
Intracranial Aneurysms and Genetics: An Extensive Overview of Genomic Variations, Underlying Molecular Dynamics, Inflammatory Indicators, and Forward-Looking Insights
 , ,
, ,
Abstract
:1. Introduction
2. Risk Factors Associated with IAs
2.1. Genetic Syndromes with Increased Intracranial Aneurysm Incidence
2.2. Genetic Implications
2.3. Genome-Wide Association Studies
3. Molecular and Physiopathological Mechanisms Implicated in IA Formation and Progression
4. Adherens Junction, MAPK, and Notch Signaling, Which Are Functionally Relevant to the Pathogenesis of an IA
Inflammation Contribution
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brown, R. Unruptured Intracranial Aneurysms. Semin. Neurol. 2010, 30, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Rinkel, G.J.E.; Djibuti, M.; Algra, A.; Van Gijn, J. Prevalence and Risk of Rupture of Intracranial Aneurysms: A Systematic Review. Stroke 1998, 29, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.L.; Levy, D.M.; Manna, B. Pediatric Cerebral Aneurysm. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: http://www.ncbi.nlm.nih.gov/books/NBK537085/ (accessed on 10 August 2023).
- Menghini, V.V.; Brown, R.D.; Sicks, J.D.; O’Fallon, W.M.; Wiebers, D.O. Incidence and prevalence of intracranial aneurysms and hemorrhage in Olmsted County, Minnesota, 1965 to 1995. Neurology 1998, 51, 405–411. [Google Scholar] [CrossRef]
- Ujiie, H.; Sato, K.; Onda, H.; Oikawa, A.; Kagawa, M.; Takakura, K.; Kobayashi, N. Clinical analysis of incidentally discovered unruptured aneurysms. Stroke 1993, 24, 1850–1856. [Google Scholar] [CrossRef] [PubMed]
- The International Study of Unruptured Intracranial Aneurysms Investigators. Unruptured Intracranial Aneurysms—Risk of Rupture and Risks of Surgical Intervention. N. Engl. J. Med. 1998, 339, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Schackert, H.K.; Schackert, G.; Krex, D. Genesis of Cerebral Aneurysms—An Update. Acta Neurochir. 2001, 143, 429–449. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Hashi, K. The incidence and treatment of asymptomatic, unruptured cerebral aneurysms. J. Neurosurg. 1994, 80, 217–223. [Google Scholar] [CrossRef]
- De Rooij, N.K.; Linn, F.H.H.; Van Der Plas, J.A.; Algra, A.; Rinkel, G.J.E. Incidence of subarachnoid haemorrhage: A systematic review with emphasis on region, age, gender and time trends. J. Neurol. Neurosurg. Psychiatry 2007, 78, 1365–1372. [Google Scholar] [CrossRef]
- Johnston, S.C.; Selvin, S.; Gress, D.R. The burden, trends, and demographics of mortality from subarachnoid hemorrhage. Neurology 1998, 50, 1413–1418. [Google Scholar] [CrossRef]
- Grobelny, T.J. Brain Aneurysms: Epidemiology, Treatment Options, and Milestones of Endovascular Treatment Evolution. Dis. Mon. 2011, 57, 647–655. [Google Scholar] [CrossRef]
- Kubota, M.; Yamaura, A.; Ono, J. Prevalence of risk factors for aneurysmal subarachnoid haemorrhage: Results of a Japanese multicentre case control study for stroke. Br. J. Neurosurg. 2001, 15, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M. Systematic review of reviews of risk factors for intracranial aneurysms. Neuroradiology 2008, 50, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Kernan, W.N.; Viscoli, C.M.; Brass, L.M.; Broderick, J.P.; Brott, T.; Feldmann, E.; Morgenstern, L.B.; Wilterdink, J.L.; Horwitz, R.I. Phenylpropanolamine and the Risk of Hemorrhagic Stroke. N. Engl. J. Med. 2000, 343, 1826–1832. [Google Scholar] [CrossRef] [PubMed]
- Greving, J.P.; Wermer, M.J.H.; Brown, R.D.; Morita, A.; Juvela, S.; Yonekura, M.; Ishibashi, T.; Torner, J.C.; Nakayama, T.; Rinkel, G.J.E.; et al. Development of the PHASES score for prediction of risk of rupture of intracranial aneurysms: A pooled analysis of six prospective cohort studies. Lancet Neurol. 2014, 13, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Raaymakers, T.W.M. Aneurysms in relatives of patients with subarachnoid hemorrhage: Frequency and risk factors. Neurology 1999, 53, 982. [Google Scholar] [CrossRef] [PubMed]
- Nahed, B.V.; Bydon, M.; Ozturk, A.K.; Bilguvar, K.; Bayrakli, F.; Gunel, M. Genetics of intracranial aneurysms. Neurosurgery 2007, 60, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Cagnazzo, F.; Gambacciani, C.; Morganti, R.; Perrini, P. Intracranial aneurysms in patients with autosomal dominant polycystic kidney disease: Prevalence, risk of rupture, and management. A systematic review. Acta Neurochir. 2017, 159, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Pirson, Y. Extrarenal Manifestations of Autosomal Dominant Polycystic Kidney Disease. Adv. Chronic Kidney Dis. 2010, 17, 173–180. [Google Scholar] [CrossRef]
- Vlak, M.H.; Algra, A.; Brandenburg, R.; Rinkel, G.J. Prevalence of unruptured intracranial aneurysms, with emphasis on sex, age, comorbidity, country, and time period: A systematic review and meta-analysis. Lancet Neurol. 2011, 10, 626–636. [Google Scholar] [CrossRef]
- Harburger, D.S.; Calderwood, D.A. Integrin signalling at a glance. J. Cell Sci. 2009, 122, 159–163. [Google Scholar] [CrossRef]
- Duker, A.; Jackson, A.; Bober, M.B. Microcephalic Osteodysplastic Primordial Dwarfism Type II. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK575926/ (accessed on 10 August 2023).
