
TNFα-Induced Oxidative Stress and Mitochondrial Dysfunction Alter Hypothalamic Neurogenesis and Promote Appetite Versus Satiety Neuropeptide Expression in Mice

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. NPC Cultures
2.3. NPC Mitochondrial Function
2.4. In Vitro Treatment with TNFα and H2O2
2.5. In Vitro Treatment with Inhibitors
2.6. ROS Assay
2.7. Proliferation Assay
2.8. Western Blot
2.9. Data Analysis
3. Results
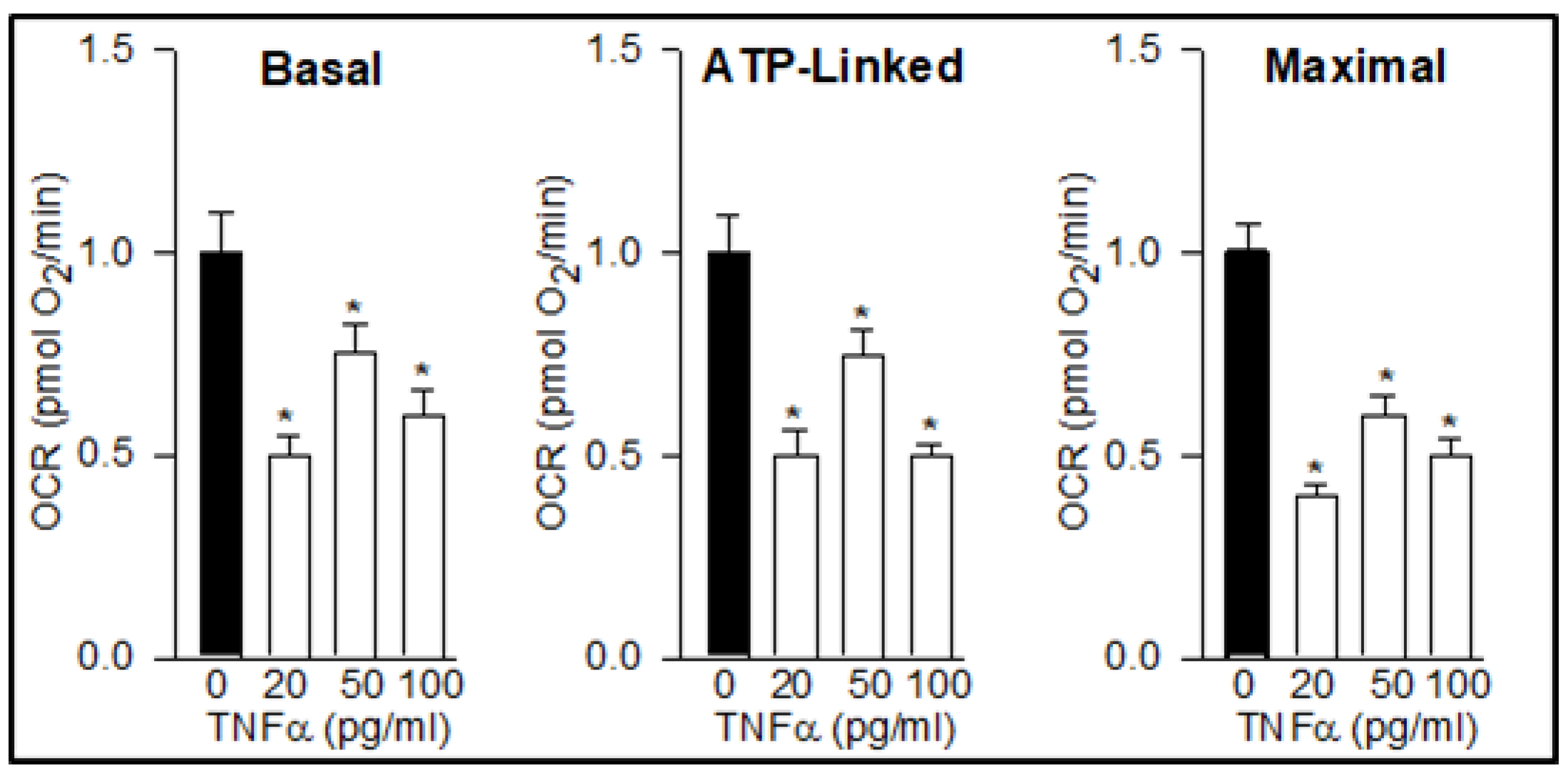
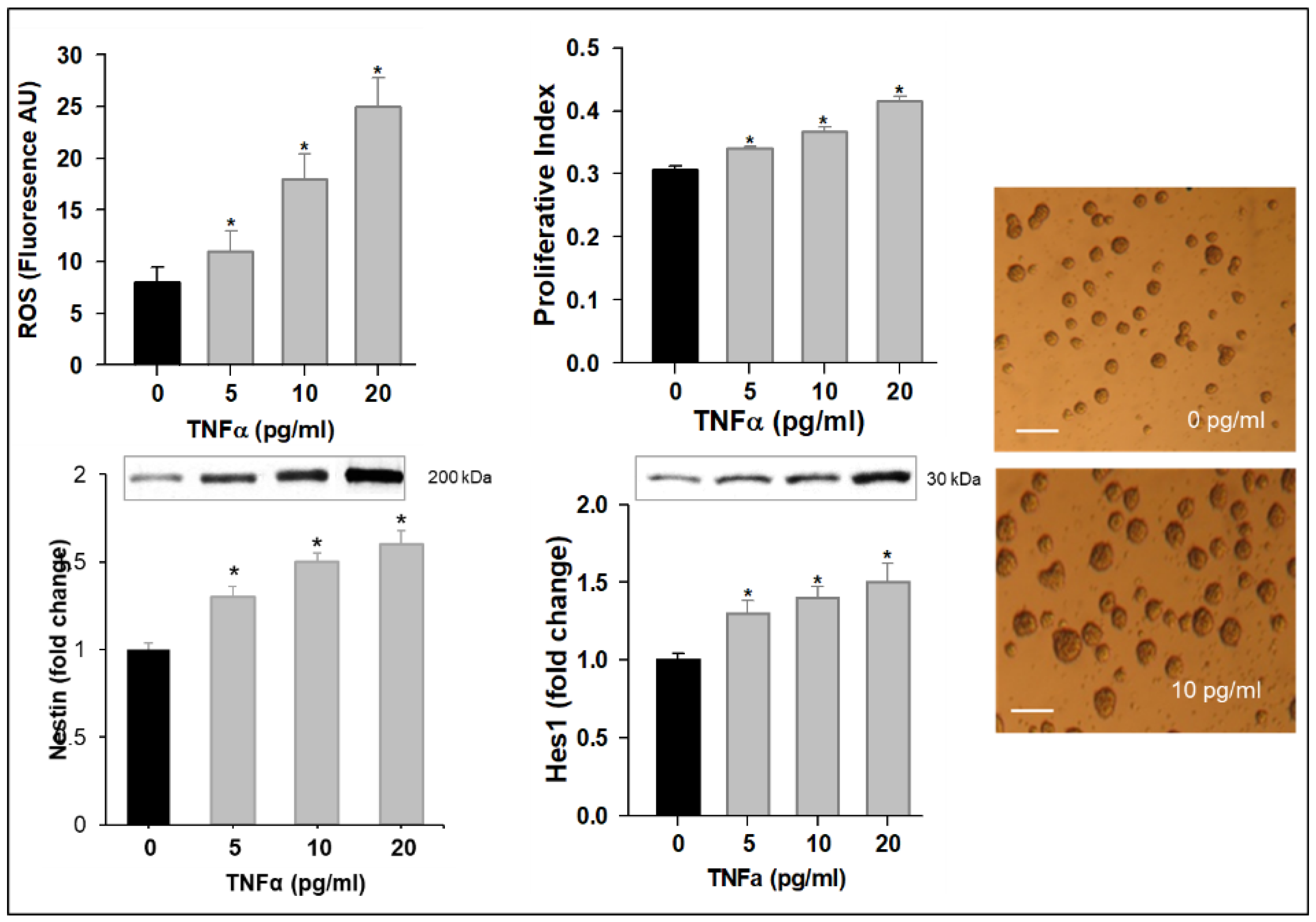
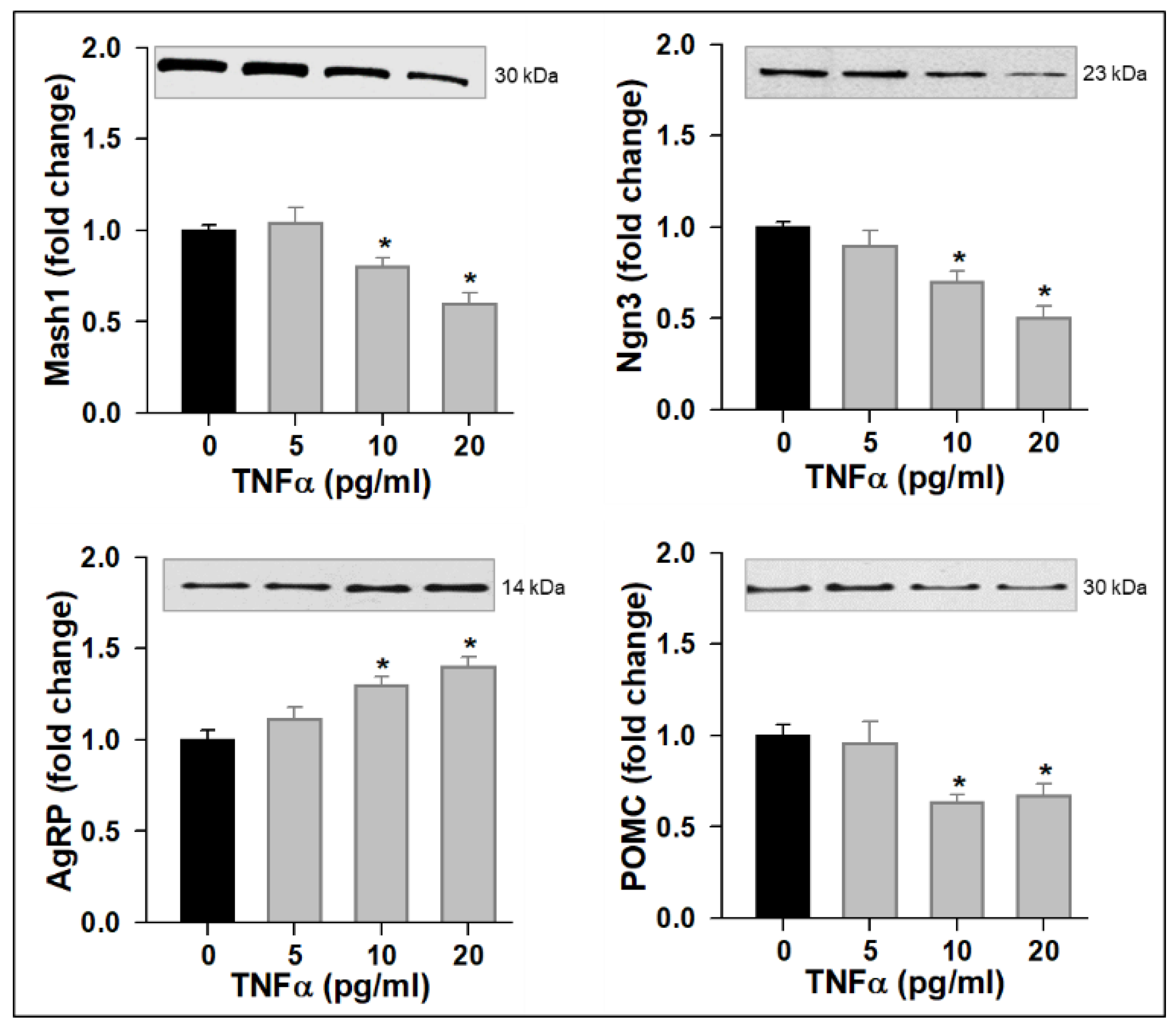
3.1. TNFα Impairs NPC Mitochondrial Function, Promotes ROS Production, and Alters Neuropeptide Expression
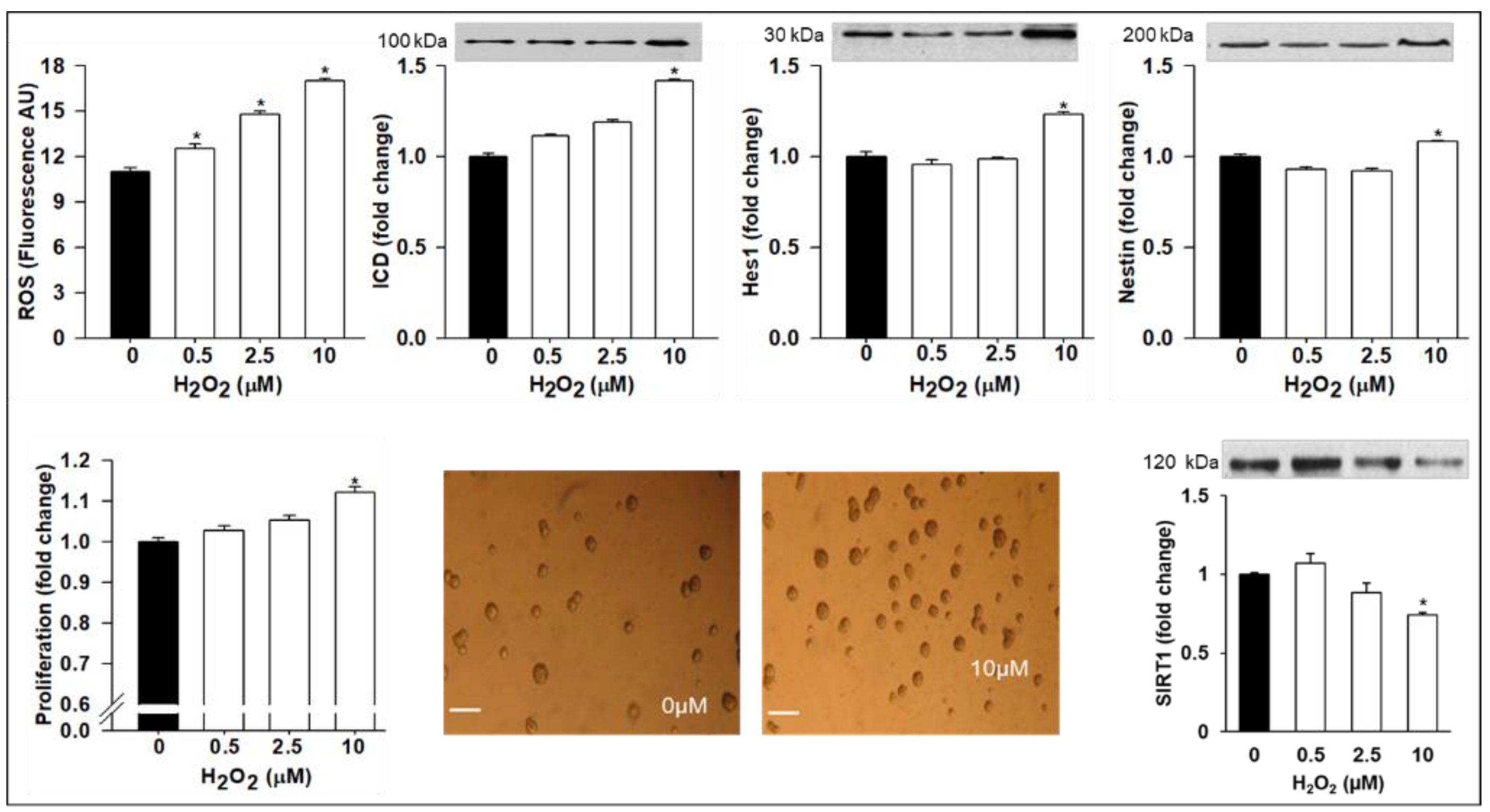
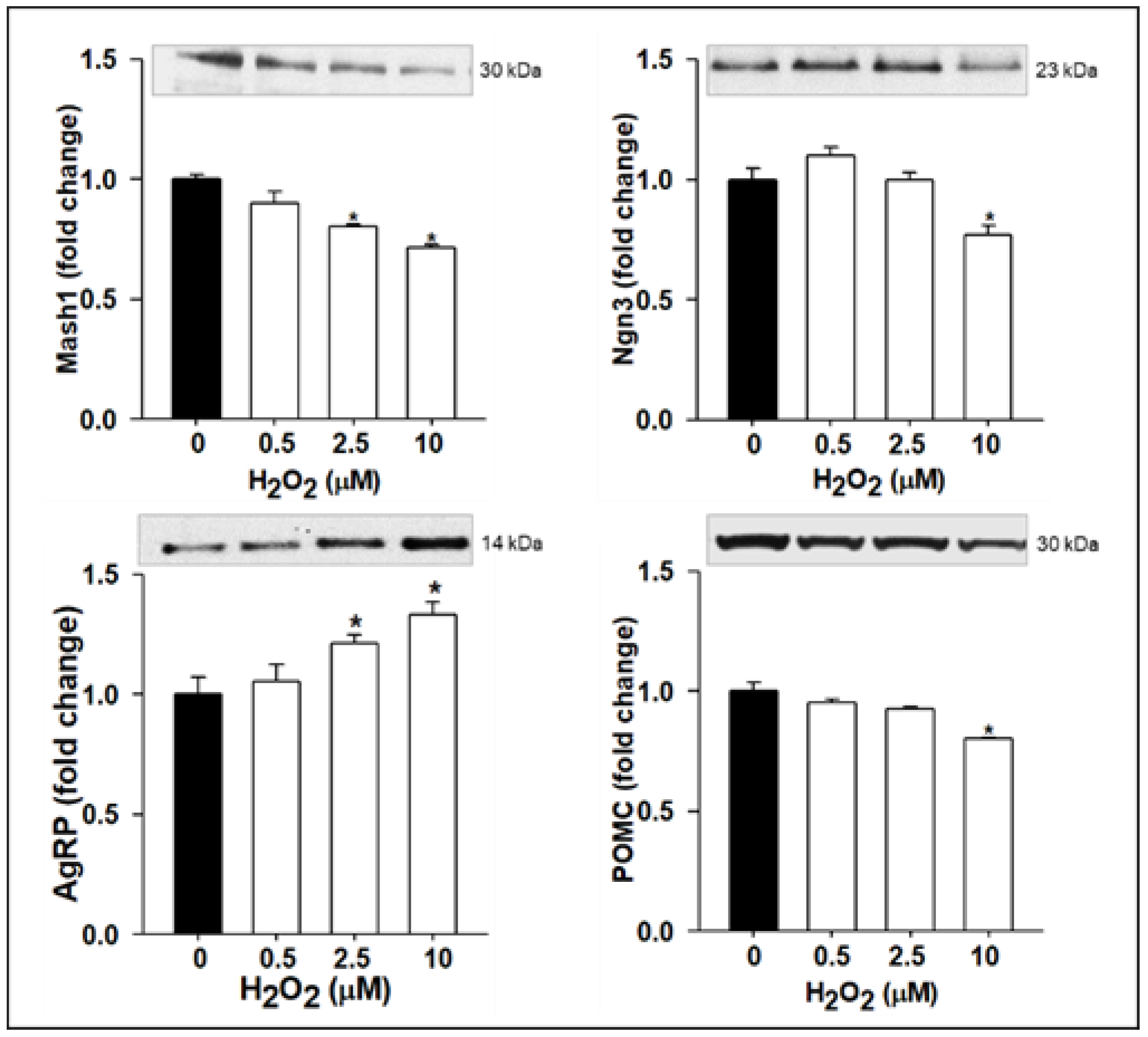
3.2. ROS Suppresses Antioxidant SIRT1 and Alters NPC Proliferation/Differentiation and Neuropeptide Expression
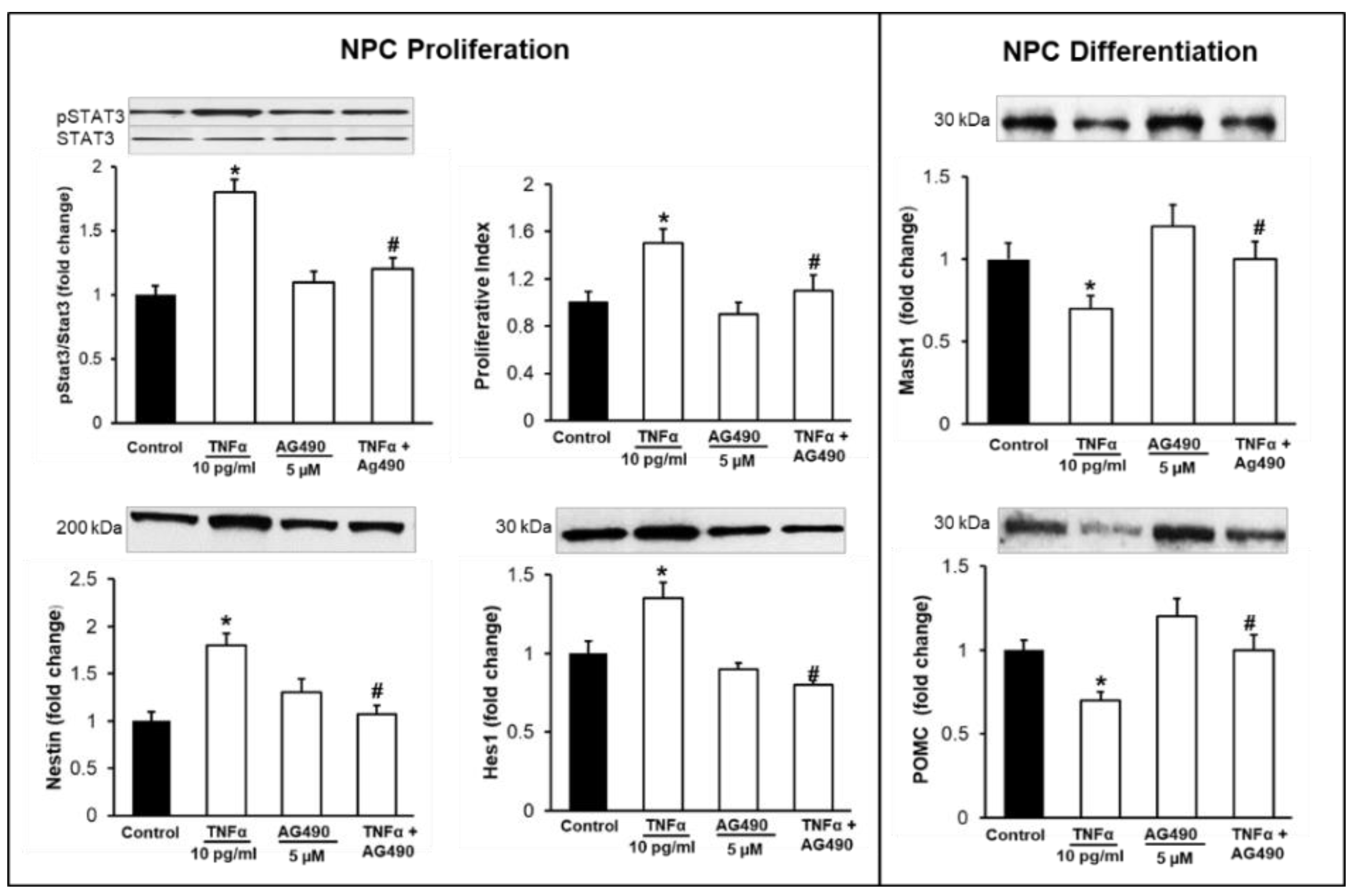
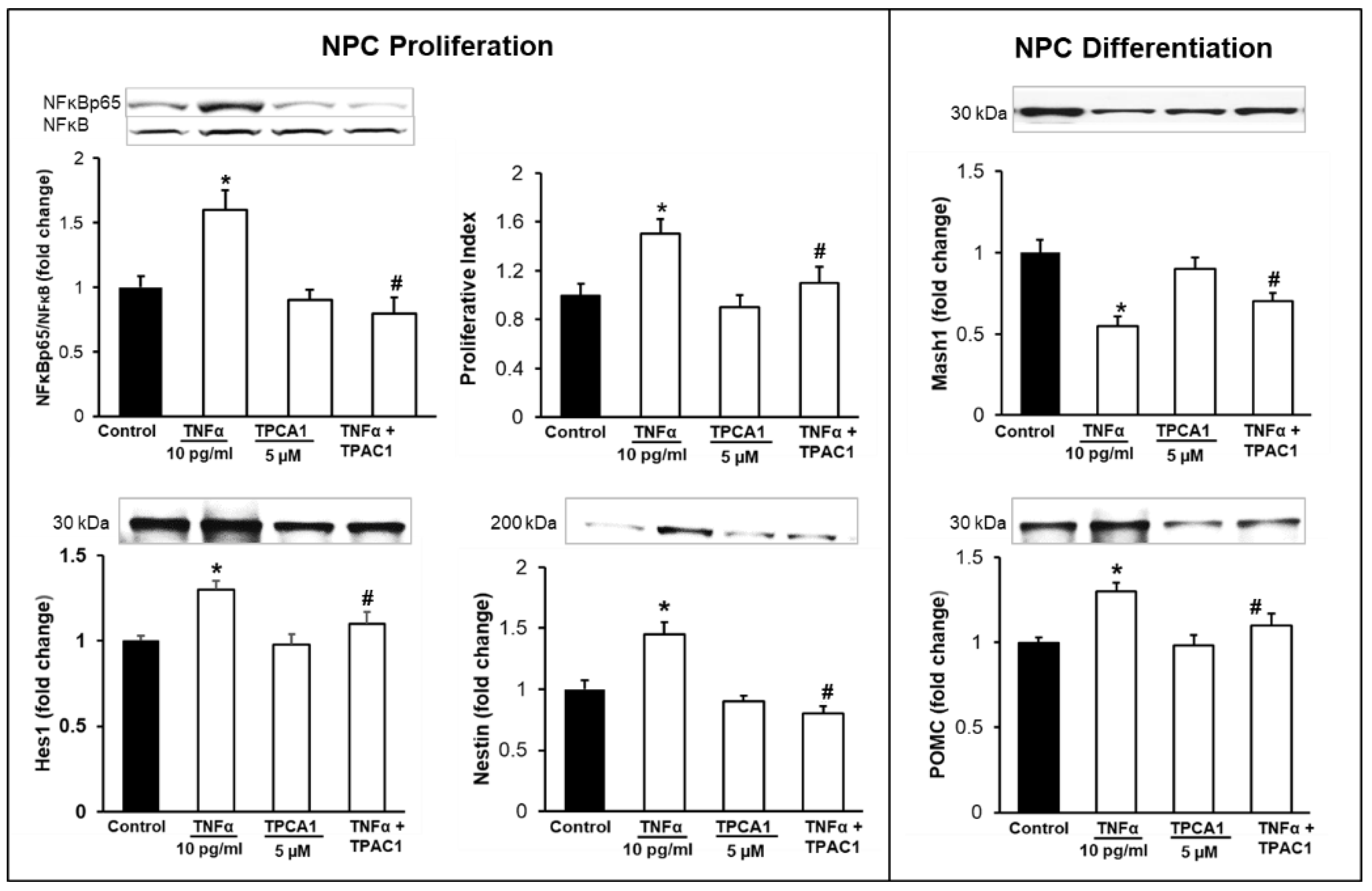
3.3. Inhibition of TNFα-Mediated STAT3/NFκB Signaling Normalizes NPC Proliferation/Differentiation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Satpathy, H.K.; Fleming, A.; Frey, D.; Barsoom, M.; Satpathy, C.; Khandalavala, J. Maternal obesity and pregnancy. Postgrad. Med. 2008, 120, E01–E09. [Google Scholar] [CrossRef] [PubMed]
- Catalano, P.; Demouzon, S.H. Maternal obesity and metabolic risk to the offspring: Why lifestyle interventions may have not achieved the desired outcomes. Int. J. Obes. 2015, 39, 642–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oken, E.; Rifas-Shiman, S.L.; Field, A.E.; Frazier, A.L.; Gillman, M.W. Maternal gestational weight gain and offspring weight in adolescence. Obstet. Gynecol. 2008, 112, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Hermann, G.M.; Dallas, L.M.; Haskell, S.E.; Roghair, R.D. Neonatal macrosomia is an independent risk factor for adult metabolic syndrome. Neonatology 2010, 98, 238–244. [Google Scholar] [CrossRef] [Green Version]
