
The Role of Superoxide Dismutase 1 in Amyotrophic Lateral Sclerosis: Identification of Signaling Pathways, Regulators, Molecular Interaction Networks, and Biological Functions through Bioinformatics


Abstract
:1. Introduction
2. Materials and Methods
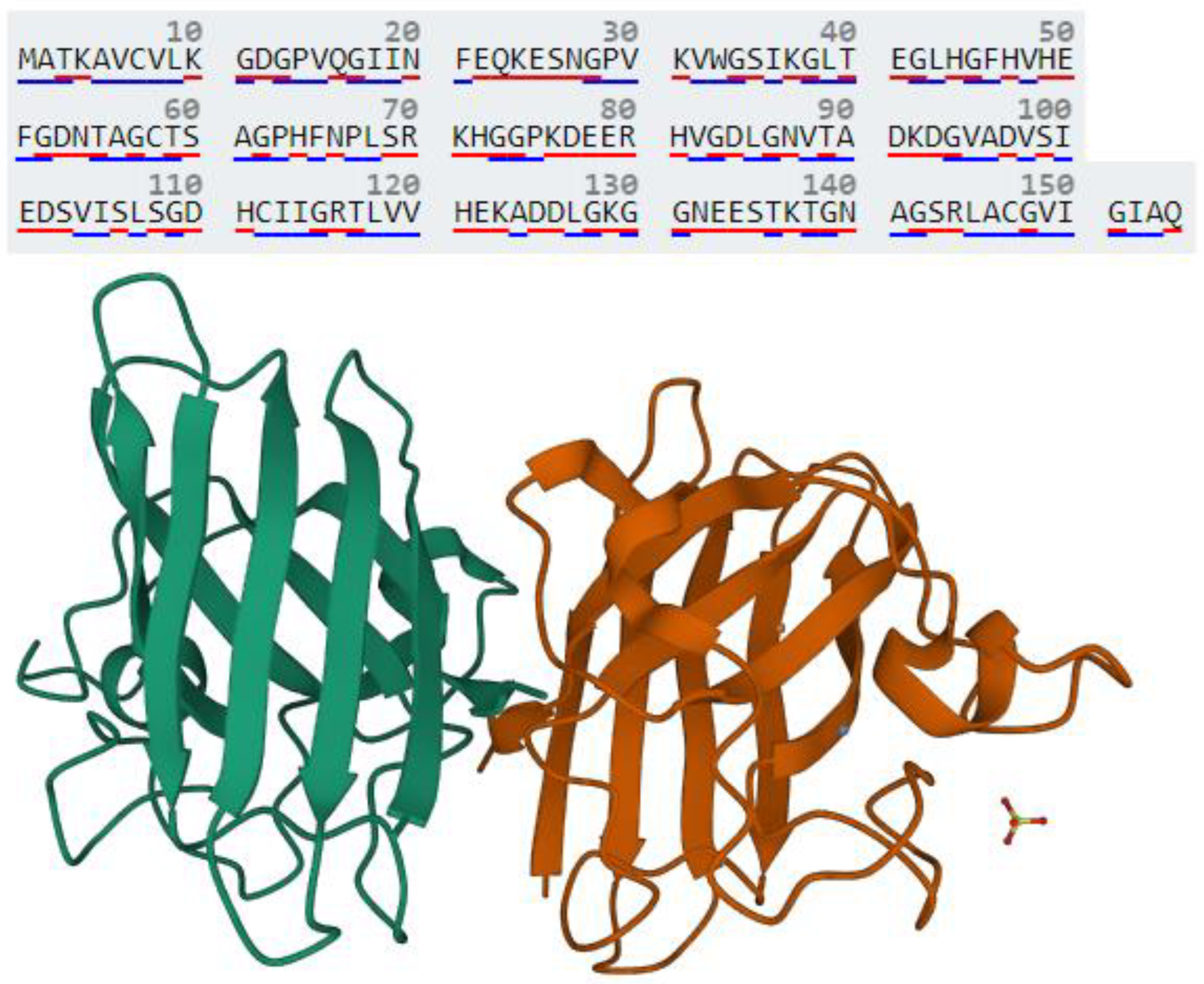
2.1. IPA Toxicity Analysis of SOD1
2.2. IPA Exploration of the Pathway/Pathway Molecules between SOD1 and ALS
2.3. Gene Ontology Analysis of SOD1-ALS Pathway Genes
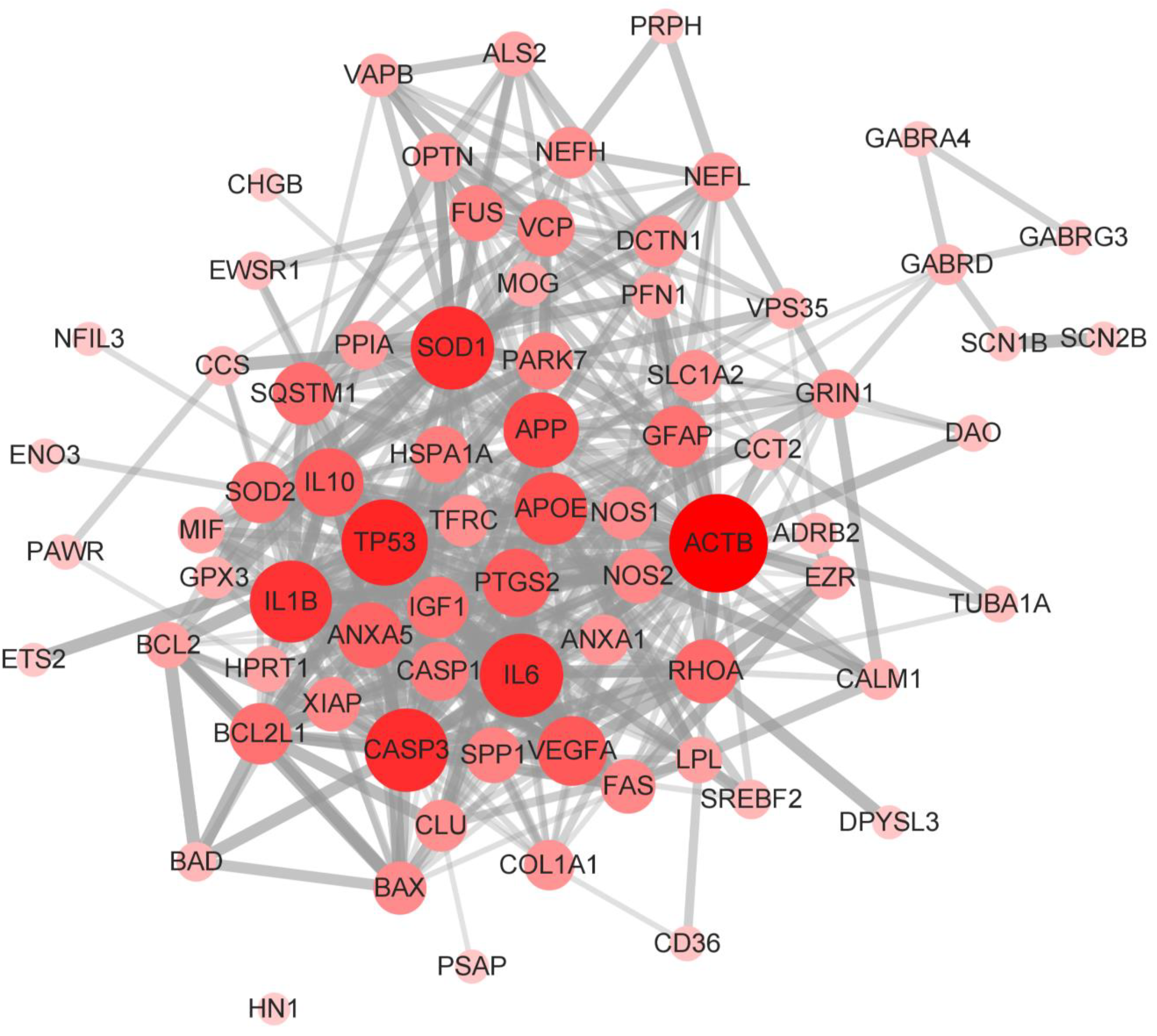
2.4. Construction of Molecular Interaction Network of SOD1-ALS Pathway Molecules
3. Results
3.1. Identification of Canonical Pathways, Regulatory Molecules, Biological Functions and Toxicity Outcome of SOD1
3.2. Identification of SOD1-ALS Pathway Molecules
3.3. GO Analysis of SOD1-ALS Pathway Genes
3.4. Construction and Analysis of Interaction Network of SOD1-ALS Pathway Molecules
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McCord, J.M.; Fridovich, I. Superoxide Dismutase. An Enzymic Function for Erythrocuprein (Hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef] [PubMed]
- Sturtz, L.A.; Diekert, K.; Jensen, L.T.; Lill, R.; Culotta, V.C. A Fraction of Yeast Cu,Zn-Superoxide Dismutase and its Metallochaperone, CCS, Localize to the Intermembrane Space of Mitochondria. A Physiological Role for SOD1 in Guarding Against Mitochondrial Oxidative Damage. J. Biol. Chem. 2001, 276, 38084–38089. [Google Scholar] [CrossRef]
- Mesecke, N.; Terziyska, N.; Kozany, C.; Baumann, F.; Neupert, W.; Hell, K.; Herrmann, J.M. A Disulfide Relay System in the Intermembrane Space of Mitochondria that Mediates Protein Import. Cell 2005, 121, 1059–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, L.S.; Furukawa, Y.; O’Halloran, T.V.; Culotta, V.C. Factors Controlling the Uptake of Yeast Copper/Zinc Superoxide Dismutase into Mitochondria. J. Biol. Chem. 2003, 278, 28052–28059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montllor-Albalate, C.; Kim, H.; Thompson, A.E.; Jonke, A.P.; Torres, M.P.; Reddi, A.R. SOD1 Integrates Oxygen Availability to Redox Regulate NADPH Production and the Thiol Redoxome. Proc. Natl. Acad. Sci. USA 2022, 119, e2023328119. [Google Scholar] [CrossRef] [PubMed]