- Duker, A.L.; Kinderman, D.; Jordan, C.; Niiler, T.; Baker-Smith, C.M.; Thompson, L.; Parry, D.A.; Carroll, R.S.; Bober, M.B. Microcephalic osteodysplastic primordial dwarfism type II is associated with global vascular disease. Orphanet J. Rare Dis. 2021, 16, 231. [Google Scholar] [CrossRef] [PubMed]
- De Paepe, A.; Malfait, F. The Ehlers-Danlos syndrome, a disorder with many faces. Clin. Genet. 2012, 82, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Superti-Furga, A.; Gugler, E.; Gitzelmann, R.; Steinmann, B. Ehlers-Danlos syndrome type IV: A multi-exon deletion in one of the two COL3A1 alleles affecting structure, stability, and processing of type III procollagen. J. Biol. Chem. 1988, 263, 6226–6232. [Google Scholar] [CrossRef] [PubMed]
- Bober, M.B.; Khan, N.; Kaplan, J.; Lewis, K.; Feinstein, J.A.; Scott, C.I.; Steinberg, G.K. Majewski Osteodysplastic Primordial Dwarfism Type II (MOPD II): Expanding the vascular phenotype. Am. J. Med. Genet. A 2010, 152A, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Li, F.-F.; Wang, X.-D.; Zhu, M.-W.; Lou, Z.-H.; Zhang, Q.; Zhu, C.-Y.; Feng, H.-L.; Lin, Z.-G.; Liu, S.-L. Identification of two novel critical mutations in PCNT gene resulting in microcephalic osteodysplastic primordial dwarfism type II associated with multiple intracranial aneurysms. Metab. Brain Dis. 2015, 30, 1387–1394. [Google Scholar] [CrossRef]
- Jurczyk, A.; Gromley, A.; Redick, S.; Agustin, J.S.; Witman, G.; Pazour, G.J.; Peters, D.J.M.; Doxsey, S. Pericentrin forms a complex with intraflagellar transport proteins and polycystin-2 and is required for primary cilia assembly. J. Cell Biol. 2004, 166, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Betancor, O.; Blackburn, P.R.; Edwards, E.; Vázquez-do-Campo, R.; Klee, E.W.; Labbé, C.; Hodges, K.; Glover, P.; Sigafoos, A.N.; Soto, A.I.; et al. PCNT point mutations and familial intracranial aneurysms. Neurology 2018, 91, e2170–e2181. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Brinjikji, W.; Kallmes, D.F. Prevalence of Intracranial Aneurysms in Patients with Connective Tissue Diseases: A Retrospective Study. Am. J. Neuroradiol. 2016, 37, 1422–1426. [Google Scholar] [CrossRef]
- Arnaud, P.; Milleron, O.; Hanna, N.; Ropers, J.; Ould Ouali, N.; Affoune, A.; Langeois, M.; Eliahou, L.; Arnoult, F.; Renard, P.; et al. Clinical relevance of genotype–phenotype correlations beyond vascular events in a cohort study of 1500 Marfan syndrome patients with FBN1 pathogenic variants. Genet. Med. 2021, 23, 1296–1304. [Google Scholar] [CrossRef]
- Conway, J.E.; Hutchins, G.M.; Tamargo, R.J. Marfan Syndrome Is Not Associated With Intracranial Aneurysms. Stroke 1999, 30, 1632–1636. [Google Scholar] [CrossRef]
- Van Den Berg, J.S.P.; Limburg, M.; Hennekam, R.C.M. Is Marfan Syndrome Associated With Symptomatic Intracranial Aneurysms? Stroke 1996, 27, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Le, C.; Bedocs, P.M. Neurofibromatosis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: http://www.ncbi.nlm.nih.gov/books/NBK459329/ (accessed on 10 August 2023).
- Schievink, W.I.; Riedinger, M.; Maya, M.M. Frequency of incidental intracranial aneurysms in neurofibromatosis type 1. Am. J. Med. Genet. Part A 2005, 134A, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Broadbent, H.M.; Peden, J.F.; Lorkowski, S.; Goel, A.; Ongen, H.; Green, F.; Clarke, R.; Collins, R.; Franzosi, M.G.; Tognoni, G.; et al. Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum. Mol. Genet. 2008, 17, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Dion, P.A.; Rouleau, G.A. Genetics of Intracranial Aneurysms. Stroke 2018, 49, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Salvi, E.; Kutalik, Z.; Glorioso, N.; Benaglio, P.; Frau, F.; Kuznetsova, T.; Arima, H.; Hoggart, C.; Tichet, J.; Nikitin, Y.P.; et al. Genomewide Association Study Using a High-Density Single Nucleotide Polymorphism Array and Case-Control Design Identifies a Novel Essential Hypertension Susceptibility Locus in the Promoter Region of Endothelial NO Synthase. Hypertension 2012, 59, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Jiang, H.; Fan, Y.; Xu, Y.; Wang, N. The association of CDKN2BAS gene polymorphisms and intracranial aneurysm: A meta-analysis. Medicine 2020, 99, e23209. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Kudo, M.; Chen, M.N.; Nakazawa, S.; Wakabayashi, I. Cerebral Aneurysm Associated with Multiple Endocrine Neoplasia, Type 1—Case Report. Neurol. Med. Chir. 1993, 33, 309–311. [Google Scholar] [CrossRef]