- Desai, M.; Jellyman, J.K.; Han, G.; Beall, M.; Lane, R.H.; Ross, M.G. Maternal obesity and high-fat diet program offspring metabolic syndrome. Am. J. Obstet. Gynecol. 2014, 211, 237.e1–237.e13. [Google Scholar] [CrossRef] [Green Version]
- Desai, M.; Ferrini, M.G.; Han, G.; Narwani, K.; Ross, M.G. Maternal High Fat Diet Programs Male Mice Offspring Hyperphagia and Obesity: Mechanism of Increased Appetite Neurons via Altered Neurogenic Factors and Nutrient Sensor AMPK. Nutrients 2020, 12, 3326. [Google Scholar] [CrossRef]
- Lemes, S.F.; de Souza, A.C.P.; Payolla, T.B.; Versutti, M.D.; de Fatima da Silva Ramalho, A.; Mendes-da-Silva, C.; Souza, C.M.; Milanski, M.; Torsoni, A.S.; Torsoni, M.A. Maternal Consumption of High-fat Diet in Mice Alters Hypothalamic Notch Pathway, NPY Cell Population and Food Intake in Offspring. Neuroscience 2018, 371, 1–15. [Google Scholar] [CrossRef]
- Desai, M.; Li, T.; Ross, M.G. Hypothalamic neurosphere progenitor cells in low birth-weight rat newborns: Neurotrophic effects of leptin and insulin. Brain Res. 2011, 1378, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Miller, F.D.; Gauthier, A.S. Timing is everything: Making neurons versus glia in the developing cortex. Neuron 2007, 54, 357–369. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, I.; Johansen, J.E.; Schalling, M.; Hökfelt, T.; Fetissov, S.O. Maturation of the hypothalamic arcuate agouti-related protein system during postnatal development in the mouse. Brain Res. Dev. Brain Res. 2005, 155, 147–154. [Google Scholar] [CrossRef]