- Tsang, C.K.; Liu, Y.; Thomas, J.; Zhang, Y.; Zheng, X.F.S. Superoxide Dismutase 1 Acts as a Nuclear Transcription Factor to Regulate Oxidative Stress Resistance. Nat. Commun. 2014, 5, 3446. [Google Scholar] [CrossRef] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn Superoxide Dismutase Gene are Associated with Familial Amyotrophic Lateral Sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Mulder, D.W.; Kurland, L.T.; Offord, K.P.; Beard, C.M. Familial Adult Motor Neuron Disease: Amyotrophic Lateral Sclerosis. Neurology 1986, 36, 511–517. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Miller, T.M.; Cleveland, D.W. Unraveling the Mechanisms Involved in Motor Neuron Degeneration in ALS. Annu. Rev. Neurosci. 2004, 27, 723–749. [Google Scholar] [CrossRef] [Green Version]
- ALSoD—Amyotrophic Lateral Sclerosis Online Database. Available online: https://alsod.ac.uk/output/gene.php/SOD1 (accessed on 10 November 2022).
- Berdyński, M.; Miszta, P.; Safranow, K.; Andersen, P.M.; Morita, M.; Filipek, S.; Żekanowski, C.; Kuźma-Kozakiewicz, M. SOD1 Mutations Associated with Amyotrophic Lateral Sclerosis Analysis of Variant Severity. Sci. Rep. 2022, 12, 103. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Shaw, B.F.; Durazo, A.; Sohn, S.H.; Doucette, P.A.; Nersissian, A.M.; Faull, K.F.; Eggers, D.K.; Tiwari, A.; Hayward, L.J.; et al. Destabilization of Apoprotein is Insufficient to Explain Cu,Zn-Superoxide Dismutase-linked ALS Pathogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 10516–10521. [Google Scholar] [CrossRef] [Green Version]
