
Frontotemporal-TDP and LATE Neurocognitive Disorders: A Pathophysiological and Genetic Approach
 ,
,  , and
, and
Abstract
:1. Biology of TDP-43
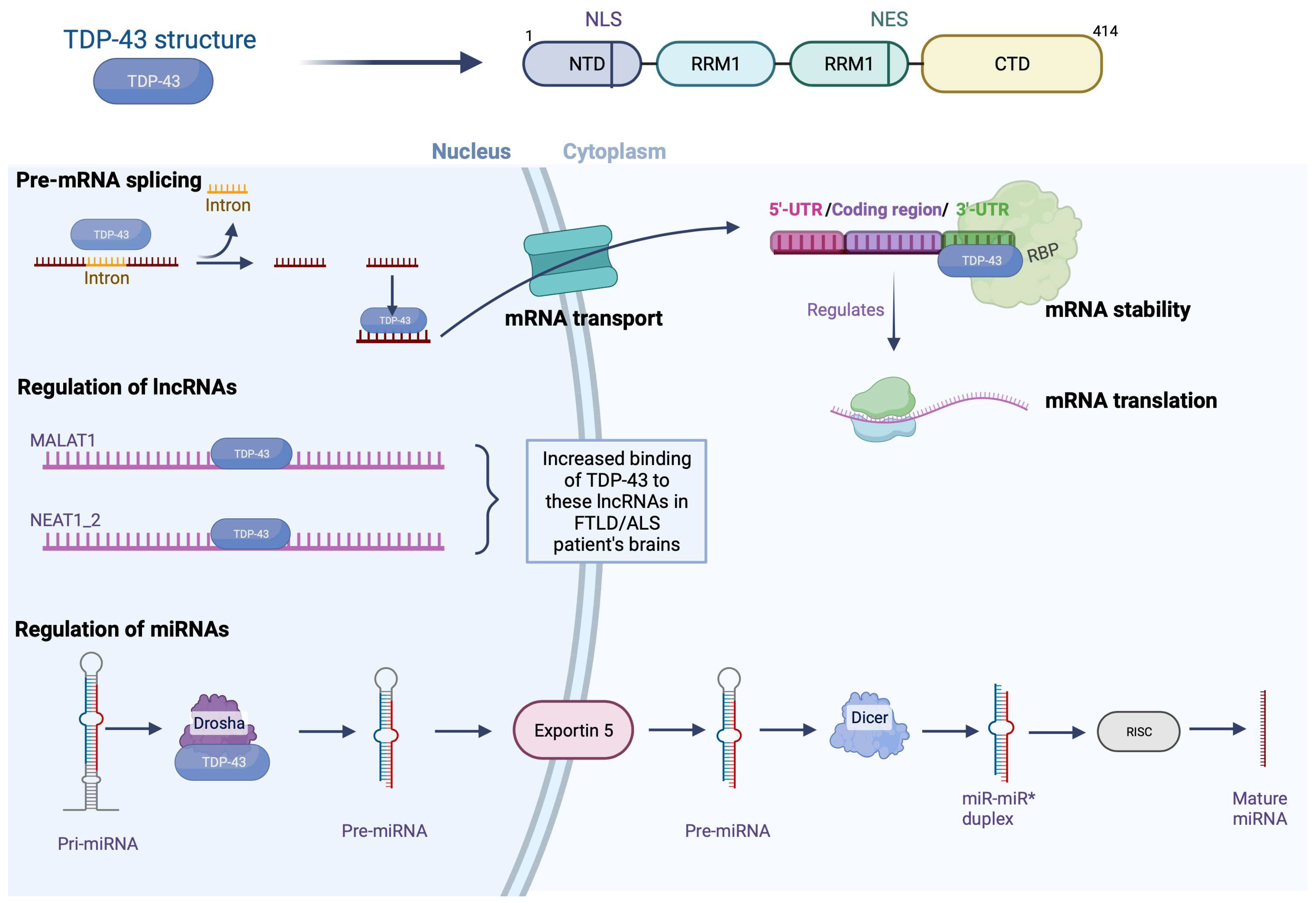
1.1. Structure of TDP-43
1.2. Physiological Functions of TDP-43
1.2.1. Nuclear Functions: Modulation of Pre-mRNA Splicing by TDP-43
1.2.2. Nuclear Functions: Regulation of Micro-RNAs
1.2.3. Nuclear Functions: Regulation of the Expression of Long Non-Coding RNAs
1.2.4. Cytosolic Functions: Stabilization of mRNA
1.2.5. Cytosolic Functions: Transport of mRNA
1.2.6. Cytosolic Functions: Regulation of mRNA Translation
1.3. Pathophysiology of TDP-43
2. The Different Histopathological Subtypes of FTLD-TDP
2.1. FTLD-TDP Type A
2.2. FTLD-TDP Type B
2.3. FTLD-TDP Type C
2.4. FTLD-TDP Type D
3. TDP-43 Mutations
3.1. Post-Translational Modifications
3.2. Phosphorylation
3.3. Ubiquitination
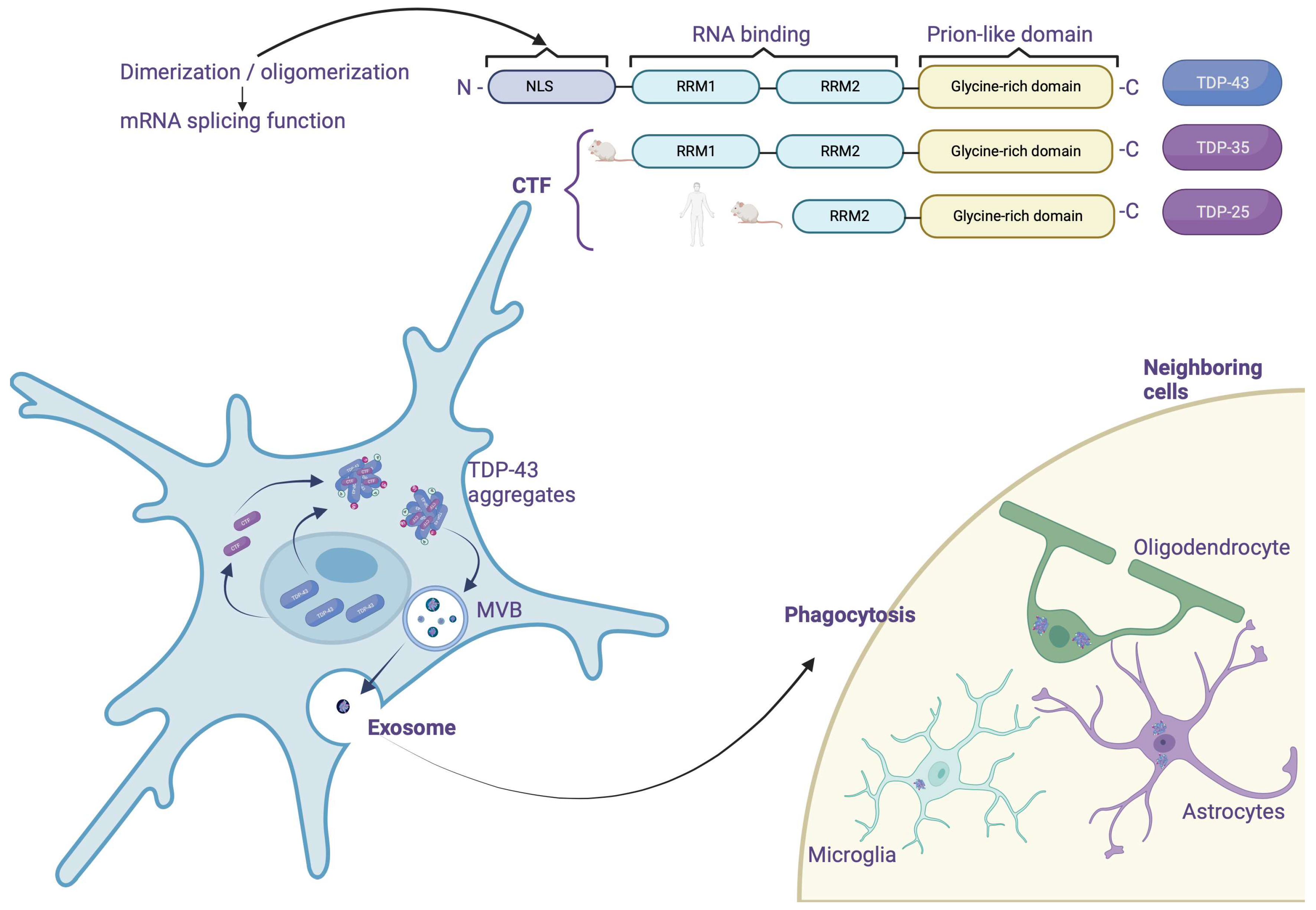
3.4. Truncation
3.5. TDP-43 Relocation
3.6. Aggregation
4. TDP-43 Considerations about Being a Prion Protein
- Most of the TDP-43 mutations found in ALS target the C-terminal part.
