
DEAD-Box Helicase 17 Promotes Amyloidogenesis by Regulating BACE1 Translation
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. Antibodies and Reagents
2.3. Cell Lines and Chemicals
2.4. Plasmid and Transfection
2.5. Western Blotting
2.6. Luciferase Activity Assay
2.7. RNA Pulldown Assay
2.8. Enzyme-Linked Immunosorbent Assay (ELISA)
2.9. Statistical Analysis
3. Results
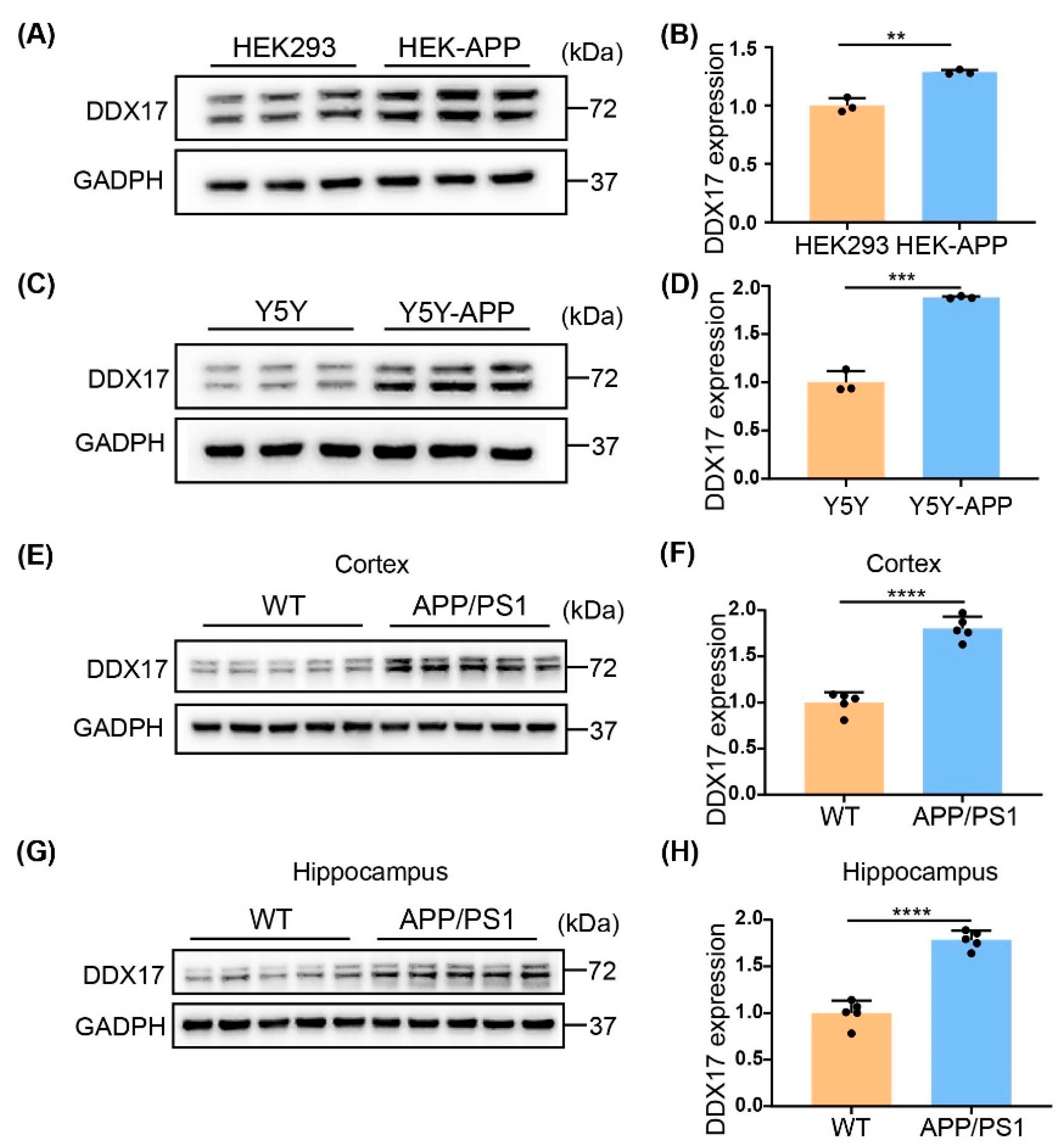
3.1. DDX17 Protein Levels Were Increased in Cellular and Animal Models of AD
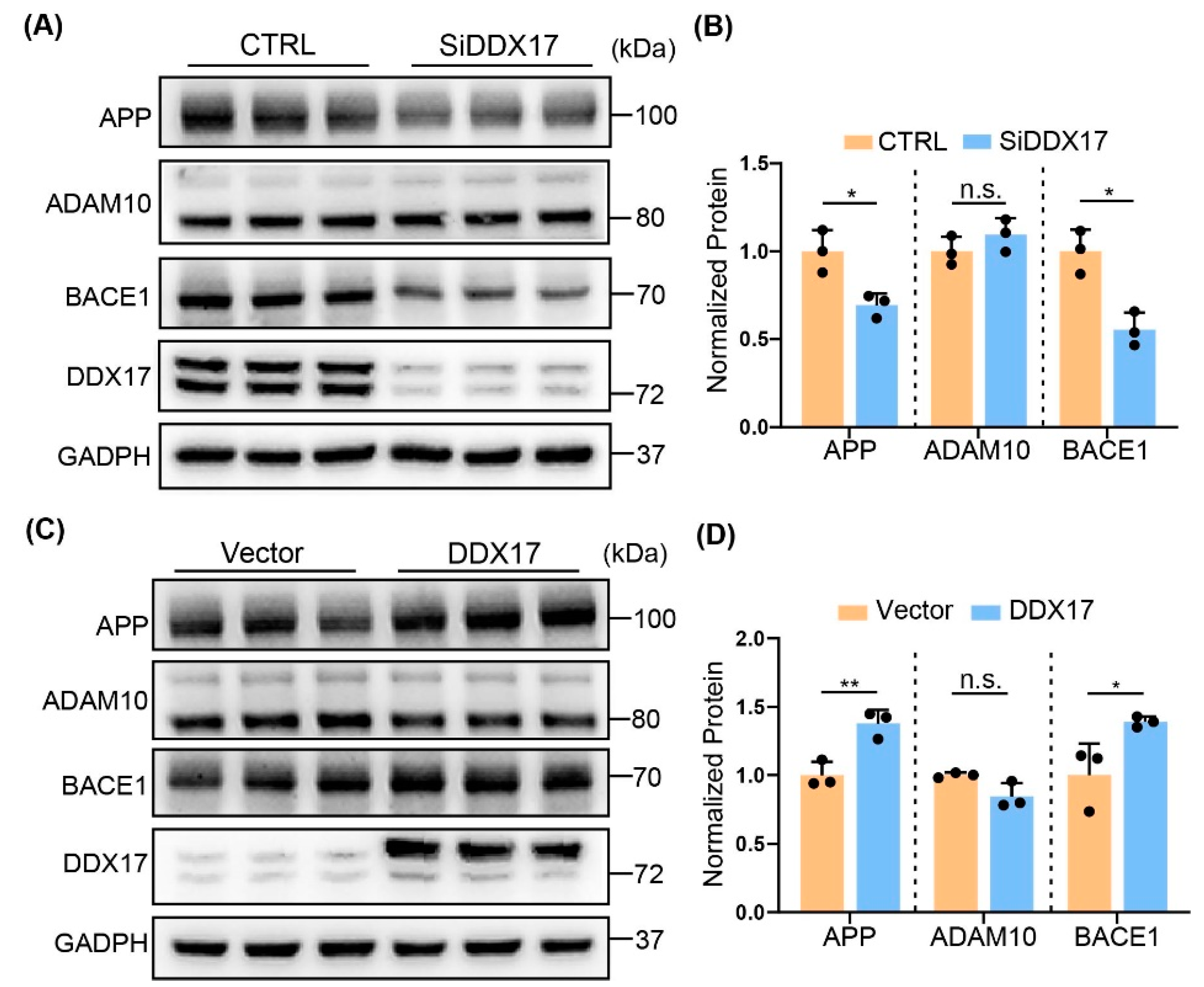
3.2. DDX17 Regulated APP Processing by Enhancing BACE1 Protein Level
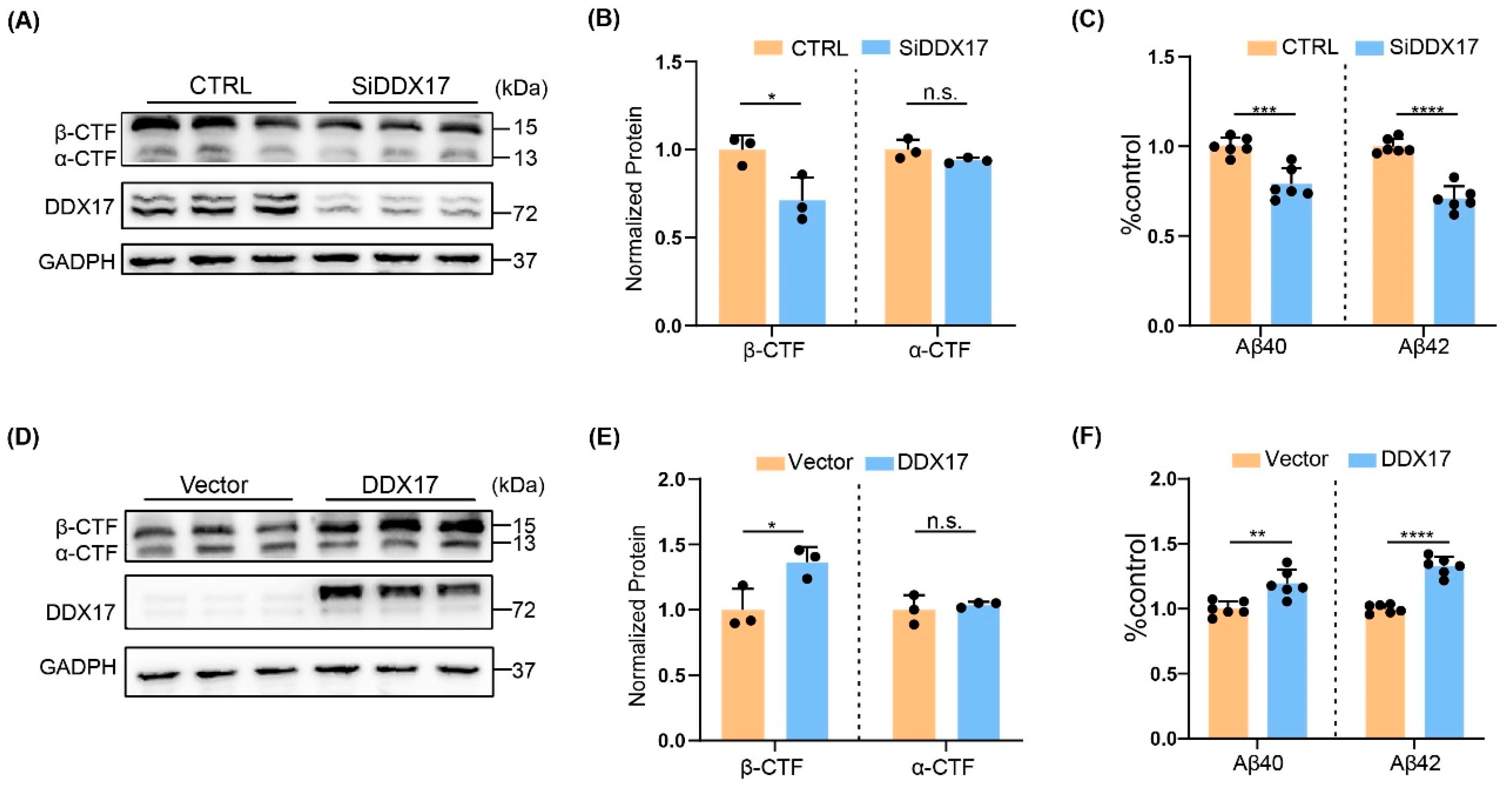
3.3. DDX17 Regulated Amyloidogenesis
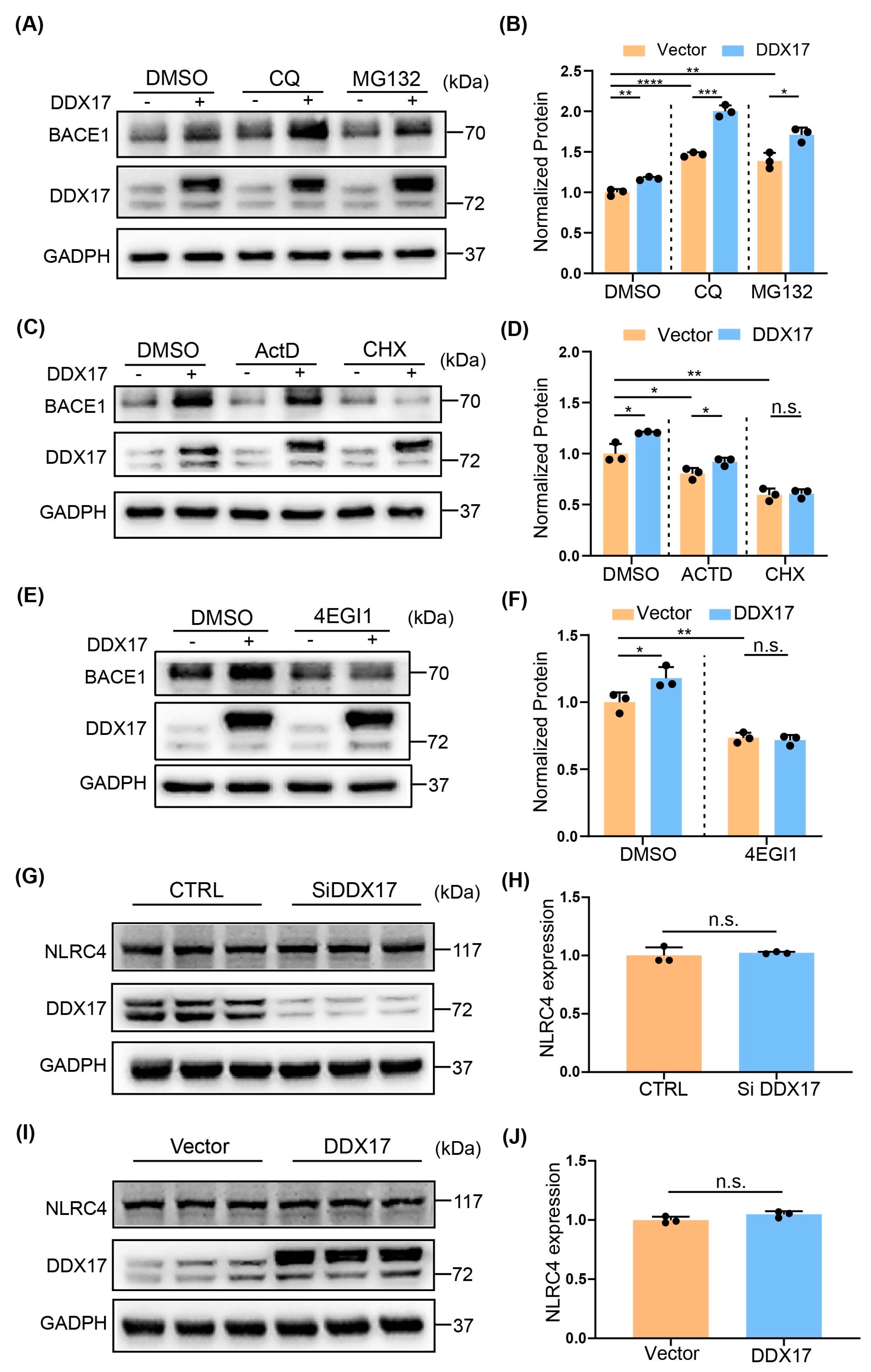
3.4. DDX17-Mediated Regulation of BACE1 Involved a Translational Mechanism
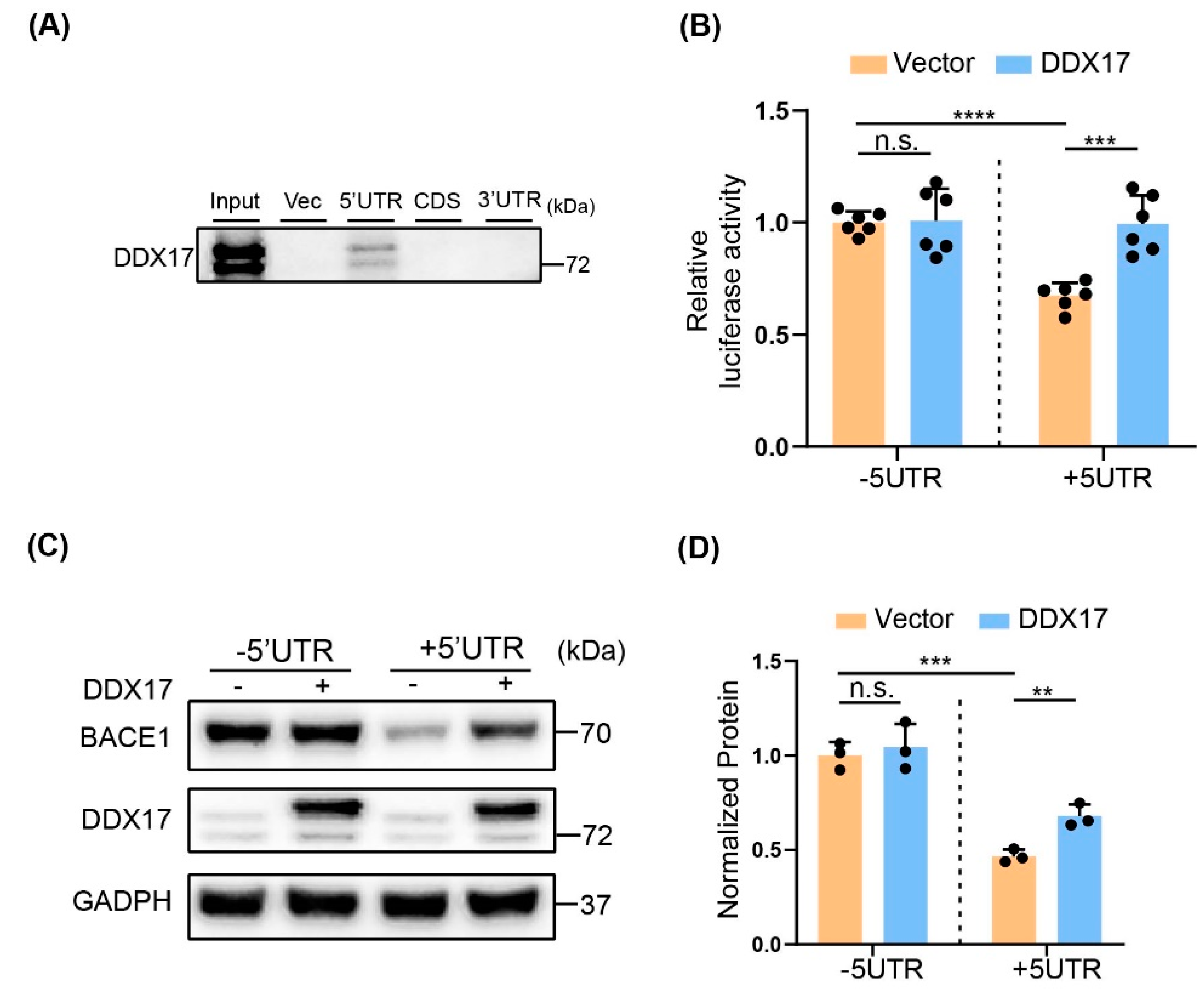
3.5. The Effect of DDX17 on BACE1 Translation Was Dependent on the 5′UTR
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Villain, N.; Dubois, B. Alzheimer’s Disease Including Focal Presentations. Semin. Neurol. 2019, 39, 213–226. [Google Scholar] [CrossRef]
- Soria Lopez, J.A.; Gonzalez, H.M.; Leger, G.C. Alzheimer’s disease. Handb. Clin. Neurol. 2019, 167, 231–255. [Google Scholar] [CrossRef]
- Battaglia, S.; Cardellicchio, P.; Di Fazio, C.; Nazzi, C.; Fracasso, A.; Borgomaneri, S. Stopping in (e)motion: Reactive action inhibition when facing valence-independent emotional stimuli. Front. Behav. Neurosci. 2022, 16, 998714. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, J.; Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 2018, 7, 1161. [Google Scholar] [CrossRef] [Green Version]
- Oboudiyat, C.; Glazer, H.; Seifan, A.; Greer, C.; Isaacson, R.S. Alzheimer’s Disease. Semin. Neurol. 2013, 33, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Walter, J.; Saido, T.C.; Fändrich, M. Neuropathology and biochemistry of Aβ and its aggregates in Alzheimer’s disease. Acta Neuropathol. 2015, 129, 167–182. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A.; et al. The β-Secretase BACE1 in Alzheimer’s Disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef]
- Postina, R. Activation of α-secretase cleavage. J. Neurochem. 2012, 120, 46–54. [Google Scholar] [CrossRef]
- Hrabinova, M.; Pejchal, J.; Kucera, T.; Jun, D.; Schmidt, M.; Soukup, O. Is It the Twilight of BACE1 Inhibitors? Curr. Neuropharmacol. 2021, 19, 61–77. [Google Scholar] [CrossRef]
