
A Novel Necroptosis-Related Prognostic Signature of Glioblastoma Based on Transcriptomics Analysis and Single Cell Sequencing Analysis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
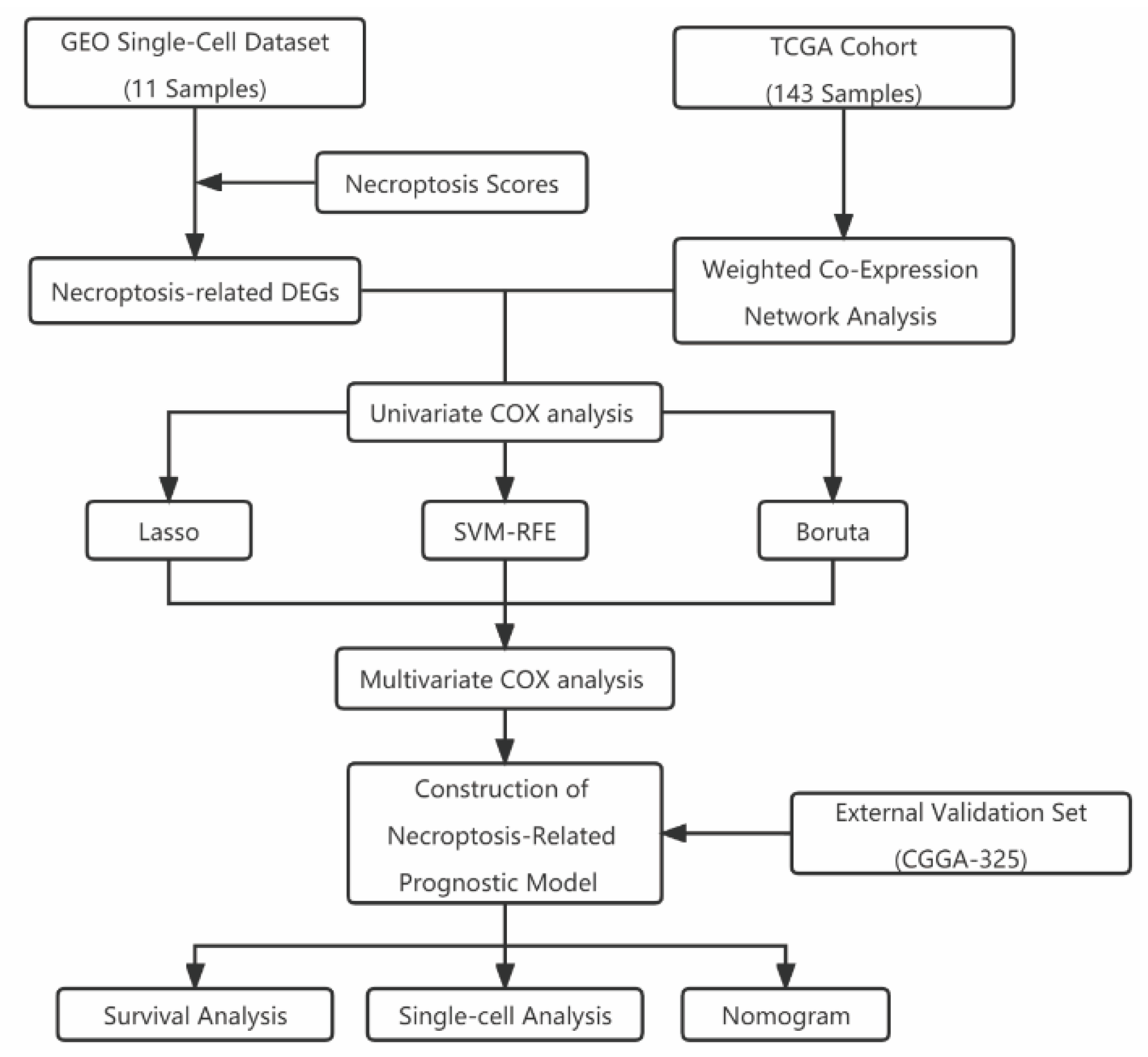
2. Materials and Methods
2.1. Transcriptome Data Download and Processing
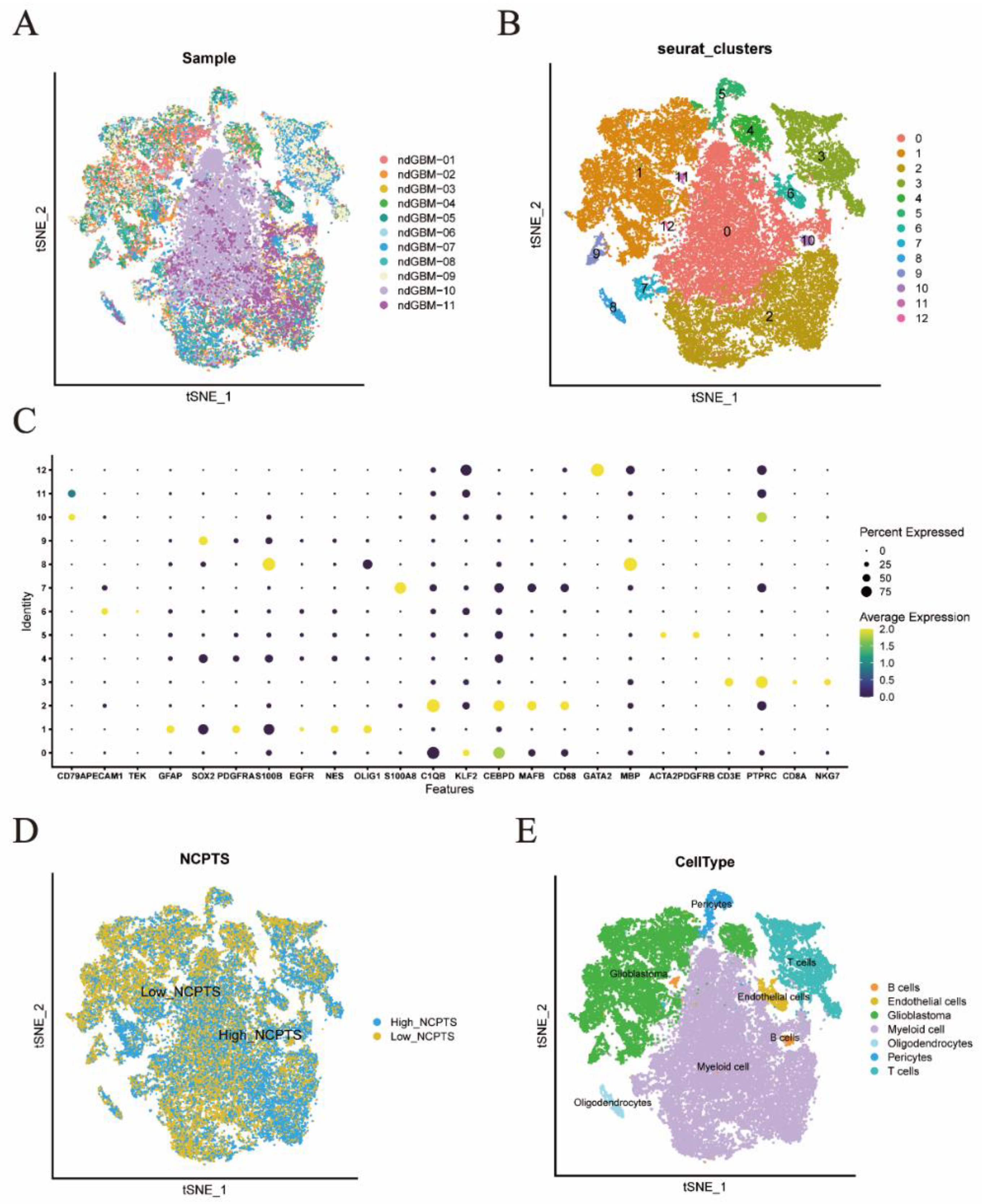
2.2. Single-Cell Data Download and Processing
2.3. The Acquisition of NCPS-Related Genes
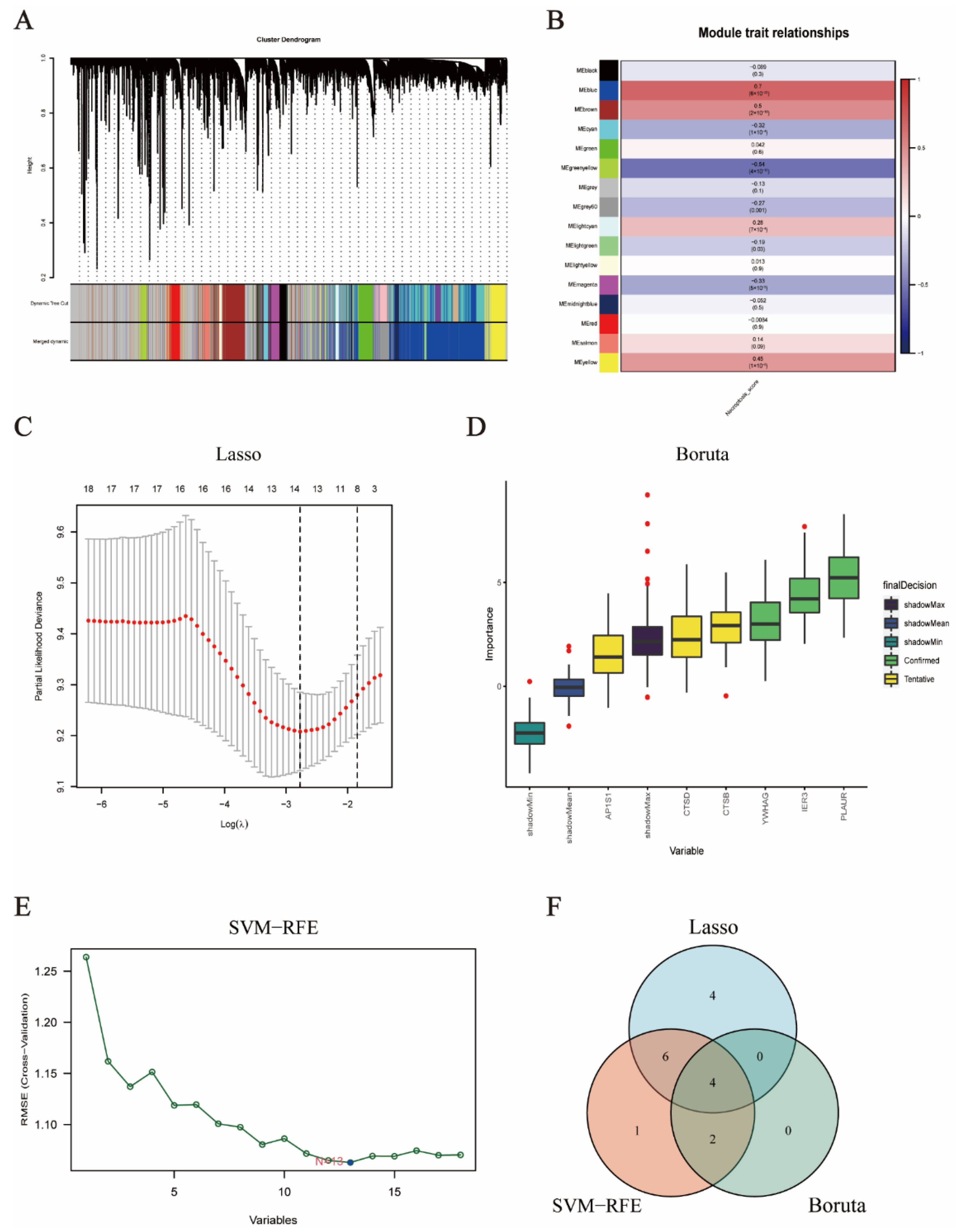
2.4. Weighted Co-Expression Network Analysis (WGCNA) and Single Sample Gene Set Enrichment Analysis (ssGSEA)
2.5. Construction of the Prognostic Model Associated with NCPS
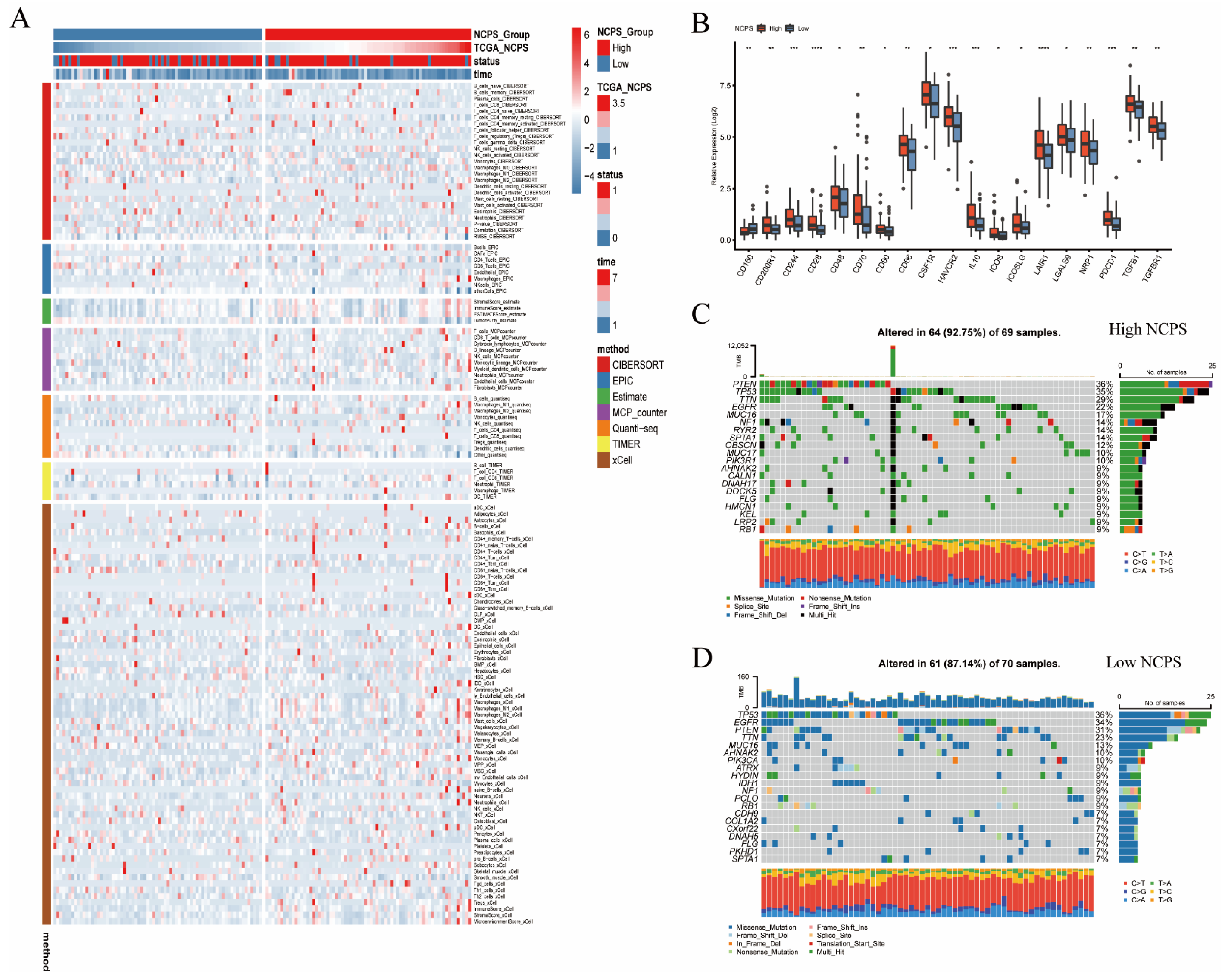
2.6. Immunological Function and Mutation Analysis
2.7. Location and Expression of Prognostic Models in Single-Cell Sequencing Analysis
2.8. Building a Predictive Nomogram
3. Results
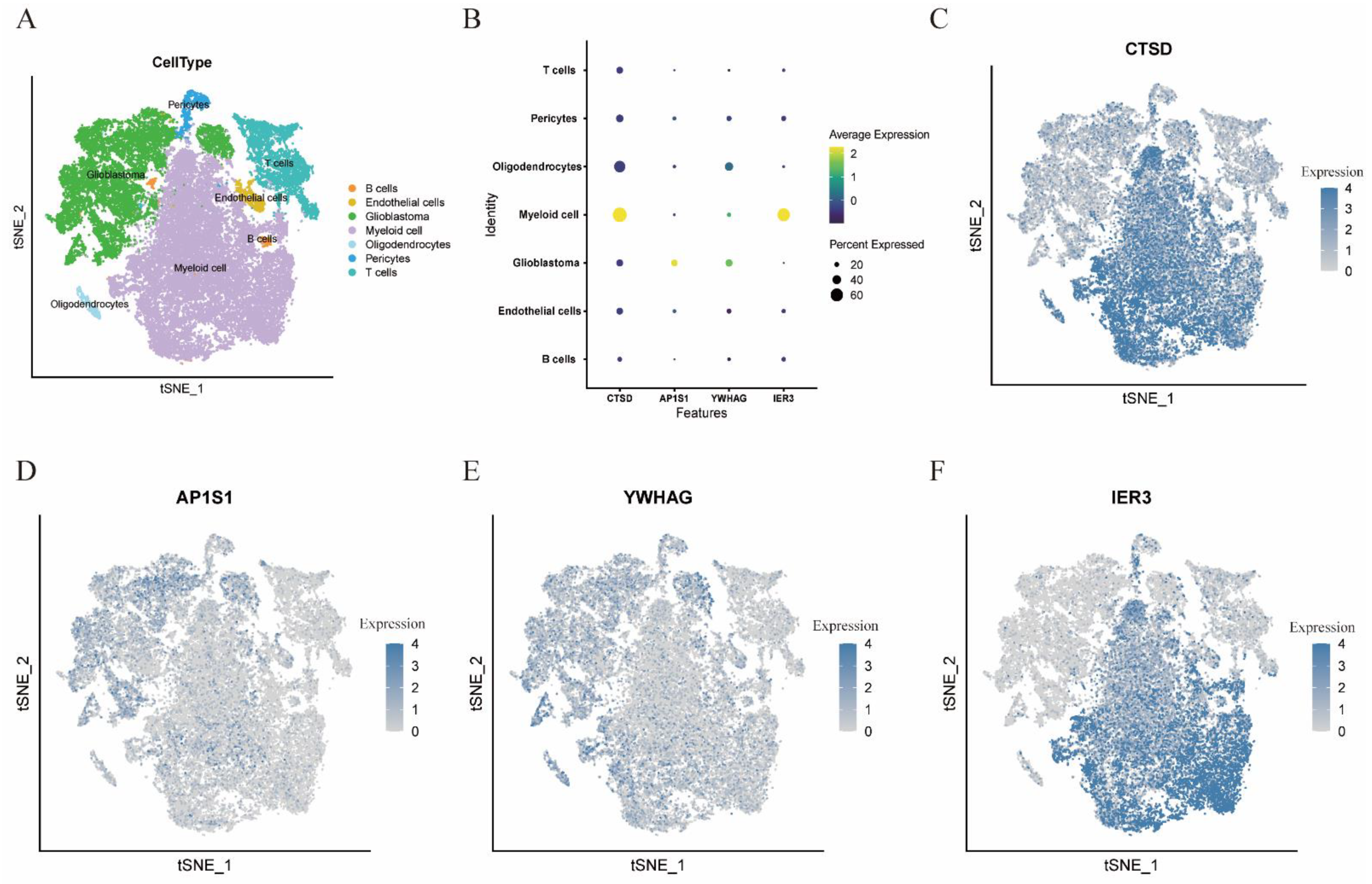
3.1. Single Cell Sequencing Data Analysis
3.2. Weighted Co-Expression Network Analysis
3.3. Selection of Prognostic Candidate Genes by Machine Learning Algorithms
3.4. Construction and Validation of NCPS-Related Prognostic Model
3.5. Immune Infiltration and Mutation Landscape
3.6. Cell Localization of Four Modeling Genes
3.7. The Construction of a Nomogram
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pagano, C.; Navarra, G.; Coppola, L.; Avilia, G.; Pastorino, O.; Della Monica, R.; Buonaiuto, M.; Torelli, G.; Caiazzo, P.; Bifulco, M.; et al. N6-isopentenyladenosine induces cell death through necroptosis in human glioblastoma cells. Cell Death Discov. 2022, 8, 173. [Google Scholar] [CrossRef] [PubMed]
- Pasqualetti, F.; Montemurro, N.; Desideri, I.; Loi, M.; Giannini, N.; Gadducci, G.; Malfatti, G.; Cantarella, M.; Gonnelli, A.; Montrone, S.; et al. Impact of recurrence pattern in patients undergoing a second surgery for recurrent glioblastoma. Acta Neurol. Belg. 2022, 122, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Suchorska, B.; Weller, M.; Tabatabai, G.; Senft, C.; Hau, P.; Sabel, M.C.; Herrlinger, U.; Ketter, R.; Schlegel, U.; Marosi, C.; et al. Complete resection of contrast-enhancing tumor volume is associated with improved survival in recurrent glioblastoma-results from the DIRECTOR trial. Neuro Oncol. 