- CTFs, which essentially contain this prion domain, appear to promote aggregation in vivo [40].
5. FTLD-TDP Genetics
5.1. Granular Precurson (GRN) Gene
5.2. Progranulin (GRN) Structure
5.3. Expression in the CNS of Progranulin
5.4. Modulation of Expression by Transmembrane Protein 106B (TMEM106B)
5.5. Biological Function of Progranulin
5.6. Progranulin Neuronal Function and Growth of Neural Tracts
5.7. Synaptic Plasticity
5.8. Microglial Functions
5.9. Progranulin on TDP-43 Aggregation
5.10. Neuropathology and Associated Clinical Signs
5.11. White Matter Abnormalities
5.12. Nucleotide C9ORF72
5.13. Pathophysiological Mechanisms Related to GGGGCC Repeats
- The presence of many repeats could cause a reduction in the expression of the C9ORF72 gene leading to a loss of its physiological function. It would seem that the expression of the C9ORF72 gene is reduced in patients carrying this type of mutation [112].
- It has been shown that brain and spinal cord tissue from FTLD/ALS patients is not distinguished by the presence of nuclear foci composed of GGGGCC RNA but also GGCCCC antisense RNA [115,116], similar to the mechanism described in myotonic dystrophy type I (DM1) [117]; these foci could sequester certain RBPs or splicing factors and lead to a loss of their physiological functions.
- The transcripts (sense and antisense) produced from the repeated sequences would be the target of an unconventional translation mechanism that does not depend on the presence of an ATG codon. This mechanism, called translation-associated repeat not initiated by ATG (RAN), would be responsible for the production of a series of dipeptides (dipeptide repeat, DPR; glycine-alanine, GA; glycine-proline, GP; glycine-arginine, GR; proline-alanine, PA; and proline-arginine, PR) [116]. These DPRs, located throughout the entire CNS, have the characteristic of being pro-aggregative, and, therefore, could participate in the neurodegenerative process [116]. Several studies carried out in cell culture models of Drosophila demonstrate the toxicity of these DPRs [115,118] (Figure 7).
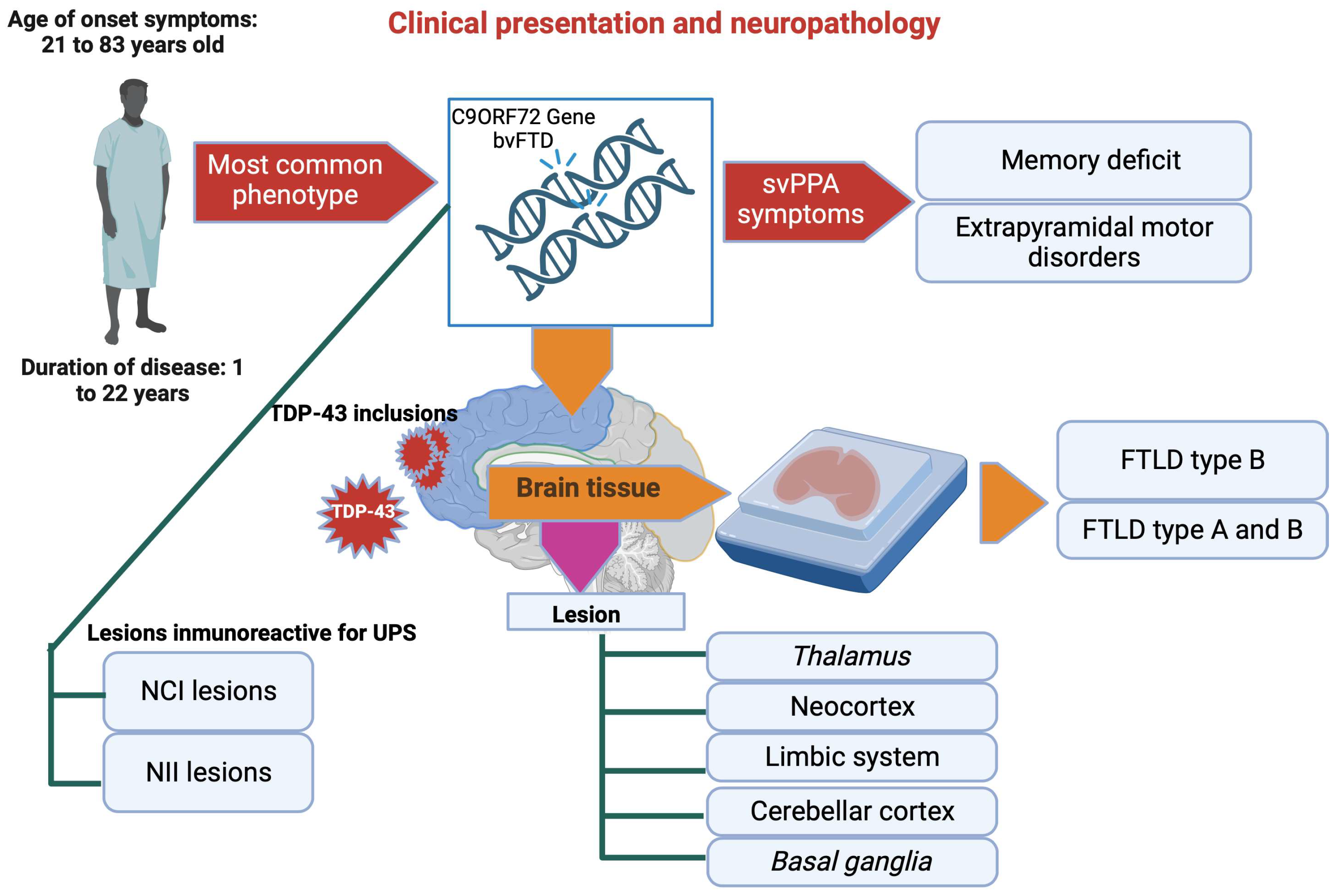
6. Clinical Presentation and Neuropathology
7. Other Genes Involved
7.1. TARDBP Gene
7.2. VCP Gene
8. Minority Subclasses of FTDL and Related Syndromes
9. Frontotemporal Lobar Degeneration with FUS (FTLD-FET)
- First, a dysfunction of the Trn1 protein, resulting from a genetic variation of the TNPO1 gene or a post-translational modification, could decrease the efficiency of FET protein transport to the nucleus. However, the absence of other proteins carried by Trn1 within the aggregates, such as heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1), makes this hypothesis less plausible [124].