- Padilla, S.L.; Carmody, J.S.; Zeltser, L.M. Pomc-expressing progenitors give rise to antagonistic neuronal populations in hypothalamic feeding circuits. Nat. Med. 2010, 16, 403–405. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, R.; Ohtsuka, T.; Kobayashi, T. Roles of Hes genes in neural development. Dev. Growth Differ. 2008, 50 (Suppl. 1), S97–S103. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.; Han, G.; Ross, M.G. Programmed hyperphagia in offspring of obese dams: Altered expression of hypothalamic nutrient sensors, neurogenic factors and epigenetic modulators. Appetite 2016, 99, 193–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, M.J.; Chen, H. Established maternal obesity in the rat reprograms hypothalamic appetite regulators and leptin signaling at birth. Int. J. Obes. 2009, 33, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stofkova, A.; Skurlova, M.; Kiss, A.; Zelezna, B.; Zorad, S.; Jurcovicova, J. Activation of hypothalamic NPY, AgRP, MC4R, AND IL-6 mRNA levels in young Lewis rats with early-life diet-induced obesity. Endocr. Regul. 2009, 43, 99–106. [Google Scholar]
- Anthwal, N.; Pelling, M.; Claxton, S.; Mellitzer, G.; Collin, C.; Kessaris, N.; Richardson, W.D.; Gradwohl, G.; Ang, S.-L. Conditional deletion of neurogenin-3 using Nkx2.1iCre results in a mouse model for the central control of feeding, activity and obesity. Dis. Model. Mech. 2013, 6, 1133–1145. [Google Scholar]
- MacKay, H.; Abizaid, A. Embryonic development of the hypothalamic feeding circuitry: Transcriptional, nutritional, and hormonal influences. Mol. Metab. 2014, 3, 813–822. [Google Scholar] [CrossRef]