2016, 18, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Lah, T.T.; Novak, M.; Breznik, B. Brain malignancies: Glioblastoma and brain metastases. Semin. Cancer Biol. 2020, 60, 262–273. [Google Scholar] [CrossRef]
- Stupp, R.; Brada, M.; van den Bent, M.J.; Tonn, J.C.; Pentheroudakis, G. High-grade glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25 (Suppl. S3), iii93–iii101. [Google Scholar] [CrossRef]
- Akyerli, C.B.; Yüksel, Ş.; Can, Ö.; Erson-Omay, E.Z.; Oktay, Y.; Coşgun, E.; Ülgen, E.; Erdemgil, Y.; Sav, A.; von Deimling, A.; et al. Use of telomerase promoter mutations to mark specific molecular subsets with reciprocal clinical behavior in IDH mutant and IDH wild-type diffuse gliomas. J. Neurosurg. 2018, 128, 1102–1114. [Google Scholar] [CrossRef] [Green Version]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Peñaranda Fajardo, N.M.; Meijer, C.; Kruyt, F.A.E. The endoplasmic reticulum stress/unfolded protein response in gliomagenesis, tumor progression and as a therapeutic target in glioblastoma. Biochem. Pharmacol. 2016, 118, 1–8. [Google Scholar] [CrossRef]
- Zheng, P.; Ding, B.; Zhu, G.; Li, C.; Lin, J. Biodegradable Ca2+ Nanomodulators Activate Pyroptosis through Mitochondrial Ca2+ Overload for Cancer Immunotherapy. Angew. Chem. Int. Ed. Engl. 2022. [Google Scholar] [CrossRef]
- de Vasconcelos, N.M.; Van Opdenbosch, N.; Van Gorp, H.; Parthoens, E.; Lamkanfi, M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. 2019, 26, 146–161. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Kroemer, G. Necroptosis: A specialized pathway of programmed necrosis. Cell 2008, 135, 1161–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol. Cancer 2015, 14, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krysko, O.; Aaes, T.L.; Kagan, V.E.; D’Herde, K.; Bachert, C.; Leybaert, L.; Vandenabeele, P.; Krysko, D.V. Necroptotic cell death in anti-cancer therapy. Immunol. Rev. 