, G.; Gastaldi, M.; Businaro, P.; Chemkhi, Z.; Carrozza, C.; Mascagna, G.; Falso, S.; Scaranzin, S.; Franciotta, D.; Evoli, A.; et al. Comparison of Fixed and Live Cell-Based Assay for the Detection of AChR and MuSK Antibodies in Myasthenia Gravis. Neurol. Neuroimmunol. Neuroinflamm. 2022, 10, e200038. [Google Scholar] [CrossRef] [PubMed]
- Mirian, A.; Nicolle, M.W.; Edmond, P.; Budhram, A. Comparison of fixed cell-based assay to radioimmunoprecipitation assay for acetylcholine receptor antibody detection in myasthenia gravis. J. Neurol. Sci. 2022, 432, 120084. [Google Scholar] [CrossRef]
- Gambino, C.M.; Agnello, L.; Sasso, B.L.; Scazzone, C.; Giglio, R.V.; Candore, G.; Ciaccio, A.M.; Di Stefano, V.; Brighina, F.; Vidali, M.; et al. Comparative Analysis of BIOCHIP Mosaic-Based Indirect Immunofluorescence with Enzyme-Linked Immunosorbent Assay for Diagnosing Myasthenia Gravis. Diagnostics 2021, 11, 2098. [Google Scholar] [CrossRef]
- Yang, A.; Xuan, R.; Melbourne, W.; Hashimoto, T.; Uzun, S.; Daneshpazhooh, M.; Yamagami, J.; Di Zenzo, G.; Mascaro, J.; Mahmoudi, H.; et al. Inter-rater reliability of the BIOCHIP indirect immunofluorescence dermatology mosaic in bullous pemphigoid and pemphigus patients. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 2327–2333. [Google Scholar] [CrossRef]
- Gambino, C.M.; Agnello, L.; Ciaccio, A.M.; Scazzone, C.; Vidali, M.; Di Stefano, V.; Milano, S.; Brighina, F.; Candore, G.; Sasso, B.L.; et al. Detection of Antibodies against the Acetylcholine Receptor in Patients with Myasthenia Gravis: A Comparison of Two Enzyme Immunoassays and a Fixed Cell-Based Assay. J. Clin. Med. 2023, 12, 4781. [Google Scholar] [CrossRef]
- Huda, S.; Waters, P.; Woodhall, M.; Leite, M.I.; Jacobson, L.; De Rosa, A.; Maestri, M.; Ricciardi, R.; Heckmann, J.M.; Maniaol, A.; et al. IgG-specific cell-based assay detects potentially pathogenic MuSK-Abs in seronegative MG. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e357. [Google Scholar] [CrossRef]
- Tzartos, J.S.; Zisimopoulou, P.; Rentzos, M.; Karandreas, N.; Zouvelou, V.; Evangelakou, P.; Tsonis, A.; Thomaidis, T.; Lauria, G.; Andreetta, F.; et al. LRP4 antibodies in serum and CSF from amyotrophic lateral sclerosis patients. Ann. Clin. Transl. Neurol. 2014, 1, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Rivner, M.H.; Quarles, B.M.; Pan, J.; Yu, Z.; Howard, J.F.; Corse, A.; Dimachkie, M.M.; Jackson, C.; Vu, T.; Small, G.; et al. Clinical features of LRP4/agrin-antibody-positive myasthenia gravis: A multicenter study. Muscle Nerve 2020, 62, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; McConville, J.; Farrugia, M.E.; Newsom-Davis, J. Seronegative myasthenia gravis. Semin. Neurol. 2004, 24, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Padua, L.; Stalberg, E.; LoMonaco, M.; Evoli, A.; Batocchi, A.; Tonali, P. SFEMG in ocular myasthenia gravis. Clin. Neurophysiol. 2000, 111, 1203–1207. [Google Scholar] [CrossRef]
- Hamel, J.I. Myotonic Dystrophy. Continuum 2022, 28, 1715–1734. [Google Scholar] [CrossRef]
- Manousakis, G. Inflammatory Myopathies. Continuum 2022, 28, 1643–1662. [Google Scholar] [CrossRef]
- Rowland, L.P.; Hirano, M.; DiMauro, S.; Schon, E.A. Oculopharyngeal muscular dystrophy, other ocular myopathies, and progressive external ophthalmoplegia. Neuromuscul. Disord. 1997, 7 (Suppl. 1), S15–S21. [Google Scholar] [CrossRef]