- Bock, A.; Schwegler, G. Intracerebral haemorrhage as first manifestation of Pseudoxanthoma elasticum. Clin. Neurol. Neurosurg. 2008, 110, 262–264. [Google Scholar] [CrossRef]
- Willemse, R.B.; Mager, J.J.; Westermann, C.J.J.; Overtoom, T.T.C.; Mauser, H.; Wolbers, J.G. Bleeding risk of cerebral vascular malformations in hereditary hemorrhagic telangiectasia. J. Neurosurg. 2000, 92, 779–784. [Google Scholar] [CrossRef]
- Hitchcock, E.; Gibson, W.T. A Review of the Genetics of Intracranial Berry Aneurysms and Implications for Genetic Counseling. J. Genet. Couns. 2017, 26, 21–31. [Google Scholar] [CrossRef]
- Tromp, G.; Weinsheimer, S.; Ronkainen, A.; Kuivaniemi, H. Molecular basis and genetic predisposition to intracranial aneurysm. Ann. Med. 2014, 46, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, J.H. Modifier genes in mice and humans. Nat. Rev. Genet. 2001, 2, 165–174. [Google Scholar] [CrossRef] [PubMed]
- You, C.; Chen, Z.; Ma, J.; Cen, Y.; Liu, Y. The angiotensin converting enzyme insertion/deletion polymorphism and intracranial aneurysm: A meta-analysis of case-control studies. Neurol. India 2013, 61, 293. [Google Scholar] [CrossRef] [PubMed]
- Yasuno, K.; Bilguvar, K.; Bijlenga, P.; Low, S.-K.; Krischek, B.; Auburger, G.; Simon, M.; Krex, D.; Arlier, Z.; Nayak, N.; et al. Genome-wide association study of intracranial aneurysm identifies three new risk loci. Nat. Genet. 2010, 42, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Moitra, K.; Garcia, S.; Jaldin, M.; Etoundi, C.; Cooper, D.; Roland, A.; Dixon, P.; Reyes, S.; Turan, S.; Terry, S.; et al. ABCC6 and Pseudoxanthoma Elasticum: The Face of a Rare Disease from Genetics to Advocacy. Int. J. Mol. Sci. 2017, 18, 1488. [Google Scholar] [CrossRef] [PubMed]
- Samuel, N.; Radovanovic, I. Genetic basis of intracranial aneurysm formation and rupture: Clinical implications in the postgenomic era. Neurosurg. Focus 2019, 47, E10. [Google Scholar] [CrossRef] [PubMed]
- Vergouwen, M.D.I.; Frijns, C.J.M.; Roos, Y.B.W.E.M.; Rinkel, G.J.E.; Baas, F.; Vermeulen, M. Plasminogen Activator Inhibitor-1 4G Allele in the 4G/5G Promoter Polymorphism Increases the Occurrence of Cerebral Ischemia After Aneurysmal Subarachnoid Hemorrhage. Stroke 2004, 35, 1280–1283. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.; Liu, S.; Fan, Y.; Wang, X.; Jia, Y.; Li, L.; Cong, S.; Han, F. Single nucleotide polymorphisms, variable number tandem repeats and allele influence on serotonergic enzyme modulators for aggressive and suicidal behaviors: A review. Pharmacol. Biochem. Behav. 2019, 180, 74–82. [Google Scholar] [CrossRef]
- Concannon, P.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Pociot, F.; Todd, J.A.; Rich, S.S. Type 1 Diabetes: Evidence for Susceptibility Loci from Four Genome-Wide Linkage Scans in 1,435 Multiplex Families. Diabetes 2005, 54, 2995–3001. [Google Scholar] [CrossRef]
- Wilson, F.H.; Disse-Nicodème, S.; Choate, K.A.; Ishikawa, K.; Nelson-Williams, C.; Desitter, I.; Gunel, M.; Milford, D.V.; Lipkin, G.W.; Achard, J.-M.; et al. Human Hypertension Caused by Mutations in WNK Kinases. Science 2001, 293, 1107–1112. [Google Scholar] [CrossRef]
- Nahed, B.V.; Seker, A.; Guclu, B.; Ozturk, A.K.; Finberg, K.; Hawkins, A.A.; DiLuna, M.L.; State, M.; Lifton, R.P.; Gunel, M. Mapping a Mendelian Form of Intracranial Aneurysm to 1p34.3-p36.13. Am. J. Hum. Genet. 2005, 76, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Utsunomiya, M.; Inoue, K.; Nozaki, K.; Inoue, S.; Takenaka, K.; Hashimoto, N.; Koizumi, A. Genome-Wide Scan for Japanese Familial Intracranial Aneurysms: Linkage to Several Chromosomal Regions. Circulation 2004, 110, 3727–3733. [Google Scholar] [CrossRef] [PubMed]