- Sun, X.; Bromley-Brits, K.; Song, W. Regulation of β-site APP-cleaving enzyme 1 gene expression and its role in Alzheimer’s Disease. J. Neurochem. 2012, 120 (Suppl. 1), 62–70. [Google Scholar] [CrossRef] [PubMed]
- Chami, L.; Checler, F. BACE1 is at the crossroad of a toxic vicious cycle involving cellular stress and β-amyloid production in Alzheimer’s disease. Mol. Neurodegener. 2012, 7, 52. [Google Scholar] [CrossRef] [Green Version]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [Green Version]
- Linder, P.; Jankowsky, E. From unwinding to clamping—the DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 2011, 12, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Godbout, R.; Squire, J. Amplification of a DEAD box protein gene in retinoblastoma cell lines. Proc. Natl. Acad. Sci. USA 1993, 90, 7578–7582. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Pelletier, J. General and Target-Specific DExD/H RNA Helicases in Eukaryotic Translation Initiation. Int. J. Mol. Sci. 2020, 21, 4402. [Google Scholar] [CrossRef]
- Jankowsky, E. RNA helicases at work: Binding and rearranging. Trends Biochem. Sci. 2011, 36, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Moy, R.H.; Cole, B.S.; Yasunaga, A.; Gold, B.; Shankarling, G.; Varble, A.; Molleston, J.M.; Tenoever, B.R.; Lynch, K.W.; Cherry, S. Stem-Loop Recognition by DDX17 Facilitates miRNA Processing and Antiviral Defense. Cell 2014, 158, 764–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minton, K. DDX17 identified as inflammasome sensor for retrotransposon RNA. Nat. Rev. Immunol. 2022, 22, 73. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-B.; Narendran, S.; Hirahara, S.; Varshney, A.; Pereira, F.; Apicella, I.; Ambati, M.; Ambati, V.L.; Yerramothu, P.; Ambati, K.; et al. DDX17 is an essential mediator of sterile NLRC4 inflammasome activation by retrotransposon RNAs. Sci. Immunol. 2021, 6, eabi4493. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, T.R.; Kour, S.; Anderson, E.N.; Ward, C.; Rajasundaram, D.; Donnelly, C.J.; Hermann, A.; Wyne, H.; Shewmaker, F.; Pandey, U.B. DDX17 is involved in DNA damage repair and modifies FUS toxicity in an RGG-domain dependent manner. Acta Neuropathol. 2021, 142, 515–536. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.