- Post-translational modifications, absent under physiological conditions, can also affect the FET proteins. Interestingly, hypomethylation of arginine residues in a region close to the NLS of FET proteins is observed.
10. FTLD-FET Subtypes
10.1. aFTLD-U
10.2. NIFID
10.3. BIBD
11. Genetics in FTLD-FET
12. FTLD-UPS
13. Other Pathologies
14. LATE
- -
- Stage 1: toxic clumps are formed in the amygdala, a region of the brain involved in managing emotions;
- -
- Stage 2: protein aggregates spread inside the hippocampus, which plays an essential role in memory processes;
- -
14.1. LATE Symptoms
14.2. Causes of LATE
15. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ou, S.H.; Wu, F.; Harrich, D.; Garcia-Martinez, L.F.; Gaynor, R.B. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 1995, 69, 3584–3596. [Google Scholar] [CrossRef] [PubMed]
- Fuentealba, R.A.; Udan, M.; Bell, S.; Wegorzewska, I.; Shao, J.; Diamond, M.I.; Weihl, C.C.; Baloh, R.H. Interaction with polyglutamine aggregates reveals a Q/N-rich domain in TDP-43. J. Biol. Chem. 2010, 285, 26304–26314. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, F.E. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J. Biol. Chem. 2001, 276, 36337–36343. [Google Scholar] [CrossRef] [PubMed]
- Sephton, C.F.; Cenik, C.; Kucukural, A.; Dammer, E.B.; Cenik, B.; Han, Y.; Dewey, C.M.; Roth, F.P.; Herz, J.; Peng, J.; et al. Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J. Biol. Chem. 2011, 286, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; Konig, J.; Hortobagyi, T.; Nishimura, A.L.; Zupunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef]
- Ayala, Y.M.; De Conti, L.; Avendaño-Vázquez, S.E.; Dhir, A.; Romano, M.; D’Ambrogio, A.; Tollervey, J.; Ule, J.; Baralle, M.; Buratti, E.; et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011, 30, 277–288. [Google Scholar] [CrossRef]
- Nelson, P.T.; Keller, J.N. RNA in brain disease: No longer just “the messenger in the middle”. J. Neuropathol. Exp. Neurol. 2007, 66, 461–468. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Radmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef]
- Cullen, B.R. Derivation and function of small interfering RNAs and microRNAs. Virus Res. 2004, 102, 3–9. [Google Scholar] [CrossRef]
- Casafont, I.; Bengoechea, R.; Tapia, O.; Berciano, M.T.; Lafarga, M. TDP-43 localizes in mRNA transcription and processing sites in mammalian neurons. J. Struct. Biol. 2009, 167, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Colombrita, C.; Onesto, E.; Buratti, E.; de la Grange, P.; Gumina, V.; Baralle, F.E.; Silani, V.; Ratti, A. From transcriptomic to protein level changes in TDP-43 and FUS loss-of-function cell models. Biochim. Biophys. Acta 2015, 1849, 1398–1410. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Morris, K.V.; Wood, M.J.A. The role of long non-coding RNAs in neurodevelopment, brain function and neurological disease. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130507. [Google Scholar] [CrossRef]
- Nishimoto, Y.; Nakagawa, S.; Hirose, T.; Okano, H.J.; Takao, M.; Shibata, S.; Suyama, S.; Kuwako, K.; Imai, T.; Murayama, S.; et al. The long non-coding RNA nuclear-enriched abundant transcript 1_2 induces paraspeckle formation in the motor neuron during the early phase of amyotrophic lateral sclerosis. Mol. Brain 2013, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, M.; Boerkoel, C.F. The role of nuclear bodies in gene expression and disease. Biology 2013, 2, 976–1033. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, G.F.; Janitz, M.; Huang, Y.; Halliday, G.M. Long noncoding RNAs in TDP-43 and FUS/TLS-related frontotemporal lobar degeneration (FTLD). Neurobiol. Dis. 2015, 82, 445–454. [Google Scholar] [CrossRef]
- Fiesel, F.C.; Schurr, C.; Weber, S.S.; Kahle, P.J. TDP-43 knockdown impairs neurite outgrowth dependent on its target histone deacetylase 6. Mol. Neurodegener. 2011, 6, 64. [Google Scholar] [CrossRef]
- Costessi, L.; Porro, F.; Iaconcig, A.; Muro, A.F. TDP-43 regulates β-adducin (Add2) transcript stability. RNA Biol. 2014, 11, 1280–1290. [Google Scholar] [CrossRef]
- Colombrita, C.; Onesto, E.; Megiorni, F.; Pizzuti, A.; Baralle, F.E.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J. Biol. Chem. 2012, 287, 15635–15647. [Google Scholar] [CrossRef]
- Lee, S.; Lee, T.A.; Lee, E.; Kang, S.; Park, A.; Kim, S.W.; Park, H.J.; Yoon, J.H.; Ha, S.J.; Park, T.; et al. Identification of a subnuclear body involved in sequence-specific cytokine RNA processing. Nat. Commun. 2015, 6, 5791. [Google Scholar] [CrossRef] [PubMed]
- Liu-Yesucevitz, L.; Bassell, G.J.; Gitler, A.D.; Hart, A.C.; Klann, E.; Richter, J.D.; Warren, S.T.; Wolozin, B. Local RNA translation at the synapse and in disease. J. Neurosci. 2011, 31, 16086–16093. [Google Scholar] [CrossRef] [PubMed]
- Fallini, C.; Bassell, G.J.; Rossoll, W. The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum. Mol. Genet. 2012, 21, 3703–3718. [Google Scholar] [CrossRef] [PubMed]
- Alami, N.H.; Smith, R.B.; Carrasco, M.A.; Williams, L.A.; Winborn, C.S.; Han, S.S.W.; Kiskinis, E.; Winborn, B.; Freibaum, B.D.; Kanagaraj, A.; et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 2014, 81, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Sun, M.Y.; Hou, Q.; Parpura, V. Hyposmolality differentially and spatiotemporally modulates levels of glutamine synthetase and serine racemase in rat supraoptic nucleus. Glia 2013, 61, 529–538. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Chitta, R.K.; High, A.A.; Taylor, J.P. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J. Proteome Res. 2010, 9, 1104–1120. [Google Scholar] [CrossRef]