- Banci, L.; Bertini, I.; Boca, M.; Calderone, V.; Cantini, F.; Girotto, S.; Vieru, M. Structural and Dynamic Aspects Related to Oligomerization of Apo SOD1 and its Mutants. Proc. Natl. Acad. Sci. USA 2009, 106, 6980–6985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Prakash, A.; Pandey, P.; Lynn, A.M.; Hassan, M.I. TFE-induced local unfolding and fibrillation of SOD1: Bridging the experiment and simulation studies. Biochem. J. 2018, 475, 1701–1719. [Google Scholar] [CrossRef]
- Prakash, A.; Kumar, V.; Pandey, P.; Bharti, D.R.; Vishwakarma, P.; Singh, R.; Hassan, M.I.; Lynn, A.M. Solvent sensitivity of protein aggregation in Cu, Zn superoxide dismutase: A molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2018, 36, 2605–2617. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, M.; Craig, L.; Huff, M.E.; Thayer, M.M.; Cardoso, R.M.F.; Kassmann, C.J.; Lo, T.P.; Bruns, C.K.; Powers, E.T.; Kelly, J.W.; et al. ALS Mutants of Human Superoxide Dismutase Form Fibrous Aggregates via Framework Destabilization. J. Mol. Biol. 2003, 332, 601–615. [Google Scholar] [CrossRef]
- Stathopulos, P.B.; Rumfeldt, J.A.O.; Scholz, G.A.; Irani, R.A.; Frey, H.E.; Hallewell, R.A.; Lepock, J.R.; Meiering, E.M. Cu/Zn Superoxide Dismutase Mutants Associated with Amyotrophic Lateral Sclerosis Show Enhanced Formation of Aggregates In Vitro. Proc. Natl. Acad. Sci. USA 2003, 100, 7021–7026. [Google Scholar] [CrossRef] [Green Version]
- Trist, B.G.; Hilton, J.B.; Hare, D.J.; Crouch, P.J.; Double, K.L. Superoxide Dismutase 1 in Health and Disease: How a Frontline Antioxidant Becomes Neurotoxic. Angew. Chem. Int. Ed. Engl. 2021, 60, 9215–9246. [Google Scholar] [CrossRef] [PubMed]
- Peggion, C.; Scalcon, V.; Massimino, M.L.; Nies, K.; Lopreiato, R.; Rigobello, M.P.; Bertoli, A. SOD1 in ALS: Taking Sock in Pathogenic Mechanisms and the Role of Glial and Muscle Cells. Antioxidants 2022, 11, 614. [Google Scholar] [CrossRef]
- NCBI (2022). National Center for Biotechnology (NCBI). Available online: https://www.ncbi.nlm.nih.gov/gene (accessed on 30 September 2022).