-J. The role of miRNA biogenesis and DDX17 in tumorigenesis and cancer stemness. Biomed. J. 2020, 43, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Fuller-Pace, F.V.; Moore, H.C. RNA helicases p68 and p72: Multifunctional proteins with important implications for cancer development. Futur. Oncol. 2011, 7, 239–251. [Google Scholar] [CrossRef]
- Xue, Y.; Jia, X.; Li, C.; Zhang, K.; Li, L.; Wu, J.; Yuan, J.; Li, Q. DDX17 promotes hepatocellular carcinoma progression via inhibiting Klf4 transcriptional activity. Cell Death Dis. 2019, 10, 814. [Google Scholar] [CrossRef] [Green Version]
- Mori, M.; Triboulet, R.; Mohseni, M.; Schlegelmilch, K.; Shrestha, K.; Camargo, F.D.; Gregory, R.I. Hippo Signaling Regulates Microprocessor and Links Cell-Density-Dependent miRNA Biogenesis to Cancer. Cell 2014, 156, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Dardenne, E.; Pierredon, S.; Driouch, K.; Gratadou, L.; Lacroix-Triki, M.; Espinoza, M.P.; Zonta, E.; Germann, S.; Mortada, H.; Villemin, J.-P.; et al. Splicing switch of an epigenetic regulator by RNA helicases promotes tumor-cell invasiveness. Nat. Struct. Mol. Biol. 2012, 19, 1139–1146. [Google Scholar] [CrossRef]
- Hönig, A.; Auboeuf, D.; Parker, M.M.; O’Malley, B.W.; Berget, S.M. Regulation of Alternative Splicing by the ATP-Dependent DEAD-Box RNA Helicase p72. Mol. Cell. Biol. 2002, 22, 5698–5707. [Google Scholar] [CrossRef] [Green Version]
- Luo, Q.; Que, T.; Luo, H.; Meng, Y.; Chen, X.; Huang, H.; Hu, R.; Luo, K.; Zheng, C.; Yan, P.; et al. Upregulation of DEAD box helicase 5 and 17 are correlated with the progression and poor prognosis in gliomas. Pathol. Res. Pract. 2020, 216, 152828. [Google Scholar] [CrossRef]
- Nel, M.; Dashti, M.J.S.; Gamieldien, J.; Heckmann, J.M. Exome sequencing identifies targets in the treatment-resistant ophthalmoplegic subphenotype of myasthenia gravis. Neuromuscul. Disord. 2017, 27, 816–825. [Google Scholar] [CrossRef]
- Masuda, K.; Ripley, B.; Nishimura, R.; Mino, T.; Takeuchi, O.; Shioi, G.; Kiyonari, H.; Kishimoto, T. Arid5a controls IL-6 mRNA stability, which contributes to elevation of IL-6 level in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 9409–9414. [Google Scholar] [CrossRef] [Green Version]