- Yamashita, S. Recent Progress in Oculopharyngeal Muscular Dystrophy. J. Clin. Med. 2021, 10, 1375. [Google Scholar] [CrossRef]
- Hirano, M.; Pitceathly, R.D.S. Progressive external ophthalmoplegia. Handb. Clin. Neurol. 2023, 194, 9–21. [Google Scholar]
- Kesner, V.G.; Oh, S.J.; Dimachkie, M.M.; Barohn, R.J. Lambert-Eaton Myasthenic Syndrome. Neurol. Clin. 2018, 36, 379–394. [Google Scholar] [CrossRef]
- Ohno, K.; Ohkawara, B.; Shen, X.M.; Selcen, D.; Engel, A.G. Clinical and Pathologic Features of Congenital Myasthenic Syndromes Caused by 35 Genes-A Comprehensive Review. Int. J. Mol. Sci. 2023, 24, 3730. [Google Scholar] [PubMed]
- Finsterer, J. Congenital myasthenic syndromes. Orphanet J. Rare Dis. 2019, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.J. Horner Syndrome: A Clinical Review. ACS Chem. Neurosci. 2018, 9, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; Yang, J.; Xu, C.; Wei, D.; Luo, L. Myasthenia-like paraneoplastic syndrome with multiple cranial nerve tumor infiltration: A case report and literature review. Medicine 2023, 102, e33774. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Ul-Haq, M.A.; Emsley, H.C. Myasthenia gravis as a ‘stroke mimic’—it’s all in the history. Clin. Med. 2014, 14, 640–642. [Google Scholar]
- Greim, B.; Engel, C.; Apel, A.; Zettl, U.K. Fatigue in neuroimmunological diseases. J. Neurol. 2007, 254 (Suppl. 2), II102-6, Erratum in J. Neurol. 2008, 255, 309–310. [Google Scholar] [CrossRef]
- Tanovska, N.; Novotni, G.; Sazdova-Burneska, S.; Kuzmanovski, I.; Boshkovski, B.; Kondov, G.; Jovanovski-Srceva, M.; Kokareva, A.; Isjanovska, R. Myasthenia Gravis and Associated Diseases. Open Access Maced. J. Med. Sci. 2018, 6, 472–478. [Google Scholar] [CrossRef]
- Zouvelou, V.; Zisimopoulou, P.; Rentzos, M.; Karandreas, N.; Evangelakou, P.; Stamboulis, E.; Tzartos, S.J. Double seronegative myasthenia gravis with anti-LRP 4 antibodies. Neuromuscul. Disord. 2013, 23, 568–570. [Google Scholar] [CrossRef]
- Evoli, A.; Bianchi, M.R.; Riso, R.; Minicuci, G.M.; Batocchi, A.P.; Servidei, S.; Scuderi, F.; Bartoccioni, E. Response to therapy in myasthenia gravis with anti-MuSK antibodies. Ann. N. Y. Acad. Sci. 2008, 1132, 76–83. [Google Scholar] [CrossRef]
- Deymeer, F. Myasthenia gravis: muSK-MG, late-onset MG and ocular MG. Acta Myol. 2020, 39, 345–352. [Google Scholar]
- Sanders, D.B.; Wolfe, G.I.; Benatar, M.; Evoli, A.; Gilhus, N.E.; Illa, I.; Kuntz, N.; Massey, J.M.; Melms, A.; Murai, H.; et al. International consensus guidance for management of myasthenia gravis: Executive summary. Neurology 2016, 87, 419–425. [Google Scholar] [PubMed]


{kind=link}
| SnMG Mimics and Chamaleons | Etiology | Sharing Features with SnMG | Distinguishing Features | Clue for Diagnosis |
|---|---|---|---|---|
| Myositis | Autoimmune, iatrogenic, Viral infectious, idiopathic | Ptosis, diplopia, dysphagia, response to steroids and immunosuppressants | Proximal weakness of upper and lower limbs | Serum CPK, EMG (myopathic features, pseudomyotonic discharges), Normal RNS. Muscle biopsy (myopathic features). |