- Higashi, S.; Kabuta, T.; Nagai, Y.; Tsuchiya, Y.; Akiyama, H.; Wada, K. TDP-43 associates with stalled ribosomes and contributes to cell survival during cellular stress. J. Neurochem. 2013, 126, 288–300. [Google Scholar] [CrossRef]
- Coyne, A.N.; Siddegowda, B.B.; Estes, P.S.; Johannesmeyer, J.; Kovalik, T.; Daniel, S.G.; Pearson, A.; Bowser, R.; Zarnescu, D.C. Futsch/MAP1B mRNA is a translational target of TDP-43 and is neuroprotective in a Drosophila model of amyotrophic lateral sclerosis. J. Neurosci. 2014, 34, 15962–15974. [Google Scholar] [CrossRef]
- Majumder, P.; Chen, Y.T.; Bose, J.K.; Wu, C.C.; Cheng, W.C.; Cheng, S.J.; Fang, Y.H.; Chen, Y.L.; Tsai, K.J.; Lien, C.C.; et al. TDP-43 regulates the mammalian spinogenesis through translational repression of Rac1. Acta Neuropathol. 2012, 124, 231–245. [Google Scholar] [CrossRef]
- MacNair, L.; Xiao, S.; Miletic, D.; Ghani, M.; Julien, J.P.; Keith, J.; Zinman, L.; Rogaeva, E.; Robertson, J. MTHFSD and DDX58 are novel RNA-binding proteins abnormally regulated in amyotrophic lateral sclerosis. Brain 2016, 139, 86–100. [Google Scholar] [CrossRef]
- Lee, E.B.; Lee, V.M.; Trojanowski, J.Q. Gains or losses: Molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 2011, 13, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Neumann, M. Molecular neuropathology of frontotemporal dementia: Insights into disease mechanisms from post-mortem studies. J. Neurochem. 2016, 138 (Suppl. S1), 54–70. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Stroh, A.; Dugger, B.; Dickson, D.W. Evaluation of subcortical pathology and clinical correlations in FTLD-U subtypes. Acta Neuropathol. 2009, 118, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Neumann, M.; Baborie, A.; Sampathu, D.M.; Du Plessis, D.; Jaros, E.; Perry, R.H.; Trojanowski, J.Q.; Mann, D.M.; Lee, V.M. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011, 122, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Arai, T.; Nonaka, T.; Kametani, F.; Yoshida, M.; Hashizume, Y.; Beach, T.G.; Buratti, E.; Baralle, F.; Morita, M.; et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 2008, 64, 60–70. [Google Scholar] [CrossRef]
- Neumann, M.; Kwong, L.K.; Lee, E.B.; Kremmer, E.; Flatley, A.; Xu, Y.; Forman, M.S.; Troost, D.; Kretzschmar, H.A.; Trojanowski, J.Q.; et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009, 117, 137–149. [Google Scholar] [CrossRef]
- Strong, M.J.; Volkening, K.; Hammond, R.; Yang, W.; Strong, W.; Leystra-Lantz, C.; Shoesmith, C. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol. Cell. Neurosci. 2007, 35, 320–327. [Google Scholar] [CrossRef]
- Kametani, F.; Nonaka, T.; Suzuki, T.; Arai, T.; Dohmae, N.; Akiyama, H.; Hasegawa, M. Identification of casein kinase-1 phosphorylation sites on TDP-43. Biochem. Biophys. Res. Commun. 2009, 382, 405–409. [Google Scholar] [CrossRef]
- Igaz, L.M.; Kwong, L.K.; Chen-Plotkin, A.; Winton, M.J.; Unger, T.L.; Xu, Y.; Neumann, M.; Trojanowski, J.Q.; Lee, V.M. Expression of TDP-43 C-terminal Fragments in Vitro Recapitulates Pathological Features of TDP-43 Proteinopathies. J. Biol. Chem. 2009, 284, 8516–8524. [Google Scholar] [CrossRef]
- Nonaka, T.; Kametani, F.; Arai, T.; Akiyama, H.; Hasegawa, M. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum. Mol. Genet. 2009, 18, 3353–3364. [Google Scholar] [CrossRef]
- Winton, M.J.; Igaz, L.M.; Wong, M.M.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 2008, 283, 13302–13309. [Google Scholar] [CrossRef] [PubMed]
- Wegorzewska, I.; Bell, S.; Cairns, N.J.; Miller, T.M.; Baloh, R.H. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 18809–18814. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.F.; Gendron, T.F.; Zhang, Y.J.; Lin, W.L.; D’Alton, S.; Sheng, H.; Casey, M.C.; Tong, J.; Knight, J.; Yu, X.; et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci. 2010, 30, 10851–10859. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Gendron, T.F.; Xu, Y.F.; Ko, L.W.; Yen, S.H.; Petrucelli, L. Phosphorylation regulates proteasomal-mediated degradation and solubility of TAR DNA binding protein-43 C-terminal fragments. Mol. Neurodegener. 2010, 5, 33. [Google Scholar] [CrossRef]
- Liachko, N.F.; McMillan, P.J.; Guthrie, C.R.; Bird, T.D.; Leverenz, J.B.; Kraemer, B.C. CDC7 inhibition blocks pathological TDP-43 phosphorylation and neurodegeneration. Ann. Neurol. 2013, 74, 39–52. [Google Scholar] [CrossRef]
- Liachko, N.F.; McMillan, P.J.; Strovas, T.J.; Loomis, E.; Greenup, L.; Murrell, J.R.; Ghetti, B.; Raskind, M.A.; Montine, T.J.; Bird, T.D.; et al. The tau tubulin kinases TTBK1/2 promote accumulation of pathological TDP-43. PLoS Genet. 2014, 10, e1004803. [Google Scholar] [CrossRef]
- Li, H.Y.; Yeh, P.A.; Chiu, H.C.; Tang, C.Y.; Tu, B.P. Hyperphosphorylation as a defense mechanism to reduce TDP-43 aggregation. PLoS ONE 2011, 6, e23075. [Google Scholar] [CrossRef]
- Dormann, D.; Capell, A.; Carlson, A.M.; Shankaran, S.S.; Rodde, R.; Neumann, M.; Kremmer, E.; Matsuwaki, T.; Yamanouchi, K.; Nishihara, M.; et al. Proteolytic processing of TAR DNA binding protein-43 by caspases produces C-terminal fragments with disease defining properties independent of progranulin. J. Neurochem. 2009, 110, 1082–1094. [Google Scholar] [CrossRef]
- Iguchi, Y.; Katsuno, M.; Takagi, S.; Ishigaki, S.; Niwa, J.; Hasegawa, M.; Tanaka, F.; Sobue, G. Oxidative stress induced by glutathione depletion reproduces pathological modifications of TDP-43 linked to TDP-43 proteinopathies. Neurobiol. Dis. 2012, 45, 862–870. [Google Scholar] [CrossRef]