2017, 280, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhang, L.; Yu, H.; Song, K.; Shi, J.; Chen, L.; Cheng, J. Necrostatin-1 Improves Long-term Functional Recovery Through Protecting Oligodendrocyte Precursor Cells After Transient Focal Cerebral Ischemia in Mice. Neuroscience 2018, 371, 229–241. [Google Scholar] [CrossRef]
- Liu, Z.-M.; Chen, Q.-X.; Chen, Z.-B.; Tian, D.-F.; Li, M.-C.; Wang, J.-M.; Wang, L.; Liu, B.-H.; Zhang, S.-Q.; Li, F.; et al. RIP3 deficiency protects against traumatic brain injury (TBI) through suppressing oxidative stress, inflammation and apoptosis: Dependent on AMPK pathway. Biochem. Biophys. Res. Commun. 2018, 499, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Branca, C.; Piras, I.S.; Ferreira, E.; Huentelman, M.J.; Liang, W.S.; Readhead, B.; Dudley, J.T.; Spangenberg, E.E.; Green, K.N.; et al. Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1236–1246. [Google Scholar] [CrossRef]
- Picelli, S.; Björklund, Å.K.; Faridani, O.R.; Sagasser, S.; Winberg, G.; Sandberg, R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 2013, 10, 1096–1098. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef]
- Lee, J.-K.; Wang, J.; Sa, J.K.; Ladewig, E.; Lee, H.-O.; Lee, I.-H.; Kang, H.J.; Rosenbloom, D.S.; Camara, P.G.; Liu, Z.; et al. Spatiotemporal genomic architecture informs precision oncology in glioblastoma. Nat. Genet. 2017, 49, 594–599. [Google Scholar] [CrossRef]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degenhardt, F.; Seifert, S.; Szymczak, S. Evaluation of variable selection methods for random forests and omics data sets. Brief. Bioinform. 2019, 20, 492–503. [Google Scholar] [CrossRef] [Green Version]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Ng, K.L.S.; Mishra, S.K. De novo SVM classification of precursor microRNAs from genomic pseudo hairpins using global and intrinsic folding measures. Bioinformatics 2007, 23, 1321–1330. [Google Scholar] [CrossRef] [Green Version]