- Onda, H.; Kasuya, H.; Yoneyama, T.; Takakura, K.; Hori, T.; Takeda, J.; Nakajima, T.; Inoue, I. Genomewide-Linkage and Haplotype-Association Studies Map Intracranial Aneurysm to Chromosome 7q11. Am. J. Hum. Genet. 2001, 69, 804–819. [Google Scholar] [CrossRef] [PubMed]
- Alg, V.S.; Sofat, R.; Houlden, H.; Werring, D.J. Genetic risk factors for intracranial aneurysms: A meta-analysis in more than 116,000 individuals. Neurology 2013, 80, 2154–2165. [Google Scholar] [CrossRef] [PubMed]
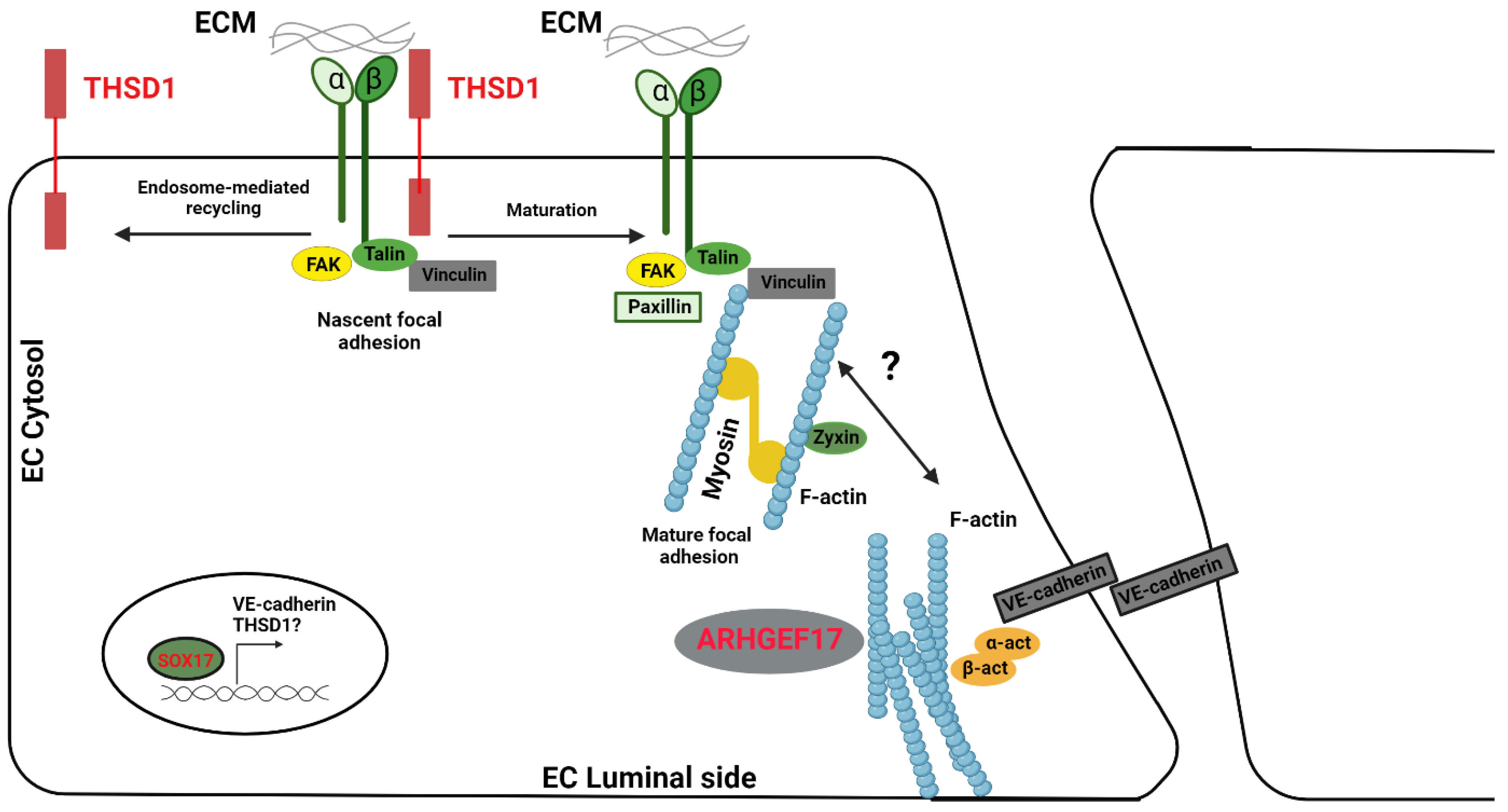
- Foroud, T.; Koller, D.L.; Lai, D.; Sauerbeck, L.; Anderson, C.; Ko, N.; Deka, R.; Mosley, T.H.; Fornage, M.; Woo, D.; et al. Genome-Wide Association Study of Intracranial Aneurysms Confirms Role of Anril and SOX17 in Disease Risk. Stroke 2012, 43, 2846–2852. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Douglas, S.A.; Louden, C.; Vickery-Clark, L.M.; Feuerstein, G.Z.; Ohlstein, E.H. Expression of Endothelin-1, Endothelin-3, Endothelin-Converting Enzyme-1, and Endothelin-A and Endothelin-B Receptor mRNA After Angioplasty-Induced Neointimal Formation in the Rat. Circ. Res. 1996, 78, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Low, S.-K.; Takahashi, A.; Cha, P.-C.; Zembutsu, H.; Kamatani, N.; Kubo, M.; Nakamura, Y. Genome-wide association study for intracranial aneurysm in the Japanese population identifies three candidate susceptible loci and a functional genetic variant at EDNRA. Hum. Mol. Genet. 2012, 21, 2102–2110. [Google Scholar] [CrossRef] [PubMed]
- Yasuno, K.; Bakırcıoğlu, M.; Low, S.-K.; Bilgüvar, K.; Gaál, E.; Ruigrok, Y.M.; Niemelä, M.; Hata, A.; Bijlenga, P.; Kasuya, H.; et al. Common variant near the endothelin receptor type A (EDNRA) gene is associated with intracranial aneurysm risk. Proc. Natl. Acad. Sci. USA 2011, 108, 19707–19712. [Google Scholar] [CrossRef]
- Keedy, A. An overview of intracranial aneurysms. McGill J. Med. MJM Int. Forum Adv. Med. Sci. Stud. 2006, 9, 141–146. [Google Scholar] [CrossRef]
- Krings, T.; Mandell, D.M.; Kiehl, T.-R.; Geibprasert, S.; Tymianski, M.; Alvarez, H.; terBrugge, K.G.; Hans, F.-J. Intracranial aneurysms: From vessel wall pathology to therapeutic approach. Nat. Rev. Neurol. 2011, 7, 547–559. [Google Scholar] [CrossRef]
- Chalouhi, N.; Ali, M.S.; Jabbour, P.M.; Tjoumakaris, S.I.; Gonzalez, L.F.; Rosenwasser, R.H.; Koch, W.J.; Dumont, A.S. Biology of Intracranial Aneurysms: Role of Inflammation. J. Cereb. Blood Flow Metab. 2012, 32, 1659–1676. [Google Scholar] [CrossRef] [PubMed]
- Fennell, V.S.; Kalani, M.Y.S.; Atwal, G.; Martirosyan, N.L.; Spetzler, R.F. Biology of Saccular Cerebral Aneurysms: A Review of Current Understanding and Future Directions. Front. Surg. 2016, 3, 43. [Google Scholar] [CrossRef] [PubMed]
- Tromp, G.; Kuivaniemi, H. Developments in Genomics to Improve Understanding, Diagnosis and Management of Aneurysms and Peripheral Artery Disease. Eur. J. Vasc. Endovasc. Surg. 2009, 38, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Weinsheimer, S.; Lenk, G.M.; Van Der Voet, M.; Land, S.; Ronkainen, A.; Alafuzoff, I.; Kuivaniemi, H.; Tromp, G. Integration of expression profiles and genetic mapping data to identify candidate genes in intracranial aneurysm. Physiol. Genom. 2007, 32, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, G.; Dejana, E. Endothelial Cell-to-Cell Junctions: Molecular Organization and Role in Vascular Homeostasis. Physiol. Rev. 2004, 84, 869–901. [Google Scholar] [CrossRef]
- Verlaan, D.J. A new locus for autosomal dominant intracranial aneurysm, ANIB4, maps to chromosome 5p15.2-14.3. J. Med. Genet. 2006, 43, e31. [Google Scholar] [CrossRef] [PubMed]