- Hebron, M.L.; Lonskaya, I.; Sharpe, K.; Weerasinghe, P.P.; Algarzae, N.K.; Shekoyan, A.R.; Moussa, C.E. Parkin ubiquitinates Tar-DNA binding protein-43 (TDP-43) and promotes its cytosolic accumulation via interaction with histone deacetylase 6 (HDAC6). J. Biol. Chem. 2013, 288, 4103–4115. [Google Scholar] [CrossRef]
- Pesiridis, G.S.; Tripathy, K.; Tanik, S.; Trojanowski, J.Q.; Lee, V.M. A “two-hit” hypothesis for inclusion formation by carboxyl-terminal fragments of TDP-43 protein linked to RNA depletion and impaired microtubule-dependent transport. J. Biol. Chem. 2011, 286, 18845–18855. [Google Scholar] [CrossRef] [PubMed]
- Low, P. The role of ubiquitin-proteasome system in ageing. Gen. Comp. Endocrinol. 2011, 172, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Hans, F.; Fiesel, F.C.; Strong, J.C.; Jäckel, S.; Rasse, T.M.; Geisler, S.; Springer, W.; Schulz, J.B.; Voigt, A.; Kahle, P.J. UBE2E ubiquitin-conjugating enzymes and ubiquitin isopeptidase Y regulate TDP-43 protein ubiquitination. J. Biol. Chem. 2014, 289, 19164–19179. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Lee, M.J.; Park, S.; Oh, D.C.; Elsasser, S.; Chen, P.C.; Gartner, C.; Dimova, N.; Hanna, J.; Gygi, S.P.; et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010, 467, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Xu, Y.F.; Dickey, C.A.; Buratti, E.; Baralle, F.; Bailey, R.; Pickering-Brown, S.; Dickson, D.; Petrucelli, L. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J. Neurosci. 2007, 27, 10530–10534. [Google Scholar] [CrossRef]
- Suzuki, H.; Lee, K.; Matsuoka, M. TDP-43-induced death is associated with altered regulation of BIM and Bcl-xL and attenuated by caspase-mediated TDP-43 cleavage. J. Biol. Chem. 2011, 286, 13171–13183. [Google Scholar] [CrossRef]
- Voigt, A.; Herholz, D.; Fiesel, F.C.; Kaur, K.; Müller, D.; Karsten, P.; Weber, S.S.; Kahle, P.J.; Marquardt, T.; Schulz, J.B. TDP-43-mediated neuron loss in vivo requires RNA-binding activity. PLoS ONE 2010, 5, e12247. [Google Scholar] [CrossRef]
- Colombrita, C.; Zennaro, E.; Fallini, C.; Weber, M.; Sommacal, A.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 2009, 111, 1051–1061. [Google Scholar] [CrossRef]
- Yamashita, M.; Nonaka, T.; Hirai, S.; Miwa, A.; Okado, H.; Arai, T.; Hosokawa, M.; Akiyama, H.; Hasegawa, M. Distinct pathways leading to TDP-43-induced cellular dysfunctions. Hum. Mol. Genet. 2014, 23, 4345–4356. [Google Scholar] [CrossRef]
- Igaz, L.M.; Kwong, L.K.; Lee, E.B.; Chen-Plotkin, A.; Swanson, E.; Unger, T.; Malunda, J.; Xu, Y.; Winton, M.J.; Trojanowski, J.Q.; et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J. Clin. Investig. 2011, 121, 726–738. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.S.; Tsai, K.J.; Chang, Y.J.; Kao, P.; Woods, R.; Kuo, P.H.; Wu, C.C.; Liao, J.Y.; Chou, S.C.; Lin, V.; et al. Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat. Commun. 2014, 5, 4824. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; Del Tredici, K.; Irwin, D.J.; Grossman, M.; Robinson, J.L.; Toledo, J.B.; Fang, L.; Van Deerlin, V.M.; Ludolph, A.C.; Lee, V.M.; et al. Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol. 2014, 127, 423–439. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kaneko, K.; Nukina, N. Molecular properties of TAR DNA binding protein-43 fragments are dependent upon its cleavage site. Biochim. Biophys. Acta 2011, 1812, 1577–1583. [Google Scholar] [CrossRef] [PubMed]
- Kleinberger, G.; Capell, A.; Haass, C.; Van Broeckhoven, C. Mechanisms of granulin deficiency: Lessons from cellular and animal models. Mol. Neurobiol. 2013, 47, 337–360. [Google Scholar] [CrossRef]
- Bateman, A.; Belcourt, D.; Bennett, H.; Lazure, C.; Solomon, S. Granulins, a novel class of peptide from leukocytes. Biochem. Biophys. Res. Commun. 1990, 173, 1161–1168. [Google Scholar] [CrossRef]
- Ahmed, Z.; Sheng, H.; Xu, Y.F.; Lin, W.L.; Innes, A.E.; Gass, J.; Yu, X.; Hou, H.; Chiba, S.; Yamanouchi, K.; et al. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am. J. Pathol. 2010, 177, 311–324. [Google Scholar] [CrossRef]
- Naphade, S.B.; Kigerl, K.A.; Jakeman, L.B.; Kostyk, S.K.; Popovich, P.G.; Kuret, J. Progranulin expression is upregulated after spinal contusion in mice. Acta Neuropathol. 2010, 119, 123–133. [Google Scholar] [CrossRef]
- Philips, T.; De Muynck, L.; Thu, H.N.; Weynants, B.; Vanacker, P.; Dhondt, J.; Sleegers, K.; Schelhaas, H.J.; Verbeek, M.; Vandenberghe, R.; et al. Microglial upregulation of progranulin as a marker of motor neuron degeneration. J. Neuropathol. Exp. Neurol. 2010, 69, 1191–1200. [Google Scholar] [CrossRef]
- Finch, N.; Carrasquillo, M.M.; Baker, M.; Rutherford, N.J.; Coppola, G.; Dejesus-Hernandez, M.; Crook, R.; Hunter, T.; Ghidoni, R.; Benussi, L.; et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology 2011, 76, 467–474. [Google Scholar] [CrossRef]
- Cruchaga, C.; Graff, C.; Chiang, H.H.; Wang, J.; Hinrichs, A.L.; Spiegel, N.; Bertelsen, S.; Mayo, K.; Norton, J.B.; Morris, J.C.; et al. Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch Neurol. 2011, 68, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.M.; Fellerer, K.; Schwenk, B.M.; Kuhn, P.H.; Kremmer, E.; Edbauer, D.; Capell, A.; Haass, C. Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J. Biol. Chem. 2012, 287, 19355–19365. [Google Scholar] [CrossRef] [PubMed]
- Chen-Plotkin, A.S.; Unger, T.L.; Gallagher, M.D.; Bill, E.; Kwong, L.K.; Volpicelli-Daley, L.; Busch, J.I.; Akle, S.; Grossman, M.; Van Deerlin, V.; et al. TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J. Neurosci. 2012, 32, 11213–11227. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Michon, A. Démences; Léger, J.M., Mas, J.L., Eds.; Doin-John Libbey Eurotext: Esher, UK, 2015; ISBN 978-2-7040-1429-3. [Google Scholar]