- Rice, S.B.; Nenadic, G.; Stapley, B.J. Mining protein function from text using term-based support vector machines. BMC Bioinform. 2005, 6 (Suppl. S1), S22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habermann, J.K.; Doering, J.; Hautaniemi, S.; Roblick, U.J.; Bündgen, N.K.; Nicorici, D.; Kronenwett, U.; Rathnagiriswaran, S.; Mettu, R.K.R.; Ma, Y.; et al. The gene expression signature of genomic instability in breast cancer is an independent predictor of clinical outcome. Int. J. Cancer 2009, 124, 1552–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, S.; Floegel, A.; Weikert, C.; Prehn, C.; Adamski, J.; Pischon, T.; Boeing, H.; Drogan, D. Identification of Serum Metabolites Associated With Incident Hypertension in the European Prospective Investigation into Cancer and Nutrition-Potsdam Study. Hypertension 2016, 68, 471–477. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Kursa, M.B.; Rudnicki, W.R. Feature selection with the Boruta package. J. Stat. Softw. 2010, 36, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Meng, F.; Wang, W.; Wang, Z.; Zhang, C.; Jiang, T. Comprehensive RNA-seq transcriptomic profiling in the malignant progression of gliomas. Sci. Data 2017, 4, 170024. [Google Scholar] [CrossRef]
- Abdelfattah, N.; Kumar, P.; Wang, C.; Leu, J.-S.; Flynn, W.F.; Gao, R.; Baskin, D.S.; Pichumani, K.; Ijare, O.B.; Wood, S.L.; et al. Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nat. Commun. 2022, 13, 767. [Google Scholar] [CrossRef] [PubMed]
- Guyon, I.; Weston, J.; Barnhill, S.; Vapnik, V. Gene selection for cancer classification using support vector machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Kursa, M.B. Robustness of Random Forest-based gene selection methods. BMC Bioinform. 2014, 15, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, D.; Ye, Z.; Shen, R.; Yu, G.; Wu, J.; Xiong, Y.; Zhou, R.; Qiu, W.; Huang, N.; Sun, L.; et al. IOBR: Multi-Omics Immuno-Oncology Biological Research to Decode Tumor Microenvironment and Signatures. Front. Immunol. 2021, 12, 687975. [Google Scholar] [CrossRef]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.-M.; Guz-Montgomery, K.; Lowe, D.B.; Saha, D. Pathogenetic Features and Current Management of Glioblastoma. Cancers 2021, 13, 856. [Google Scholar] [CrossRef]