- Nakajima-Takagi, Y.; Osawa, M.; Oshima, M.; Takagi, H.; Miyagi, S.; Endoh, M.; Endo, T.A.; Takayama, N.; Eto, K.; Toyoda, T.; et al. Role of SOX17 in hematopoietic development from human embryonic stem cells. Blood 2013, 121, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Haasdijk, R.A.; Den Dekker, W.K.; Cheng, C.; Tempel, D.; Szulcek, R.; Bos, F.L.; Hermkens, D.M.A.; Chrifi, I.; Brandt, M.M.; Van Dijk, C.; et al. THSD1 preserves vascular integrity and protects against intraplaque haemorrhaging in ApoE−/− mice. Cardiovasc. Res. 2016, 110, 129–139. [Google Scholar] [CrossRef]
- De Hoog, C.L.; Foster, L.J.; Mann, M. RNA and RNA Binding Proteins Participate in Early Stages of Cell Spreading through Spreading Initiation Centers. Cell 2004, 117, 649–662. [Google Scholar] [CrossRef]
- Khan, T.A.; Bianchi, C.; Ruel, M.; Voisine, P.; Sellke, F.W. Mitogen-activated protein kinase pathways and cardiac surgery. J. Thorac. Cardiovasc. Surg. 2004, 127, 806–811. [Google Scholar] [CrossRef]
- Joutel, A.; Andreux, F.; Gaulis, S.; Domenga, V.; Cecillon, M.; Battail, N.; Piga, N.; Chapon, F.; Godfrain, C.; Tournier-Lasserve, E. The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J. Clin. Investig. 2000, 105, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, S.; Dotti, M.T.; Federico, A. Physiology and pathology of notch signalling system. J. Cell. Physiol. 2006, 207, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Ruchoux, M.M.; Domenga, V.; Brulin, P.; Maciazek, J.; Limol, S.; Tournier-Lasserve, E.; Joutel, A. Transgenic mice expressing mutant Notch3 develop vascular alterations characteristic of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Am. J. Pathol. 2003, 162, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Brulin, P.; Godfraind, C.; Leteurtre, E.; Ruchoux, M.-M. Morphometric analysis of ultrastructural vascular changes in CADASIL: Analysis of 50 skin biopsy specimens and pathogenic implications. Acta Neuropathol. 2002, 104, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Singh, M.; Huang, J.; Prestigiacomo, C.J.; Winfree, C.J.; Solomon, R.A.; Connolly, E.S.C. Matrix Metalloproteinase-9 in Cerebral Aneurysms. Neurosurgery 1997, 41, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Peters, D.G.; Kassam, A.B.; Feingold, E.; Heidrich-O’Hare, E.; Yonas, H.; Ferrell, R.E.; Brufsky, A. Molecular Anatomy of an Intracranial Aneurysm: Coordinated Expression of Genes Involved in Wound Healing and Tissue Remodeling. Stroke 2001, 32, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Gibson, M.A.; Kumaratilake, J.S.; Cleary, E.G. Immunohistochemical and Ultrastructural Localization of MP78/70 (βig-h3) in Extracellular Matrix of Developing and Mature Bovine Tissues. J. Histochem. Cytochem. 1997, 45, 1683–1696. [Google Scholar] [CrossRef]
- Pichler, R.H.; Hugo, C.; Shankland, S.J.; Reed, M.J.; Bassuk, J.A.; Andoh, T.F.; Lombardi, D.M.; Schwartz, S.M.; Bennett, W.M.; Alpers, C.E.; et al. SPARC is expressed in renal interstitial fibrosis and in renal vascular injury. Kidney Int. 1996, 50, 1978–1989. [Google Scholar] [CrossRef]
- Iruela-Arispe, M.L.; Vernon, R.B.; Wu, H.; Jaenisch, R.; Sage, E.H. Type I collagen-deficient Mov-13 mice do not retain SPARC in the extracellular matrix: Implications for fibroblast function. Dev. Dyn. 1996, 207, 171–183. [Google Scholar] [CrossRef]
- Girard, J.-P.; Springer, T.A. Cloning from purified high endothelial venule cells of hevin, a close relative of the antiadhesive extracellular matrix protein SPARC. Immunity 1995, 2, 113–123. [Google Scholar] [CrossRef]
- Ducy, P.; Zhang, R.; Geoffroy, V.; Ridall, A.L.; Karsenty, G. Osf2/Cbfa1: A Transcriptional Activator of Osteoblast Differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, M.J.; Balbín, M.; López, J.M.; Alvarez, J.; Komori, T.; López-Otín, C. Collagenase 3 is a target of Cbfa1, a transcription factor of the runt gene family involved in bone formation. Mol. Cell. Biol. 1999, 19, 4431–4442. [Google Scholar] [CrossRef] [PubMed]
- Barrett, A.J. Human cathepsin B1. Purification and some properties of the enzyme. Biochem. J. 1973, 131, 809–822. [Google Scholar] [CrossRef] [PubMed]