- IPA (2021). Ingenuity Pathway Analysis (IPA), Qiagen. Available online: https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/ (accessed on 30 September 2022).
- Suthar, S.K.; Alam, M.M.; Lee, J.; Monga, J.; Joseph, A.; Lee, S.-Y. Bioinformatic Analyses of Canonical Pathways of TSPOAP1 and its Roles in Human Diseases. Front. Mol. Biosci. 2021, 8, 667947. [Google Scholar] [CrossRef]
- Suthar, S.K.; Lee, S.-Y. Ingenuity Pathway Analysis of α-Synuclein Predicts Potential Signaling Pathways, Network Molecules, Biological Functions, and its Role in Neurological Diseases. Front. Mol. Neurosci. 2022, 15, 1029682. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A Graphical Gene-set Enrichment Tool for Animals and Plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-Protein Interaction Networks, Integrated Over the Tree of Life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description (Term) | Gene Counts | p-Value | |
|---|---|---|---|
| BP | Response to chemical (GO:0042221) | 55 | 7.84 × 10−23 |
| Regulation of biological quality (GO:0065008) | 53 | 3.34 × 10−22 | |
| Response to organic substance (GO:0010033) | 47 | 8.62 × 10−22 | |
| Positive regulation of transport (GO:0051050) | 29 | 4.89 × 10−20 | |
| Signaling (GO:0023052) | 56 | 1.02 × 10−19 | |
| MF | Identical protein binding (GO:0042802) | 38 | 2.88 × 10−20 |
| Protein binding (GO:0005515) | 62 | 4.54 × 10−19 | |
| Protein domain specific binding (GO:0019904) | 18 | 8.00 × 10−11 | |
| Enzyme binding (GO:0019899) | 29 | 2.83 × 10−10 | |
| Signaling receptor binding (GO:0005102) | 23 | 4.65 × 10−09 | |
| CC | Vesicle (GO:0031982) | 43 | 7.79 × 10−14 |
| Extracellular space (GO:0005615) | 37 | 5.07 × 10−12 | |
| Extracellular region (GO:0005576) | 41 | 2.86 × 10−11 | |
| Cell junction (GO:0030054) | 28 | 2.63 × 10−10 | |
| Synapse (GO:0045202) | 23 | 2.30 × 10−10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suthar, S.K.; Lee, S.-Y. The Role of Superoxide Dismutase 1 in Amyotrophic Lateral Sclerosis: Identification of Signaling Pathways, Regulators, Molecular Interaction Networks, and Biological Functions through Bioinformatics. Brain Sci. 2023, 13, 151. https://doi.org/10.3390/brainsci13010151
Suthar SK, Lee S-Y. The Role of Superoxide Dismutase 1 in Amyotrophic Lateral Sclerosis: Identification of Signaling Pathways, Regulators, Molecular Interaction Networks, and Biological Functions through Bioinformatics. Brain Sciences. 2023; 13(1):151. https://doi.org/10.3390/brainsci13010151
Chicago/Turabian StyleSuthar, Sharad Kumar, and Sang-Yoon Lee. 2023. "The Role of Superoxide Dismutase 1 in Amyotrophic Lateral Sclerosis: Identification of Signaling Pathways, Regulators, Molecular Interaction Networks, and Biological Functions through Bioinformatics" Brain Sciences 13, no. 1: 151. https://doi.org/10.3390/brainsci13010151