- Jackson, C.M.; Choi, J.; Lim, M. Mechanisms of immunotherapy resistance: Lessons from glioblastoma. Nat. Immunol. 2019, 20, 1100–1109. [Google Scholar] [CrossRef]
- Christofferson, D.E.; Yuan, J. Necroptosis as an alternative form of programmed cell death. Curr. Opin. Cell Biol. 2010, 22, 263–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef]
- Zhang, D.-W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.-J.; Lin, S.-C.; Dong, M.-Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef]
- Declercq, W.; Vanden Berghe, T.; Vandenabeele, P. RIP kinases at the crossroads of cell death and survival. Cell 2009, 138, 229–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozec, D.; Iuga, A.C.; Roda, G.; Dahan, S.; Yeretssian, G. Critical function of the necroptosis adaptor RIPK3 in protecting from intestinal tumorigenesis. Oncotarget 2016, 7, 46384–46400. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Song, Q.; Yu, A.; Tang, H.; Peng, Z.; Wang, X. Receptor-interacting protein kinase 3 is a predictor of survival and plays a tumor suppressive role in colorectal cancer. Neoplasma 2015, 62, 592–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strilic, B.; Yang, L.; Albarrán-Juárez, J.; Wachsmuth, L.; Han, K.; Müller, U.C.; Pasparakis, M.; Offermanns, S. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 2016, 536, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhou, M.; Mei, L.; Ruan, J.; Hu, Q.; Peng, J.; Su, H.; Liao, H.; Liu, S.; Liu, W.; et al. Key roles of necroptotic factors in promoting tumor growth. Oncotarget 2016, 7, 22219–22233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, L.; Werba, G.; Tiwari, S.; Giao Ly, N.N.; Alothman, S.; Alqunaibit, D.; Avanzi, A.; Barilla, R.; Daley, D.; Greco, S.H.; et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 2016, 532, 245–249. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Hatanpaa, K.J.; Xie, Y.; Mickey, B.E.; Madden, C.J.; Raisanen, J.M.; Ramnarain, D.B.; Xiao, G.; Saha, D.; Boothman, D.A.; et al. The receptor interacting protein 1 inhibits p53 induction through NF-kappaB activation and confers a worse prognosis in glioblastoma. Cancer Res. 2009, 69, 2809–2816. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Xu, J.; Huang, N.; Tang, J.; Ma, P.; Cheng, Y. Clinical and Biological Significance of a Necroptosis-Related Gene Signature in Glioma. Front. Oncol. 2022, 12, 855434. [Google Scholar] [CrossRef]
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic immunity in cancer. Nat. Rev. Cancer 2021, 21, 345–359. [Google Scholar] [CrossRef]
- Wang, D.-P.; Kang, K.; Lin, Q.; Hai, J. Prognostic Significance of Preoperative Systemic Cellular Inflammatory Markers in Gliomas: A Systematic Review and Meta-Analysis. Clin. Transl. Sci. 2020, 13, 179–188. [Google Scholar] [CrossRef]
- Han, S.; Liu, Y.; Li, Q.; Li, Z.; Hou, H.; Wu, A. Pre-treatment neutrophil-to-lymphocyte ratio is associated with neutrophil and T-cell infiltration and predicts clinical outcome in patients with glioblastoma. BMC Cancer 2015, 15, 617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topkan, E.; Besen, A.A.; Ozdemir, Y.; Kucuk, A.; Mertsoylu, H.; Pehlivan, B.; Selek, U. Prognostic Value of Pretreatment Systemic Immune-Inflammation Index in Glioblastoma Multiforme Patients Undergoing Postneurosurgical Radiotherapy Plus Concurrent and Adjuvant Temozolomide. Mediat. Inflamm 2020, 2020, 4392189. [Google Scholar] [CrossRef]
- Pasqualetti, F.; Giampietro, C.; Montemurro, N.; Giannini, N.; Gadducci, G.; Orlandi, P.; Natali, E.; Chiarugi, P.; Gonnelli, A.; Cantarella, M.; et al. Old and New Systemic Immune-Inflammation Indexes Are Associated with Overall Survival of Glioblastoma Patients Treated with Radio-Chemotherapy. Genes 2022, 13, 1054. [Google Scholar] [CrossRef] [PubMed]
- Yılmaz, H.; Niğdelioğlu, B.; Oktay, E.; Meydan, N. Clinical significance of postoperatif controlling nutritional status (CONUT) score in glioblastoma multiforme. J. Clin. Neurosci. 2021, 86, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Liu, S.; Zeng, S.; Shen, H. From bench to bed: The tumor immune microenvironment and current immunotherapeutic strategies for hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 396. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Luo, Y.-B.; Wu, W.; Zhang, L.; Wang, Z.; Dai, Z.; Feng, S.; Cao, H.; Cheng, Q.; Liu, Z. The molecular feature of macrophages in tumor immune microenvironment of glioma patients. Comput. Struct. Biotechnol. J. 2021, 19, 4603–4618. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, M.; Song, S. Cathepsin D enhances breast cancer invasion and metastasis through promoting hepsin ubiquitin-proteasome degradation. Cancer Lett. 2018, 438, 105–115. [Google Scholar] [CrossRef]
- Zheng, W.; Chen, Q.; Wang, C.; Yao, D.; Zhu, L.; Pan, Y.; Zhang, J.; Bai, Y.; Shao, C. Inhibition of Cathepsin D (CTSD) enhances radiosensitivity of glioblastoma cells by attenuating autophagy. Mol. Carcinog. 2020, 59, 651–660. [Google Scholar] [CrossRef]
- Zheng, D.; Fu, W.; Jin, L.; Jiang, X.; Jiang, W.; Guan, Y.; Hao, R. The Overexpression and Clinical Significance of AP1S1 in Breast Cancer. Cancer Manag. Res. 2022, 14, 1475–1492. [Google Scholar] [CrossRef]
- Alshabi, A.M.; Vastrad, B.; Shaikh, I.A.; Vastrad, C. Identification of Crucial Candidate Genes and Pathways in Glioblastoma Multiform by Bioinformatics Analysis. Biomolecules 2019, 9, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Z.; Peng, H.; White, D.E.; Wang, P.; Lieberman, P.M.; Halazonetis, T.; Rauscher, F.J. 14-3-3 binding sites in the snail protein are essential for snail-mediated transcriptional repression and epithelial-mesenchymal differentiation. Cancer Res. 2010, 70, 4385–4393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, A.K.; Morrison, D.K. 14-3-3 Proteins: Diverse functions in cell proliferation and cancer progression. Semin. Cell Dev. Biol. 2011, 22, 681–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aitken, A. 14-3-3 proteins: A historic overview. Semin. Cancer Biol. 2006, 16, 162–172. [Google Scholar] [CrossRef]