- McNay, D.E.; Pelling, M.; Claxton, S.; Guillemot, F.; Ang, S.L. Mash1 is required for generic and subtype differentiation of hypothalamic neuroendocrine cells. Mol. Endocrinol. 2006, 20, 1623–1632. [Google Scholar] [CrossRef] [Green Version]
- Pelling, M.; Anthwal, N.; McNay, D.; Gradwohl, G.; Leiter, A.B.; Guillemot, F.; Ang, S.L. Differential requirements for neurogenin 3 in the development of POMC and NPY neurons in the hypothalamus. Dev. Biol. 2011, 349, 406–416. [Google Scholar] [CrossRef] [Green Version]
- Xavier, J.M.; Rodrigues, C.M.; Solá, S. Mitochondria: Major Regulators of Neural Development. Neuroscientist 2016, 22, 346–358. [Google Scholar] [CrossRef]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Bubici, C.; Papa, S.; Pham, C.G.; Zazzeroni, F.; Franzoso, G. NF-kappaB and JNK: An intricate affair. Cell Cycle 2004, 3, 1524–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulga, N.; Pastorino, J.G. GRIM-19-mediated translocation of STAT3 to mitochondria is necessary for TNF-induced necroptosis. J. Cell Sci. 2012, 125 Pt 12, 2995–3003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.P.; Lin, H.I.; Tzeng, S.F. Tumor necrosis factor-alpha and interleukin-18 modulate neuronal cell fate in embryonic neural progenitor culture. Brain Res. 2005, 1054, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Saben, J.; Lindsey, F.; Zhong, Y.; Thakali, K.; Badger, T.M.; Andres, A.; Gomez-Acevedo, H.; Shankar, K. Maternal obesity is associated with a lipotoxic placental environment. Placenta 2014, 35, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Segovia, S.A.; Vickers, M.H.; Gray, C.; Reynolds, C.M. Maternal obesity, inflammation, and developmental programming. Biomed. Res. Int. 2014, 2014, 418975. [Google Scholar] [CrossRef]