- Diment, S.; Martin, K.J.; Stahl, P.D. Cleavage of parathyroid hormone in macrophage endosomes illustrates a novel pathway for intracellular processing of proteins. J. Biol. Chem. 1989, 264, 13403–13406. [Google Scholar] [CrossRef] [PubMed]
- Barrett, A.J. Cathepsin D: The Lysosomal Aspartic Proteinase. In Novartis Foundation Symposia; Evered, D., Whelan, J., Eds.; Wiley: Hoboken, NJ, USA, 1980; Volume 75, pp. 37–50. [Google Scholar] [CrossRef]
- Gacko, M.; Chyczewski, L. Activity and localization of cathepsin B, D and G in aortic aneurysm. Int. Surg. 1997, 82, 398–402. [Google Scholar] [PubMed]
- Abisi, S.; Burnand, K.G.; Waltham, M.; Humphries, J.; Taylor, P.R.; Smith, A. Cysteine protease activity in the wall of abdominal aortic aneurysms. J. Vasc. Surg. 2007, 46, 1260–1266. [Google Scholar] [CrossRef] [PubMed]
- Lohoefer, F.; Reeps, C.; Lipp, C.; Rudelius, M.; Zimmermann, A.; Ockert, S.; Eckstein, H.-H.; Pelisek, J. Histopathological analysis of cellular localization of cathepsins in abdominal aortic aneurysm wall. Int. J. Exp. Pathol. 2012, 93, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, C.; Qin, Y. Cathepsins: A new culprit behind abdominal aortic aneurysm. Regen. Med. Res. 2013, 1, 5. [Google Scholar] [CrossRef]
- Frösen, J.; Piippo, A.; Paetau, A.; Kangasniemi, M.; Niemelä, M.; Hernesniemi, J.; Jääskeläinen, J. Remodeling of Saccular Cerebral Artery Aneurysm Wall Is Associated With Rupture: Histological Analysis of 24 Unruptured and 42 Ruptured Cases. Stroke 2004, 35, 2287–2293. [Google Scholar] [CrossRef]
- Kataoka, K.; Taneda, M.; Asai, T.; Kinoshita, A.; Ito, M.; Kuroda, R. Structural Fragility and Inflammatory Response of Ruptured Cerebral Aneurysms: A Comparative Study Between Ruptured and Unruptured Cerebral Aneurysms. Stroke 1999, 30, 1396–1401. [Google Scholar] [CrossRef]
- Ishibashi, R.; Aoki, T.; Nishimura, M.; Hashimoto, N.; Miyamoto, S. Contribution of Mast Cells to Cerebral Aneurysm Formation. Curr. Neurovasc. Res. 2010, 7, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Tulamo, R.; Frösen, J.; Junnikkala, S.; Paetau, A.; Pitkäniemi, J.; Kangasniemi, M.; Niemelä, M.; Jääskeläinen, J.; Jokitalo, E.; Karatas, A.; et al. Complement activation associates with saccular cerebral artery aneurysm wall degeneration and rupture. Neurosurgery 2006, 59, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Tulamo, R.; Frösen, J.; Junnikkala, S.; Paetau, A.; Kangasniemi, M.; Peláez, J.; Hernesniemi, J.; Niemelä, M.; Meri, S. Complement system becomes activated by the classical pathway in intracranial aneurysm walls. Lab. Investig. 2010, 90, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Tada, Y.; Yagi, K.; Kitazato, K.T.; Tamura, T.; Kinouchi, T.; Shimada, K.; Matsushita, N.; Nakajima, N.; Satomi, J.; Kageji, T.; et al. Reduction of endothelial tight junction proteins is related to cerebral aneurysm formation in rats. J. Hypertens. 2010, 28, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
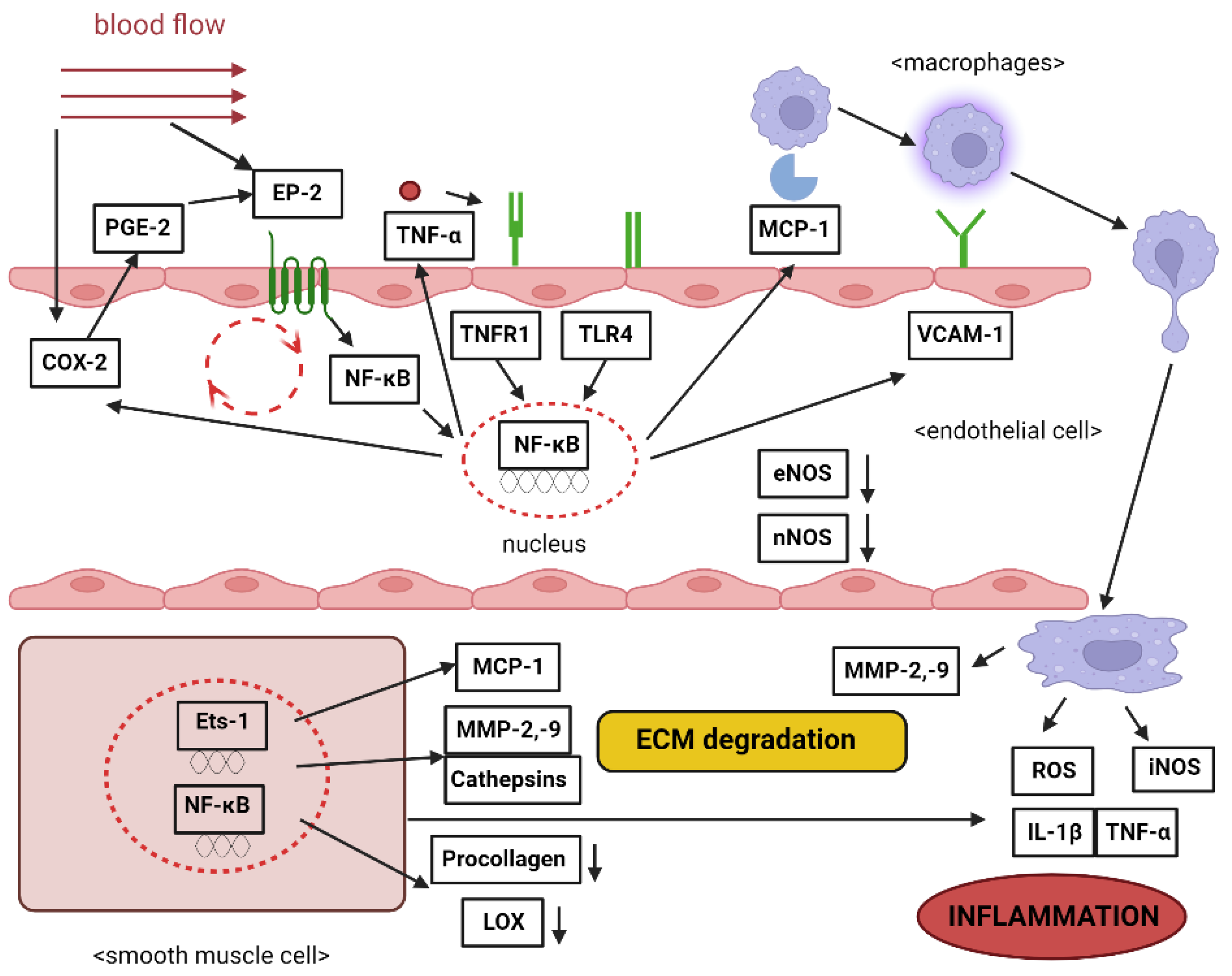
- Boring, L.; Gosling, J.; Cleary, M.; Charo, I.F. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 1998, 394, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Kataoka, H.; Ishibashi, R.; Nozaki, K.; Egashira, K.; Hashimoto, N. Impact of Monocyte Chemoattractant Protein-1 Deficiency on Cerebral Aneurysm Formation. Stroke 2009, 40, 942–951. [Google Scholar] [CrossRef]
- Kadirvel, R.; Ding, Y.-H.; Dai, D.; Lewis, D.A.; Raghavakaimal, S.; Cloft, H.J.; Kallmes, D.F. Gene Expression Profiling of Experimental Saccular Aneurysms Using Deoxyribonucleic Acid Microarrays: Fig 1. Am. J. Neuroradiol. 2008, 29, 1566–1569. [Google Scholar] [CrossRef]
- Collins, T.; Read, M.A.; Neish, A.S.; Whitley, M.Z.; Thanos, D.; Maniatis, T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1995, 9, 899–909. [Google Scholar] [CrossRef]
- Tilley, S.L.; Coffman, T.M.; Koller, B.H. Mixed messages: Modulation of inflammation and immune responses by prostaglandins and thromboxanes. J. Clin. Investig. 2001, 108, 15–23. [Google Scholar] [CrossRef]