- Lammich, S.; Schöbel, S.; Zimmer, A.; Lichtenthaler, S.F.; Haass, C. Expression of the Alzheimer protease BACE1 is suppressed via its 5′-untranslated region. EMBO Rep. 2004, 5, 620–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilkei-Gorzo, A. Genetic mouse models of brain ageing and Alzheimer’s disease. Pharmacol. Ther. 2014, 142, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.-J.; Song, L.; Deng, X.-J.; Tang, Y.; Min, Z.; Luo, B.; Wen, Q.-X.; Li, K.-Y.; Chen, J.; Ma, Y.-L.; et al. Mitochondrial methionine sulfoxide reductase B2 links oxidative stress to Alzheimer’s disease-like pathology. Exp. Neurol. 2019, 318, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.T.; Zhu, B.L.; Zhao, L.G.; Wang, J.W.; Liu, L.; Lai, Y.J.; He, L.J.; Deng, X.J.; Chen, G. Histone deacetylase inhibitor apicidin increases expression of the α-secretase ADAM10 through transcription factor USF1-mediated mechanisms. FASEB J. 2017, 31, 1482–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Q.X.; Luo, B.; Xie, X.Y.; Zhou, G.F.; Chen, J.; Song, L.; Liu, Y.; Xie, S.Q.; Chen, L.; Li, K.Y.; et al. AP2S1 regulates APP degradation through late endosome–lysosome fusion in cells and APP/PS1 mice. Traffic 2023, 24, 20–33. [Google Scholar] [CrossRef]
- Tang, Y.; Min, Z.; Xiang, X.-J.; Liu, L.; Ma, Y.-L.; Zhu, B.-L.; Song, L.; Tang, J.; Deng, X.-J.; Yan, Z.; et al. Estrogen-related receptor alpha is involved in Alzheimer’s disease-like pathology. Exp. Neurol. 2018, 305, 89–96. [Google Scholar] [CrossRef]
- Wang, Y.P.; Wang, Z.F.; Zhang, Y.C.; Tian, Q.; Wang, J.Z. Effect of amyloid peptides on serum withdrawal-induced cell differentiation and cell viability. Cell Res. 2004, 14, 467–472. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Bansoad, A.V.; Singh, R.; Khatik, G.L. BACE1: A Key Regulator in Alzheimer’s Disease Progression and Current Development of its Inhibitors. Curr. Neuropharmacol. 2022, 20, 1174–1193. [Google Scholar] [CrossRef]
- Mañucat-Tan, N.B.; Saadipour, K.; Wang, Y.-J.; Bobrovskaya, L.; Zhou, X.-F. Cellular Trafficking of Amyloid Precursor Protein in Amyloidogenesis Physiological and Pathological Significance. Mol. Neurobiol. 2018, 56, 812–830. [Google Scholar] [CrossRef]
- Wang, J.; Sun, B.-L.; Xiang, Y.; Tian, D.-Y.; Zhu, C.; Li, W.-W.; Liu, Y.-H.; Bu, X.-L.; Shen, L.-L.; Jin, W.-S.; et al. Capsaicin consumption reduces brain amyloid-beta generation and attenuates Alzheimer’s disease-type pathology and cognitive deficits in APP/PS1 mice. Transl. Psychiatry 2020, 10, 230. [Google Scholar] [CrossRef]