- De Toledo Baldi, E.; Dias Bóbbo, V.C.; Melo Lima, M.H.; Velloso, L.A.; Pereira de Araujo, E. Tumor necrosis factor-alpha levels in blood cord is directly correlated with the body weight of mothers. Obes. Sci. Pract. 2016, 2, 210–214. [Google Scholar] [CrossRef] [Green Version]
- Dosch, N.C.; Guslits, E.F.; Weber, M.B.; Murray, S.E.; Ha, B.; Coe, C.L.; Auger, A.P.; Kling, P.J. Maternal Obesity Affects Inflammatory and Iron Indices in Umbilical Cord Blood. J. Pediatr. 2016, 172, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Hoseindoost, M.; Alipour, M.R.; Farajdokht, F.; Diba, R.; Bayandor, P.; Mehri, K.; Nayebi Rad, S.; Babri, S. Effects of troxerutin on inflammatory cytokines and BDNF levels in male offspring of high-fat diet fed rats. Avicenna, J. Phytomed. 2019, 9, 597–605. [Google Scholar]
- Payolla, T.B.; Lemes, S.F.; de Fante, T.; Reginato, A.; Mendes da Silva, C.; de Oliveira Micheletti, T.; Rodrigues, H.G.; Torsoni, A.S.; Milanski, M.; Torsoni, M.A. High-fat diet during pregnancy and lactation impairs the cholinergic anti-inflammatory pathway in the liver and white adipose tissue of mouse offspring. Mol. Cell Endocrinol. 2016, 422, 192–202. [Google Scholar] [CrossRef]
- Zhu, M.J.; Du, M.; Nathanielsz, P.W.; Ford, S.P. Maternal obesity up-regulates inflammatory signaling pathways and enhances cytokine expression in the mid-gestation sheep placenta. Placenta 2010, 31, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Howell, K.R.; Powell, T.L. Effects of maternal obesity on placental function and fetal development. Reproduction 2017, 153, R97–R108. [Google Scholar] [CrossRef] [PubMed]