- Com, E.; Clavreul, A.; Lagarrigue, M.; Michalak, S.; Menei, P.; Pineau, C. Quantitative proteomic Isotope-Coded Protein Label (ICPL) analysis reveals alteration of several functional processes in the glioblastoma. J. Proteom. 2012, 75, 3898–3913. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wang, J.; Fu, Y.; Yan, C.; Zhu, M.; Jiang, Y.; Chen, J.; Ding, Y.; Fan, X.; Li, G.; et al. Functional genetic variants in centrosome-related genes CEP72 and YWHAG confer susceptibility to gastric cancer. Arch. Toxicol. 2020, 94, 2861–2872. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Schäfer, H. Role of the immediate early response 3 (IER3) gene in cellular stress response, inflammation and tumorigenesis. Eur. J. Cell Biol. 2011, 90, 545–552. [Google Scholar] [CrossRef]
- Yang, C.; Trent, S.; Ionescu-Tiba, V.; Lan, L.; Shioda, T.; Sgroi, D.; Schmidt, E.V. Identification of cyclin D1- and estrogen-regulated genes contributing to breast carcinogenesis and progression. Cancer Res. 2006, 66, 11649–11658. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Geng, L.; Liu, X.; Shi, H.; He, W.; Wu, M.X. Clinical significance of IEX-1 expression in ovarian carcinoma. Ultrastruct. Pathol. 2011, 35, 260–266. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Huang, Y.; Zhou, C.; Wang, H.; Wang, Z.; Wu, J.; Nie, S.; Deng, X.; Sun, J.; Gao, X. A Novel Necroptosis-Related Prognostic Signature of Glioblastoma Based on Transcriptomics Analysis and Single Cell Sequencing Analysis. Brain Sci. 2022, 12, 988. https://doi.org/10.3390/brainsci12080988
Wu Y, Huang Y, Zhou C, Wang H, Wang Z, Wu J, Nie S, Deng X, Sun J, Gao X. A Novel Necroptosis-Related Prognostic Signature of Glioblastoma Based on Transcriptomics Analysis and Single Cell Sequencing Analysis. Brain Sciences. 2022; 12(8):988. https://doi.org/10.3390/brainsci12080988
Chicago/Turabian StyleWu, Yiwen, Yi Huang, Chenhui Zhou, Haifeng Wang, Zhepei Wang, Jiawei Wu, Sheng Nie, Xinpeng Deng, Jie Sun, and Xiang Gao. 2022. "A Novel Necroptosis-Related Prognostic Signature of Glioblastoma Based on Transcriptomics Analysis and Single Cell Sequencing Analysis" Brain Sciences 12, no. 8: 988. https://doi.org/10.3390/brainsci12080988