- Zhang, Y.-W.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Ma, X.; Wang, J.; Wang, L.; Wang, Y. Pretreatment with the γ-secretase inhibitor DAPT sensitizes drug-resistant ovarian cancer cells to cisplatin by downregulation of Notch signaling. Int. J. Oncol. 2014, 44, 1401–1409. [Google Scholar] [CrossRef] [Green Version]
- Imbimbo, B.P.; Watling, M. Investigational BACE inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2019, 28, 967–975. [Google Scholar] [CrossRef]
- Roßner, S.; Sastre, M.; Bourne, K.; Lichtenthaler, S.F. Transcriptional and translational regulation of BACE1 expression—Implications for Alzheimer’s disease. Prog. Neurobiol. 2006, 79, 95–111. [Google Scholar] [CrossRef]
- Zhu, B.-L.; Long, Y.; Luo, W.; Yan, Z.; Lai, Y.-J.; Zhao, L.-G.; Zhou, W.-H.; Wang, Y.-J.; Shen, L.-L.; Liu, L.; et al. MMP13 inhibition rescues cognitive decline in Alzheimer transgenic mice via BACE1 regulation. Brain 2018, 142, 176–192. [Google Scholar] [CrossRef]
- Min, Z.; Tang, Y.; Hu, X.-T.; Zhu, B.L.; Ma, Y.-L.; Zha, J.-S.; Deng, X.-J.; Yan, Z.; Chen, G.-J. Cosmosiin Increases ADAM10 Expression via Mechanisms Involving 5’UTR and PI3K Signaling. Front. Mol. Neurosci. 2018, 11, 198. [Google Scholar] [CrossRef]
- Lindqvist, L.; Pelletier, J. Inhibitors of translation initiation as cancer therapeutics. Futur. Med. Chem. 2009, 1, 1709–1722. [Google Scholar] [CrossRef] [PubMed]
- De, A.; Jacobson, B.A.; Peterson, M.S.; Jay-Dixon, J.; Kratzke, M.G.; Sadiq, A.A.; Patel, M.R.; Kratzke, R.A. 4EGI-1 represses cap-dependent translation and regulates genome-wide translation in malignant pleural mesothelioma. Investig. New Drugs 2018, 36, 217–229. [Google Scholar] [CrossRef]
- Creus-Muncunill, J.; Badillos-Rodriguez, R.; Garcia-Forn, M.; Masana, M.; Garcia-Diaz Barriga, G.; Guisado-Corcoll, A.; Alberch, J.; Malagelada, C.; Delgado-García, J.M.; Gruart, A.; et al. Increased translation as a novel pathogenic mechanism in Huntington’s disease. Brain 2019, 142, 3158–3175. [Google Scholar] [CrossRef] [PubMed]
- Donsbach, P.; Klostermeier, D. Regulation of RNA helicase activity: Principles and examples. Biol. Chem. 2021, 402, 529–559. [Google Scholar] [CrossRef] [PubMed]