- Jais, A.; Brüning, J.C. Hypothalamic inflammation in obesity and metabolic disease. J. Clin. Investig. 2017, 127, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Shook, L.L.; Kislal, S.; Edlow, A.G. Fetal brain and placental programming in maternal obesity: A review of human and animal model studies. Prenat. Diagn. 2020, 40, 1126–1137. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Xu, D.; Lan, X.; Jia, B.; Sun, L.; Zheng, J.C.; Peng, H. A novel role of the STAT3 pathway in brain inflammation-induced human neural progenitor cell differentiation. Curr. Mol. Med. 2013, 13, 1474–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, W.; Ramos, E.H.; Wong, B.C.; Belsham, D.D. Beneficial Effects of Metformin and/or Salicylate on Palmitate- or TNFα-Induced Neuroinflammatory Marker and Neuropeptide Gene Regulation in Immortalized NPY/AgRP Neurons. PLoS ONE 2016, 11, e0166973. [Google Scholar] [CrossRef]
- Ramadori, G.; Lee, C.E.; Bookout, A.L.; Lee, S.; Williams, K.W.; Anderson, J.; Elmquist, J.K.; Coppari, R. Brain SIRT1: Anatomical distribution and regulation by energy availability. J. Neurosci. 2008, 28, 9989–9996. [Google Scholar] [CrossRef]
- Desai, M.; Li, T.; Ross, M.G. Fetal hypothalamic neuroprogenitor cell culture: Preferential differentiation paths induced by leptin and insulin. Endocrinology 2011, 152, 3192–3201. [Google Scholar] [CrossRef]
- Kim, M.; Jung, K.; Kim, I.S.; Lee, I.S.; Ko, Y.; Shin, J.E.; Park, K.I. TNF-α induces human neural progenitor cell survival after oxygen-glucose deprivation by activating the NF-κB pathway. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.; Xiao, H.; Li, F.; Zeng, L.; Yin, Y. The profiles of mitochondrial respiration and glycolysis using extracellular flux analysis in porcine enterocyte IPEC-J2. Anim. Nutr. 2015, 1, 239–243. [Google Scholar] [CrossRef]