| Myotonic muscular dystrophies | DMPK gene mutation (19q13.3); CNBP (ZNF9) gene mutation (3q21). | Ptosis, diplopia, dysphagia | Myotonia, adult onset; proximal weakness of upper and lower limbs with early footdrop, multisystemic involvement (diabetes, cataract, cognitive impairment, baldness). Autosomal dominant inheritance | Serum CPK, EMG (myopathic features, myotonic discharges), Normal RNS. Muscle biopsy (myopathic features). CTG repeat expansion on the DMPK gene. |
| OPMD | PABPN1 gene mutation (14q11.2) | Ptosis, dysphagia | Onset in the sixth decade. Proximal weakness of upper limbs. Autosomal dominant inheritance | Serum CPK, EMG (myopathic features), Normal RNS. Muscle biopsy (myopathic features). Trinucleotide expansion of the PABPN1 gene. |
| CPEO | Mitocondrial DNA mutations (ANT1, POLG, POLG2 and PEO1 genes) | Ptosis | Onset in the fifth-sixth decade, external ophtalmoparesis. Maternal inheritance. | Serum CPK, EMG (myopathic features), Normal RNS. Muscle biopsy (myopathic features). Mithocondrial DNA sequencing. |
| LEMS | Autoimmune, paraneoplastic, antibodies against voltage-gated calcium channels (VGCC) on presynaptic nerve terminal. | Ptosis, diplopia, dysphagia, response to steroids and immunosuppressants | Weakness of lower limbs, cramps associated with small cell lung carcinoma or thymoma, amelioration after exercise | Increased reflexes after exercise; increasing pattern at high frequency RNS; anti-presynaptic P/Q-type voltage-gated calcium channels antibodies in 60% of cases. |
| CMS | CHRNE, APSN, CHAT, COLQ, and DOK7 genes mutations. | Ptosis, diplopia | Juvenile onset; weakness of upper and lower limbs, in some patients low-set ears, skeletal abnormalities, cognitive impairment, epilepsy and a high-arched palate | Normal EMG with altered RNS. Genetic testing. |
| Horner’s syndrome | Middle ear infection to a carotid artery dissection or apical chest tumor | ptosis | Anisocoria with smaller pupil on the affected side | MRI of the brain, virus and bacteria. Carotid doppler ultrasound. |
| Third cranial nerve palsy | Extraorbital: diabetes, pituitary apoplexy, aneurysm, or carotid-cavernous fistula. Intraorbital: Trauma, tumors, and Tolosa-Hunt syndrome. | Unilateral ptosis, dyplopia | No fluctuation, diabetes, mechanic source of compression, herpes virus | MRI of the brain, virus, and bacteria. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vinciguerra, C.; Bevilacqua, L.; Lupica, A.; Ginanneschi, F.; Piscosquito, G.; Rini, N.; Rossi, A.; Barone, P.; Brighina, F.; Di Stefano, V. Diagnosis and Management of Seronegative Myasthenia Gravis: Lights and Shadows. Brain Sci. 2023, 13, 1286. https://doi.org/10.3390/brainsci13091286
Vinciguerra C, Bevilacqua L, Lupica A, Ginanneschi F, Piscosquito G, Rini N, Rossi A, Barone P, Brighina F, Di Stefano V. Diagnosis and Management of Seronegative Myasthenia Gravis: Lights and Shadows. Brain Sciences. 2023; 13(9):1286. https://doi.org/10.3390/brainsci13091286
Chicago/Turabian StyleVinciguerra, Claudia, Liliana Bevilacqua, Antonino Lupica, Federica Ginanneschi, Giuseppe Piscosquito, Nicasio Rini, Alessandro Rossi, Paolo Barone, Filippo Brighina, and Vincenzo Di Stefano. 2023. "Diagnosis and Management of Seronegative Myasthenia Gravis: Lights and Shadows" Brain Sciences 13, no. 9: 1286. https://doi.org/10.3390/brainsci13091286