- Pestova, T.V.; Kolupaeva, V.G. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev. 2002, 16, 2906–2922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dmitriev, S.E.; Terenin, I.M.; Dunaevsky, Y.E.; Merrick, W.C.; Shatsky, I.N. Assembly of 48S Translation Initiation Complexesfrom Purified Components with mRNAs That Have Some Base Pairingwithin Their 5′ UntranslatedRegions. Mol. Cell. Biol. 2003, 23, 8925–8933. [Google Scholar] [CrossRef] [Green Version]
- Mihailovich, M.; Thermann, R.; Grohovaz, F.; Hentze, M.W.; Zacchetti, D. Complex translational regulation of BACE1 involves upstream AUGs and stimulatory elements within the 5’ untranslated region. Nucleic Acids Res. 2007, 35, 2975–2985. [Google Scholar] [CrossRef]
- Xu, M.; Zhang, D.; Luo, R.; Wu, Y.; Zhou, H.; Kong, L.; Bi, R.; Yao, Y. A systematic integrated analysis of brain expression profiles reveals YAP1 and other prioritized hub genes as important upstream regulators in Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 215–229. [Google Scholar] [CrossRef]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Yin, L.; Thambisetty, M.; Troncoso, J.C.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. Deep proteomic network analysis of Alzheimer’s disease brain reveals alterations in RNA binding proteins and RNA splicing associated with disease. Mol. Neurodegener. 2018, 13, 52. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Hales, C.M.; Chen, P.-C.; Gozal, Y.; Dammer, E.B.; Fritz, J.J.; Wang, X.; Xia, Q.; Duong, D.M.; Street, C.; et al. U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2013, 110, 16562–16567. [Google Scholar] [CrossRef] [Green Version]
- Shafik, A.M.; Zhou, H.; Lim, J.; Dickinson, B.; Jin, P. Dysregulated mitochondrial and cytosolic tRNA m1A methylation in Alzheimer’s disease. Hum. Mol. Genet. 2022, 31, 1673–1680. [Google Scholar] [CrossRef]
- van der Linden, R.J.; Gerritsen, J.S.; Liao, M.; Widomska, J.; Pearse, R.V., 2nd; White, F.M.; Franke, B.; Young-Pearse, T.L.; Poelmans, G. RNA-binding protein ELAVL4/HuD ameliorates Alzheimer’s disease-related molecular changes in human iPSC-derived neurons. Prog. Neurobiol. 2022, 217, 102316. [Google Scholar] [CrossRef]
- Ngo, T.D.; Partin, A.C.; Nam, Y. RNA Specificity and Autoregulation of DDX17, a Modulator of MicroRNA Biogenesis. Cell Rep. 2019, 29, 4024–4035.e4025. [Google Scholar] [CrossRef] [Green Version]