- Hasan, D.; Hashimoto, T.; Kung, D.; Macdonald, R.L.; Winn, H.R.; Heistad, D. Upregulation of Cyclooxygenase-2 (COX-2) and Microsomal Prostaglandin E2 Synthase-1 (mPGES-1) in Wall of Ruptured Human Cerebral Aneurysms: Preliminary Results. Stroke 2012, 43, 1964–1967. [Google Scholar] [CrossRef]
- Jayaraman, T.; Berenstein, V.; Li, X.; Mayer, J.; Silane, M.; Shin, Y.S.; Niimi, Y.; Kılıç, T.; Gunel, M.; Berenstein, A. Tumor Necrosis Factor α is a Key Modulator of Inflammation in Cerebral Aneurysms. Neurosurgery 2005, 57, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Fukuda, M.; Nishimura, M.; Nozaki, K.; Narumiya, S. Critical role of TNF-alpha-TNFR1 signaling in intracranial aneurysm formation. Acta Neuropathol. Commun. 2014, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Björkbacka, H. Multiple roles of Toll-like receptor signaling in atherosclerosis. Curr. Opin. Lipidol. 2006, 17, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Nishimura, M.; Ishibashi, R.; Kataoka, H.; Takagi, Y.; Hashimoto, N. Toll-like receptor 4 expression during cerebral aneurysm formation: Laboratory investigation. J. Neurosurg. 2010, 113, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Wang, X.; Huang, Z.; Li, X.; Luo, Y. Difference in inflammation, atherosclerosis, and platelet activation between coronary artery aneurysm and coronary artery ectasia. J. Thorac. Dis. 2020, 12, 5811–5821. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Nishimura, M.; Kataoka, H.; Ishibashi, R.; Nozaki, K.; Miyamoto, S. Complementary inhibition of cerebral aneurysm formation by eNOS and nNOS. Lab. Investig. 2011, 91, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Dhillon, S.; Geary, I.; Howell, W.M.; Iannotti, F.; Day, I.N.M.; Ye, S. Polymorphisms in Matrix Metalloproteinase-1, -3, -9, and -12 Genes in Relation to Subarachnoid Hemorrhage. Stroke 2001, 32, 2198–2202. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, J.; Wu, C.; Cao, X.; He, M.; You, C. The Interleukin-6-572G/C Gene Polymorphism and the Risk of Intracranial Aneurysms in a Chinese Population. Genet. Test. Mol. Biomark. 2012, 16, 822–826. [Google Scholar] [CrossRef]
- Li, L.-J.; Pan, X.-M.; Sima, X.; Li, Z.-H.; Zhang, L.-S.; Sun, H.; Zhu, Y.; Liang, W.-B.; Gao, L.-B.; Zhang, L. Interactions of interleukin-12A and interleukin-12B polymorphisms on the risk of intracranial aneurysm. Mol. Biol. Rep. 2012, 39, 11217–11223. [Google Scholar] [CrossRef]
- Mohan, D.; Munteanu, V.; Coman, T.; Ciurea, A.V. Genetic factors involves in intracranial aneurysms–actualities. J. Med. Life 2015, 8, 336–341. [Google Scholar]
- Aoki, T.; Kataoka, H.; Shimamura, M.; Nakagami, H.; Wakayama, K.; Moriwaki, T.; Ishibashi, R.; Nozaki, K.; Morishita, R.; Hashimoto, N. NF-κB Is a Key Mediator of Cerebral Aneurysm Formation. Circulation 2007, 116, 2830–2840. [Google Scholar] [CrossRef] [PubMed]
- Pawlowska, E.; Szczepanska, J.; Wisniewski, K.; Tokarz, P.; Jaskólski, D.; Blasiak, J. NF-κB-Mediated Inflammation in the Pathogenesis of Intracranial Aneurysm and Subarachnoid Hemorrhage. Does Autophagy Play a Role? Int. J. Mol. Sci. 2018, 19, 1245. [Google Scholar] [CrossRef] [PubMed]
- Kleinloog, R.; Verweij, B.H.; Van Der Vlies, P.; Deelen, P.; Swertz, M.A.; De Muynck, L.; Van Damme, P.; Giuliani, F.; Regli, L.; Van Der Zwan, A.; et al. RNA Sequencing Analysis of Intracranial Aneurysm Walls Reveals Involvement of Lysosomes and Immunoglobulins in Rupture. Stroke 2016, 47, 1286–1293. [Google Scholar] [CrossRef] [PubMed]
- Cassis, L.A.; Gupte, M.; Thayer, S.; Zhang, X.; Charnigo, R.; Howatt, D.A.; Rateri, D.L.; Daugherty, A. ANG II infusion promotes abdominal aortic aneurysms independent of increased blood pressure in hypercholesterolemic mice. Am. J. Physiol.-Heart Circ. Physiol. 2009, 296, H1660–H1665. [Google Scholar] [CrossRef] [PubMed]