- Sonobe, Y.; Lee, S.; Krishnan, G.; Gu, Y.; Kwon, D.Y.; Gao, F.B.; Roos, R.P.; Kratsios, P. Translation of dipeptide repeat proteins in C9ORF72 ALS/FTD through unique and redundant AUG initiation codons. eLife 2023, 12, e83189. [Google Scholar] [CrossRef] [PubMed]
- Kortazar-Zubizarreta, I.; Manero-Azua, A.; Afonso-Agüera, J.; de Nanclares, G.P. C9ORF72 Gene GGGGCC Hexanucleotide Expansion: A High Clinical Variability from Amyotrophic Lateral Sclerosis to Frontotemporal Dementia. J. Pers. Med. 2023, 13, 1396. [Google Scholar] [CrossRef]
- Črnigoj, M.M.; Čerček, U.; Yin, X.; Ho, M.T.; Lampret, B.R.; Neumann, M.; Hermann, A.; Rouleau, G.; Suter, B.; Mayr, M.; et al. Phenylalanine-tRNA aminoacylation is compromised by ALS/FTD-associated C9orf72 C4G2 repeat RNA. Nat. Commun. 2023, 14, 5764. [Google Scholar] [CrossRef]
- Snowden, J.S.; Pickering-Brown, S.M.; Mackenzie, I.R.; Richardson, A.M.; Varma, A.; Neary, D.; Mann, D.M. Progranulin gene mutations associated with frontotemporal dementia and progressive non-fluent aphasia. Brain 2006, 129, 3091–3102. [Google Scholar] [CrossRef]
- Saracino, D.; Ferrieux, S.; Noguès-Lassiaille, M.; Houot, M.; Funkiewiez, A.; Sellami, L.; Deramecourt, V.; Pasquier, F.; Couratier, P.; Pariente, J.; et al. Primary Progressive Aphasia Associated With GRN Mutations: New Insights Into the Nonamyloid Logopenic Variant. Neurology 2021, 97, e88–e102. [Google Scholar] [CrossRef]
- Puoti, G.; Lerza, M.C.; Ferretti, M.G.; Bugiani, O.; Tagliavini, F.; Rossi, G. A mutation in the 5’-UTR of GRN gene associated with frontotemporal lobar degeneration: Phenotypic variability and possible pathogenetic mechanisms. J. Alzheimers Dis. 2014, 42, 939–947. [Google Scholar] [CrossRef]
- Hutton, M. Missense and splice site mutations in tau associated with FTDP-17: Multiple pathogenic mechanisms. Neurology 2001, 56, S21–S25. [Google Scholar] [CrossRef]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Pottier, C.; Ravenscroft, T.A.; Brown, P.H.; Finch, N.A.; Baker, M.; Parsons, M.; Asmann, Y.W.; Ren, Y.; Christopher, E.; Levitch, D.; et al. TYROBP genetic variants in early-onset Alzheimer’s disease. Neurobiol. Aging 2016, 48, 222.e9–222.e15. [Google Scholar] [CrossRef] [PubMed]
- Watts, G.D.; Wymer, J.; Kovach, M.J.; Mehta, S.G.; Mumm, S.; Darvish, D.; Pestronk, A.; Whyte, M.P.; Kimonis, V.E. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 2004, 36, 377–381. [Google Scholar] [CrossRef]
- Vesa, J.; Su, H.; Watts, G.D.; Krause, S.; Walter, M.C.; Martin, B.; Smith, C.; Wallace, D.C.; Kimonis, V.E. Valosin containing protein associated inclusion body myopathy: Abnormal vacuolization, autophagy and cell fusion in myoblasts. Neuromuscul. Disord. 2009, 19, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Custer, S.K.; Neumann, M.; Lu, H.; Wright, A.C.; Taylor, J.P. Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum. Mol. Genet. 2010, 19, 1741–1755. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wen, Y.; Zhao, M.; Wang, Y.; Li, P.; Wang, L.; Wang, S. A novel splice-site mutation in CHMP2B associated with frontotemporal dementia: The first report from China and literature review. Mol. Genet. Genom. Med. 2023, 11, e2222. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, G.; Parkinson, N.J.; Brown, J.M.; Chakrabarti, L.; Lloyd, S.L.; Hummerich, H.; Nielsen, J.E.; Hodges, J.R.; Spillantini, M.G.; Thusgaard, T.; et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 2005, 37, 806–808. [Google Scholar] [CrossRef]
- Rubino, E.; Rainero, I.; Chiò, A.; Rogaeva, E.; Galimberti, D.; Fenoglio, P.; Grinberg, Y.; Isaia, G.; Calvo, A.; Gentile, S.; et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology 2012, 79, 1556–1562. [Google Scholar] [CrossRef]
- Lee, S.; Jeon, Y.M.; Cha, S.J.; Kim, S.; Kwon, Y.; Jo, M.; Jang, Y.N.; Lee, S.; Kim, J.; Kim, S.R.; et al. PTK2/FAK regulates UPS impairment via SQSTM1/p62 phosphorylation in TARDBP/TDP-43 proteinopathies. Autophagy 2020, 16, 1396–1412. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Nicholson, A.M.; Sarkar, M.; Messing, J.; Purice, M.D.; Pottier, C.; Annu, K.; Baker, M.; Perkerson, R.B.; Kurti, A.; et al. TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron 2017, 95, 808–816. [Google Scholar] [CrossRef]
- Van Damme, P.; Van Hoecke, A.; Lambrechts, D.; Vanacker, P.; Bogaert, E.; van Swieten, J.; Carmeliet, P.; Van Den Bosch, L.; Robberecht, W. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J. Cell Biol. 2008, 181, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Tapia, L.; Milnerwood, A.; Guo, A.; Mills, F.; Yoshida, E.; Vasuta, C.; Mackenzie, I.R.; Raymond, L.; Cynader, M.; Jia, W.; et al. Progranulin deficiency decreases gross neural connectivity but enhances transmission at individual synapses. J. Neurosci. 2011, 31, 11126–11132. [Google Scholar] [CrossRef] [PubMed]
- Kocerha, J.; Kouri, N.; Baker, M.; Finch, N.; DeJesus-Hernandez, M.; Gonzalez, J.; Chidamparam, K.; Josephs, K.A.; Boeve, B.F.; Graff-Radford, N.R.; et al. Altered microRNA expression in frontotemporal lobar degeneration with TDP-43 pathology caused by progranulin mutations. BMC Genom. 2011, 12, 527. [Google Scholar] [CrossRef] [PubMed]
- Lui, H.; Zhang, J.; Makinson, S.R.; Cahill, M.K.; Kelley, K.W.; Huang, H.Y.; Shang, Y.; Oldham, M.C.; Martens, L.H.; Gao, F.; et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 2016, 165, 921–935. [Google Scholar] [CrossRef]