- Young, C.L.; Khoshnevis, S.; Karbstein, K. Cofactor-dependent specificity of a DEAD-box protein. Proc. Natl. Acad. Sci. USA 2013, 110, E2668–E2676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Zhang, G.; Lu, Y.; Zhou, J.; Ren, Z. DDX17 modulates the expression and alternative splicing of genes involved in apoptosis and proliferation in lung adenocarcinoma cells. PeerJ 2022, 10, e13895. [Google Scholar] [CrossRef]
- Xu, K.; Sun, S.; Yan, M.; Cui, J.; Yang, Y.; Li, W.; Huang, X.; Dou, L.; Chen, B.; Tang, W.; et al. DDX5 and DDX17—Multifaceted proteins in the regulation of tumorigenesis and tumor progression. Front. Oncol. 2022, 12, 943032. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.Z.; Li, F.; Cheng, S.T.; Xu, Y.; Deng, H.J.; Gu, D.Y.; Wang, J.; Chen, W.; Zhou, Y.; Yang, M.; et al. DDX17-regulated alternative splicing that produced an oncogenic isoform of PXN-AS1 to promote HCC metastasis. Hepatology 2022, 75, 847–865. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Mo, C.; Gong, D.; Chen, Y.; Huang, Z.; Li, Y.; Zhang, J.; Huang, L.; Li, Y.; Fuller-Pace, F.V.; et al. DDX17 nucleocytoplasmic shuttling promotes acquired gefitinib resistance in non-small cell lung cancer cells via activation of β-catenin. Cancer Lett. 2017, 400, 194–202. [Google Scholar] [CrossRef] [Green Version]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Bienkowski, M.J.; Shuck, M.E.; Miao, H.; Tory, M.C.; Pauley, A.M.; Brashler, J.R.; Stratman, N.C.; Mathews, W.R.; Buhl, A.E.; et al. Membrane-anchored aspartyl protease with Alzheimer’s disease β-secretase activity. Nature 1999, 402, 533–537. [Google Scholar] [CrossRef]
- Moerke, N.J.; Aktas, H.; Chen, H.; Cantel, S.; Reibarkh, M.Y.; Fahmy, A.; Gross, J.D.; Degterev, A.; Yuan, J.; Chorev, M.; et al. Small-Molecule Inhibition of the Interaction between the Translation Initiation Factors eIF4E and eIF4G. Cell 2007, 128, 257–267. [Google Scholar] [CrossRef]
- Pan, L.; Huang, X.; Liu, Z.-X.; Ye, Y.; Li, R.; Zhang, J.; Wu, G.; Bai, R.; Zhuang, L.; Wei, L.; et al. Inflammatory cytokine–regulated tRNA-derived fragment tRF-21 suppresses pancreatic ductal adenocarcinoma progression. J. Clin. Investig. 2021, 131, e148130. [Google Scholar] [CrossRef]