- Deckwerth, T.L.; Johnson, E.M., Jr. Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J. Cell Biol. 1993, 123, 1207–1222. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Birrell, M.A.; Wong, S.; Hardaker, E.L.; Catley, M.C.; McCluskie, K.; Collins, M.; Haj-Yahia, S.; Belvisi, M.G. IkappaB kinase-2-independent and -dependent inflammation in airway disease models: Relevance of IKK-2 inhibition to the clinic. Mol. Pharmacol. 2006, 69, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Palmer, T.D. Differential roles of TNFR1 and TNFR2 signaling in adult hippocampal neurogenesis. Brain Behav. Immun. 2013, 30, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, X.; Chen, Q.; Wang, Y.; Jia, B.; Sun, L.; Zheng, J.; Peng, H. TNF-α affects human cortical neural progenitor cell differentiation through the autocrine secretion of leukemia inhibitory factor. PLoS ONE 2012, 7, e50783. [Google Scholar] [CrossRef] [PubMed]
- Widera, D.; Mikenberg, I.; Elvers, M.; Kaltschmidt, C.; Kaltschmidt, B. Tumor necrosis factor alpha triggers proliferation of adult neural stem cells via IKK/NF-kappaB signaling. BMC Neurosci. 2006, 7, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, M.E.; Barbuio, R.; Milanski, M.; Romanatto, T.; Barbosa, H.C.; Nadruz, W.; Bertolo, M.B.; Boschero, A.C.; Saad, M.J.; Franchini, K.G.; et al. Tumor necrosis factor-alpha activates signal transduction in hypothalamus and modulates the expression of pro-inflammatory proteins and orexigenic/anorexigenic neurotransmitters. J. Neurochem. 2006, 98, 203–212. [Google Scholar] [CrossRef]
- Bette, M.; Kaut, O.; Schäfer, M.K.; Weihe, E. Constitutive expression of p55TNFR mRNA and mitogen-specific up-regulation of TNF alpha and p75TNFR mRNA in mouse brain. J. Comp. Neurol. 2003, 465, 417–430. [Google Scholar] [CrossRef]