- Gass, J.; Lee, W.C.; Cook, C.; Finch, N.; Stetler, C.; Jansen-West, K.; Lewis, J.; Link, C.D.; Rademakers, R.; Nykjaer, A.; et al. Progranulin regulates neuronal outgrowth independent of sortilin. Mol. Neurodegener. 2012, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Gass, J.; Prudencio, M.; Stetler, C.; Petrucelli, L. Progranulin: An emerging target for FTLD therapies. Brain Res. 2012, 1462, 118–128. [Google Scholar] [CrossRef]
- Hu, F.; Padukkavidana, T.; Vaegter, C.B.; Brady, O.A.; Zheng, Y.; Mackenzie, I.R.; Feldman, H.H.; Nykjaer, A.; Strittmatter, S.M. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 2010, 68, 654–667. [Google Scholar] [CrossRef]
- Martens, L.H.; Zhang, J.; Barmada, S.J.; Zhou, P.; Kamiya, S.; Sun, B.; Min, S.W.; Gan, L.; Finkbeiner, S.; Huang, E.J.; et al. Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J. Clin. Investig. 2022, 132, e157161. [Google Scholar] [CrossRef]
- Suh, H.S.; Choi, N.; Tarassishin, L.; Lee, S.C. Regulation of progranulin expression in human microglia and proteolysis of progranulin by matrix metalloproteinase-12 (MMP-12). PLoS ONE 2012, 7, e35115. [Google Scholar] [CrossRef]
- Guo, A.; Tapia, L.; Bamji, S.X.; Cynader, M.S.; Jia, W. Progranulin deficiency leads to enhanced cell vulnerability and TDP-43 translocation in primary neuronal cultures. Brain Res. 2010, 1366, 1–8. [Google Scholar] [CrossRef]
- Kleinberger, G.; Wils, H.; Ponsaerts, P.; Joris, G.; Timmermans, J.P.; Van Broeckhoven, C.; Kumar-Singh, S. Increased caspase activation and decreased TDP-43 solubility in progranulin knockout cortical cultures. J. Neurochem. 2010, 115, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Shankaran, S.S.; Capell, A.; Hruscha, A.T.; Fellerer, K.; Neumann, M.; Schmid, B.; Haass, C. Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J. Biol. Chem. 2008, 283, 1744–1753. [Google Scholar] [CrossRef] [PubMed]
- Wils, H.; Kleinberger, G.; Pereson, S.; Janssens, J.; Capell, A.; Van Dam, D.; Cuijt, I.; Joris, G.; De Deyn, P.P.; Haass, C.; et al. Cellular ageing, increased mortality and FTLD-TDP-associated neuropathology in progranulin knockout mice. J. Pathol. 2012, 228, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Dumont, M.; Banerjee, R.; Ma, Y.; Li, H.; Lin, M.T.; Beal, M.F.; Nathan, C.; Thomas, B.; Ding, A. Behavioral deficits and progressive neuropathology in progranulin-deficient mice: A mouse model of frontotemporal dementia. FASEB J. 2010, 24, 4639–4647. [Google Scholar] [CrossRef]
- Salazar, D.A.; Butler, V.J.; Argouarch, A.R.; Hsu, T.Y.; Mason, A.; Nakamura, A.; McCurdy, H.; Cox, D.; Ng, R.; Pan, G.; et al. The Progranulin Cleavage Products, Granulins, Exacerbate TDP-43 Toxicity and Increase TDP-43 Levels. J. Neurosci. 2015, 35, 9315–9328. [Google Scholar] [CrossRef]
- Mackenzie, I.R. The neuropathology and clinical phenotype of FTD with progranulin mutations. Acta Neuropathol. 2007, 114, 49–54. [Google Scholar] [CrossRef]
- Whitwell, J.L.; Weigand, S.D.; Boeve, B.F.; Senjem, M.L.; Gunter, J.L.; DeJesus-Hernandez, M.; Rutherford, N.J.; Baker, M.; Knopman, D.S.; Wszolek, Z.K.; et al. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, Tau, progranulin and sporadics. Brain 2012, 135, 794–806. [Google Scholar] [CrossRef]
- Sieben, A.; Van Langenhove, T.; Engelborghs, S.; Martin, J.J.; Boon, P.; Cras, P.; De Deyn, P.P.; Santens, P.; Van Broeckhoven, C.; Cruts, M. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol. 2012, 124, 353–372. [Google Scholar] [CrossRef]
- Hatanpaa, K.J.; Bigio, E.H.; Cairns, N.J.; Womack, K.B.; Weintraub, S.; Morris, J.C.; Foong, C.; Xiao, G.; Hladik, C.; Mantanona, T.Y.; et al. TAR DNA-binding protein 43 immunohistochemistry reveals extensive neuritic pathology in FTLD-U: A midwest-southwest consortium for FTLD study. J. Neuropathol. Exp. Neurol. 2008, 67, 271–279. [Google Scholar] [CrossRef]
- Josephs, K.A.; Ahmed, Z.; Katsuse, O.; Parisi, J.F.; Boeve, B.F.; Knopman, D.S.; Petersen, R.C.; Davies, P.; Duara, R.; Graff-Radford, N.R.; et al. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J. Neuropathol. Exp. Neurol. 2007, 66, 142–151. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Woollacott, I.O.; Mead, S. The C9ORF72 expansion mutation: Gene structure, phenotypic and diagnostic issues. Acta Neuropathol. 2014, 127, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.; Waite, A.; Rollinson, S.; Chio, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 2012, 11, 323–330. [Google Scholar] [CrossRef]
- Gendron, T.F.; Cosio, D.M.; Petrucelli, L. c9RAN translation: A potential therapeutic target for the treatment of amyotrophic lateral sclerosis and frontotemporal dementia. Expert Opin. Ther. Targets 2013, 17, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Weng, S.M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef]
- Caillet-Boudin, M.L.; Fernandez-Gomez, F.J.; Tran, H.; Dhaenens, C.M.; Buee, L.; Sergeant, N. Brain pathology in myotonic dystrophy: When tauopathy meets spliceopathy and RNAopathy. Front. Mol. Neurosci. 2014, 6, 57. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Gendron, T.F.; Grima, J.C.; Sasaguri, H.; Jansen-West, K.; Xu, Y.F.; Katzman, R.B.; Gass, J.; Murray, M.E.; Shinohara, M.; et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat. Neurosci. 2016, 19, 668–677. [Google Scholar] [CrossRef]
- Gallagher, M.D.; Suh, E.; Grossman, M.; Elman, L.; McCluskey, L.; Van Swieten, J.C.; Al-Sarraj, S.; Neumann, M.; Gelpi, E.; Ghetti, B.; et al. TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol. 2014, 127, 407–418. [Google Scholar] [CrossRef]
- Hsiung, G.Y.; DeJesus-Hernandez, M.; Feldman, H.H.; Sengdy, P.; Bouchard-Kerr, P.; Dwosh, E.; Butler, R.; Leung, B.; Fok, A.; Rutherford, N.J.; et al. Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain 2012, 135, 709–722. [Google Scholar] [CrossRef]