- Zhong, P.; Lu, Z.; Li, Z.; Li, T.; Lan, Q.; Liu, J.; Wang, Z.; Chen, S.; Huang, Q. Effect of Renin-Angiotensin-Aldosterone System Inhibitors on the Rupture Risk Among Hypertensive Patients With Intracranial Aneurysms. Hypertension 2022, 79, 1475–1486. [Google Scholar] [CrossRef] [PubMed]
- Foroud, T.; for the FIA Study Investigators. Whole Exome Sequencing of Intracranial Aneurysm. Stroke 2013, 44, S26–S28. [Google Scholar] [CrossRef] [PubMed]
- Toth, G.; Cerejo, R. Intracranial aneurysms: Review of current science and management. Vasc. Med. 2018, 23, 276–288. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
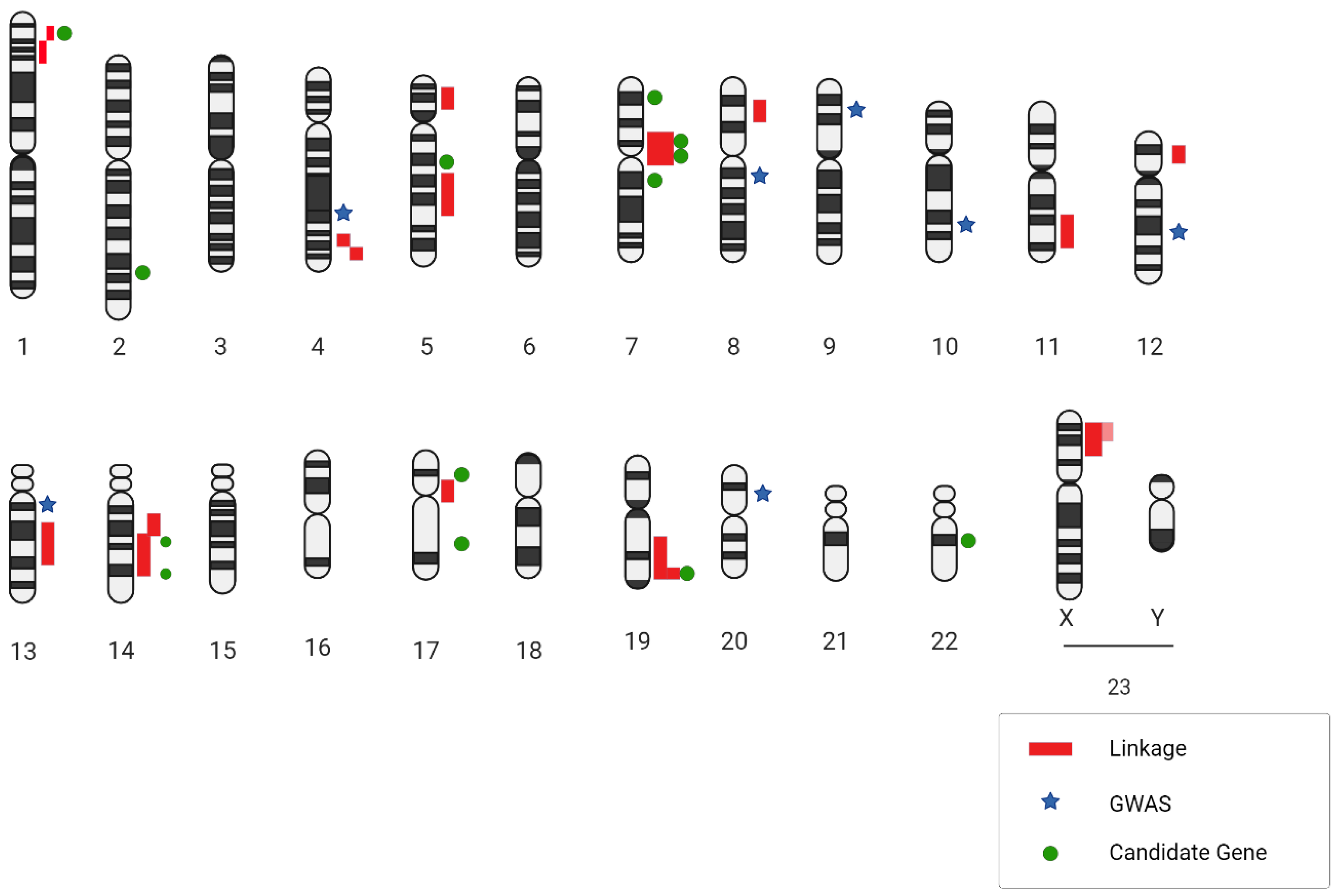
| Chromosomal Region | Study Design | LOD Score | Genetic Marker | Phenotype IDs and OMIM Locus |
|---|---|---|---|---|
| 1p36.21-p36.13 | Non-parametric | 3.18 | D1S2826-D1S234 | 609122; ANIB3 |
| 1p34.21-p36.13 | Family, AD | 4.2 | 609122; ANIB3 | |
| 4q32.2 | Non-parametric | 2.5 | Rs1458149 | |
| 4q32.3 | Parametric | 2.6 | ||
| 5p15.2-p14.3 | Family, AD | 3.57 | D5S1954 | 610213; ANIB4 |
| 5q22-q31 | Affected sib pair | 2.24 | D5S471-D5S2010 | |
| 7q11 | AR | 2.34 | D7S2421 | 105800; ANIB1 |
| 7q11 | Affected sib pair | 3.22 | D7S2415-D7S657 | 105800; ANIB1 |
| 8p22 | Family, AD | 3.61 | D8S552 | 614252; ANIB11 |
| 11q24-q25 | Family | 4.3 | rs618176-rs1940033 | 612161; ANIB7 |
| 12p12.3 | Parametric | 3.1 | ||
| 13q14.12-q21.1 | Family | 4.56 | rs7983420-rs17054625 | |
| 14q23-q31 | Family | 3.0 | rs235991-rs2373098 | 612162; ANIB8 |
| 14q22 | Affected sib pair | 2.31 | D14S258-D14S74 | |
| 17cen | Non-parametric | 3.0 | D17S921-D17S1800 | |
| 19q13 | Non-parametric | 2.15 | D19S198-D19S596 | 608542; ANIB2 |
| 19q13 | Affected sib pair with covariates | 5.70 | D19S178-D19S545 | 608542; ANIB2 |
| 19q13.3 | Affected only, parametric | 4.10 | D19S198-D19S902 | 608542; ANIB2 |
| Xp22 | Non-parametric | 2.16 | DXS987-DXS7593 | 300870; ANIB5 |
| Xp22 | Affected sib pair | 2.08 | DXS987 | 300870; ANIB5 |
| Xp22.32-p22.2 | Non-parametric | 4.54 | DXS6807-DXS1224 | 300870; ANIB5 |
| Type of Genetic Analysis | Loci | Candidate Genes | Functions |
|---|---|---|---|
| GWAS | 13q13.1 | STARD 13 | Movement of endothelial cells |
| GWAS | 18q11.2 | RBBP8 | Cell cycle |
| GWAS | 2q | PLCL1, BOLL | Similarity to phospholipase C, positioned after VEGFR2 in the signaling pathway |
| GWAS | 4q31.23 | EDNRA | Vasoconstriction |
| GWAS | 8q21.3 | SOX17 | Endothelial sprouting |
| GWAS | 9p21.3 | CDKN2A/B | Smooth muscle proliferation |
| GWAS | 10q24.32 | CNNM2 | Epithelial absorption of Mg2+ |
| GWL | 1p34.3-p36.13Xp22 | PERLECAN | Promote endothelial cell growth and renewal, maintain the endothelial barrier function, and inhibit smooth muscle cell proliferation |
| GWL | 7q11, 14q22, 5q22 | ELASTIN | Elasticity of the parietal vessel |
| GWL | 11q24, 14q23 | ||
| GWL | 19q13, Xp22 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toader, C.; Eva, L.; Bratu, B.-G.; Covache-Busuioc, R.-A.; Costin, H.P.; Dumitrascu, D.-I.; Glavan, L.-A.; Corlatescu, A.D.; Ciurea, A.V. Intracranial Aneurysms and Genetics: An Extensive Overview of Genomic Variations, Underlying Molecular Dynamics, Inflammatory Indicators, and Forward-Looking Insights. Brain Sci. 2023, 13, 1454. https://doi.org/10.3390/brainsci13101454
Toader C, Eva L, Bratu B-G, Covache-Busuioc R-A, Costin HP, Dumitrascu D-I, Glavan L-A, Corlatescu AD, Ciurea AV. Intracranial Aneurysms and Genetics: An Extensive Overview of Genomic Variations, Underlying Molecular Dynamics, Inflammatory Indicators, and Forward-Looking Insights. Brain Sciences. 2023; 13(10):1454. https://doi.org/10.3390/brainsci13101454
Chicago/Turabian StyleToader, Corneliu, Lucian Eva, Bogdan-Gabriel Bratu, Razvan-Adrian Covache-Busuioc, Horia Petre Costin, David-Ioan Dumitrascu, Luca-Andrei Glavan, Antonio Daniel Corlatescu, and Alexandru Vlad Ciurea. 2023. "Intracranial Aneurysms and Genetics: An Extensive Overview of Genomic Variations, Underlying Molecular Dynamics, Inflammatory Indicators, and Forward-Looking Insights" Brain Sciences 13, no. 10: 1454. https://doi.org/10.3390/brainsci13101454