- Buggia-Prevot, V.; Sevalle, J.; Rossner, S.; Checler, F. NFκB-dependent Control of BACE1 Promoter Transactivation by Aβ42. J. Biol. Chem. 2008, 283, 10037–10047. [Google Scholar] [CrossRef] [Green Version]
- Li, K.Y.; Xiang, X.J.; Song, L.; Chen, J.; Luo, B.; Wen, Q.X.; Zhong, B.R.; Zhou, G.F.; Deng, X.J.; Ma, Y.L.; et al. Mitochondrial TXN2 attenuates amyloidogenesis via selective inhibition of BACE1 expression. J. Neurochem. 2021, 157, 1351–1365. [Google Scholar] [CrossRef] [PubMed]
- Picón-Pagès, P.; Gutiérrez, D.A.; Barranco-Almohalla, A.; Crepin, G.; Tajes, M.; Ill-Raga, G.; Guix, F.X.; Menéndez, S.; Arumí-Uría, M.; Vicente, R.; et al. Amyloid Beta-Peptide Increases BACE1 Translation through the Phosphorylation of the Eukaryotic Initiation Factor-2α. Oxidative Med. Cell. Longev. 2020, 2020, 2739459. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.R.; Zhou, G.F.; Song, L.; Wen, Q.X.; Deng, X.J.; Ma, Y.; Hu, L.; Chen, G. TUFM is involved in Alzheimer’s disease-like pathologies that are associated with ROS. FASEB J. 2021, 35, e21445. [Google Scholar] [CrossRef]
- Chatterjee, P.; Pedrini, S.; Ashton, N.J.; Tegg, M.; Goozee, K.; Singh, A.K.; Karikari, T.K.; Simrén, J.; Vanmechelen, E.; Armstrong, N.J.; et al. Diagnostic and prognostic plasma biomarkers for preclinical Alzheimer’s disease. Alzheimer’s Dement. 2022, 18, 1141–1154. [Google Scholar] [CrossRef]
- Kavanagh, T.; Halder, A.; Drummond, E. Tau interactome and RNA binding proteins in neurodegenerative diseases. Mol. Neurodegener. 2022, 17, 66. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B.; Ivanov, P. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Zhou, G.; Song, L.; Wen, Q.; Xie, S.; Chen, L.; Wang, L.; Xie, X.; Chen, X.; Pu, Y.; et al. DEAD-Box Helicase 17 Promotes Amyloidogenesis by Regulating BACE1 Translation. Brain Sci. 2023, 13, 745. https://doi.org/10.3390/brainsci13050745
Liu Y, Zhou G, Song L, Wen Q, Xie S, Chen L, Wang L, Xie X, Chen X, Pu Y, et al. DEAD-Box Helicase 17 Promotes Amyloidogenesis by Regulating BACE1 Translation. Brain Sciences. 2023; 13(5):745. https://doi.org/10.3390/brainsci13050745
Chicago/Turabian StyleLiu, Yue, Guifeng Zhou, Li Song, Qixin Wen, Shiqi Xie, Long Chen, Lu Wang, Xiaoyong Xie, Xue Chen, Yalan Pu, and et al. 2023. "DEAD-Box Helicase 17 Promotes Amyloidogenesis by Regulating BACE1 Translation" Brain Sciences 13, no. 5: 745. https://doi.org/10.3390/brainsci13050745