- Benajiba, L.; Le Ber, I.; Camuzat, A.; Lacoste, M.; Thomas-Anterion, C.; Couratier, P.; Legallic, S.; Salachas, F.; Hannequin, D.; Decousus, M.; et al. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann. Neurol. 2009, 65, 470–473. [Google Scholar] [CrossRef]
- Ju, J.S.; Weihl, C.C. Inclusion body myopathy, Paget’s disease of the bone and fronto-temporal dementia: A disorder of autophagy. Hum. Mol. Genet. 2010, 19, R38–R45. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Neumann, M.; Mackenzie, I.R.; Cairns, N.J.; Boyer, P.J.; Markesbery, W.R.; Smith, C.D.; Taylor, J.P.; Kretzschmar, H.A.; Kimonis, V.E.; Forman, M.S. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J. Neuropathol. Exp. Neurol. 2007, 66, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Bentmann, E.; Dormann, D.; Jawaid, A.; DeJesus-Hernandez, M.; Ansorge, O.; Roeber, S.; Kretzschmar, H.A.; Munoz, D.G.; Kusaka, H.; et al. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain 2011, 134, 2595–2609. [Google Scholar] [CrossRef]
- Neumann, M.; Roeber, S.; Kretzschmar, H.A.; Rademakers, R.; Baker, M.; Mackenzie, I.R. Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol. 2009, 118, 605–616. [Google Scholar] [CrossRef]
- Schwartz, J.C.; Cech, T.R.; Parker, R.R. Biochemical Properties and Biological Functions of FET Proteins. Annu. Rev. Biochem. 2015, 84, 355–379. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.C.; Liang, T.Y.; Mazur, C.; et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef]
- Dormann, D.; Madl, T.; Valori, C.F.; Bentmann, E.; Tahirovic, S.; Abou-Ajram, C.; Kremmer, E.; Ansorge, O.; Mackenzie, I.R.; Neumann, M.; et al. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J. 2012, 31, 4258–4275. [Google Scholar] [CrossRef]
- Cairns, N.J.; Grossman, M.; Arnold, S.E.; Burn, D.J.; Jaros, E.; Perry, R.H.; Duyckaerts, C.; Stankoff, B.; Pillon, B.; Skullerud, K.; et al. Clinical and neuropathologic variation in neuronal intermediate filament inclusion disease. Neurology 2004, 63, 1376–1384. [Google Scholar] [CrossRef]
- Munoz, D.G.; Neumann, M.; Kusaka, H.; Yokota, O.; Ishihara, K.; Terada, S.; Kuroda, S.; Mackenzie, I.R. FUS pathology in basophilic inclusion body disease. Acta Neuropathol. 2009, 118, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Dormann, D.; Rodde, R.; Edbauer, D.; Bentmann, E.; Fischer, I.; Hruscha, A.; Than, M.E.; Mackenzie, I.R.; Capell, A.; Schmid, B.; et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010, 29, 2841–2857. [Google Scholar] [CrossRef] [PubMed]
- Ticozzi, N.; Vance, C.; Leclerc, A.L.; Keagle, P.; Glass, J.D.; McKenna-Yasek, D.; Sapp, P.C.; Silani, V.; Bosco, D.A.; Shaw, C.E.; et al. Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156B, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Urwin, H.; Josephs, K.A.; Rohrer, J.D.; Mackenzie, I.R.; Neumann, M.; Authier, A.; Seelaar, H.; Van Swieten, J.C.; Brown, J.M.; Johannsen, P.; et al. FUS pathology defines the majority of Tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol. 2010, 120, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Ravenscroft, T.A.; Baker, M.C.; Rutherford, N.J.; Neumann, M.; Mackenzie, I.R.; Josephs, K.A.; Boeve, B.F.; Petersen, R.; Halliday, G.M.; Kril, J.; et al. Mutations in protein N-arginine methyltransferases are not the cause of FTLD-FUS. Neurobiol. Aging 2013, 34, 2235.e11–2235.e13. [Google Scholar] [CrossRef] [PubMed]
- Paloneva, J.; Kestila, M.; Wu, J.; Salminen, A.; Bohling, T.; Ruotsalainen, V.; Hakola, P.; Bakker, A.B.; Phillips, J.H.; Pekkarinen, P.; et al. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat. Genet. 2000, 25, 357–361. [Google Scholar] [CrossRef]
- Rademakers, R.; Baker, M.; Nicholson, A.M.; Rutherford, N.J.; Finch, N.; Soto-Ortolaza, A.; Lash, J.; Wider, C.; Wojtas, A.; DeJesus-Hernandez, M.; et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat. Genet. 2011, 44, 200–205. [Google Scholar] [CrossRef]
- Wong, T.H.; Chiu, W.Z.; Breedveld, G.J.; Li, K.W.; Verkerk, A.J.; Hondius, D.; Hukema, R.K.; Seelaar, H.; Frick, P.; Severijnen, L.A.; et al. PRKAR1B mutation associated with a new neurodegenerative disorder with unique pathology. Brain 2014, 137, 1361–1373. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Gene | Protein | Reference |
|---|---|---|---|
| C9ORF72 expansion | C9ORF72 | TDP-43 | [75,76,77] |
| GRN mutation | GRN | Progranulin | [78,79,80] |
| MAPT mutation | MAPT | Tau | [81,82] |
| TBK1 mutation | TBK1 | TANK-binding kinase 1 | [83] |
| VCP mutation | VCP | Valosin-containing protein | [84,85,86] |
| CHMP2B mutation | CHMP2B | Charged multivesicular body protein 2B | [87,88] |
| SQSTM1 mutation | SQSTM1 | Sequestosome-1 (p62) | [89,90] |
| TIA1 mutation | TIA1 | TIA-1 RNA-binding protein | [91] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz, G.G.; Ramírez-Jirano, J.; Arizaga, R.L.; Delgado-Lara, D.L.C.; Torres-Sánchez, E.D. Frontotemporal-TDP and LATE Neurocognitive Disorders: A Pathophysiological and Genetic Approach. Brain Sci. 2023, 13, 1474. https://doi.org/10.3390/brainsci13101474
Ortiz GG, Ramírez-Jirano J, Arizaga RL, Delgado-Lara DLC, Torres-Sánchez ED. Frontotemporal-TDP and LATE Neurocognitive Disorders: A Pathophysiological and Genetic Approach. Brain Sciences. 2023; 13(10):1474. https://doi.org/10.3390/brainsci13101474
Chicago/Turabian StyleOrtiz, Genaro Gabriel, Javier Ramírez-Jirano, Raul L. Arizaga, Daniela L. C. Delgado-Lara, and Erandis D. Torres-Sánchez. 2023. "Frontotemporal-TDP and LATE Neurocognitive Disorders: A Pathophysiological and Genetic Approach" Brain Sciences 13, no. 10: 1474. https://doi.org/10.3390/brainsci13101474