- Iosif, R.E.; Ekdahl, C.T.; Ahlenius, H.; Pronk, C.J.; Bonde, S.; Kokaia, Z.; Jacobsen, S.E.; Lindvall, O. Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J. Neurosci. 2006, 26, 9703–9712. [Google Scholar] [CrossRef] [Green Version]
- Mauro, C.; Leow, S.C.; Anso, E.; Rocha, S.; Thotakura, A.K.; Tornatore, L.; Moretti, M.; de Smaele, E.; Beg, A.A.; Tergaonkar, V.; et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 2011, 13, 1272–1279. [Google Scholar] [CrossRef]
- Tammineni, P.; Anugula, C.; Mohammed, F.; Anjaneyulu, M.; Larner, A.C.; Sepuri, N.B. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J. Biol. Chem. 2013, 288, 4723–4732. [Google Scholar] [CrossRef] [Green Version]
- Zouein, F.A.; Duhé, R.J.; Arany, I.; Shirey, K.; Hosler, J.P.; Liu, H.; Saad, I.; Kurdi, M.; Booz, G.W. Loss of STAT3 in mouse embryonic fibroblasts reveals its Janus-like actions on mitochondrial function and cell viability. Cytokine 2014, 66, 7–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgieva, E.; Ivanova, D.; Zhelev, Z.; Bakalova, R.; Gulubova, M.; Aoki, I. Mitochondrial Dysfunction and Redox Imbalance as a Diagnostic Marker of “Free Radical Diseases”. Anticancer Res. 2017, 37, 5373–5381. [Google Scholar] [PubMed] [Green Version]
- Vona, R.; Gambardella, L.; Cittadini, C.; Straface, E.; Pietraforte, D. (Biomarkers of Oxidative Stress in Metabolic Syndrome and Associated Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 8267234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunetti, D.; Dykstra, W.; Le, S.; Zink, A.; Prigione, A. Mitochondria in neurogenesis: Implications for mitochondrial diseases. Stem Cells 2021, 39, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Poli, V. From the nucleus to the mitochondria and back: The odyssey of a multitask STAT3. Cell Cycle 2011, 10, 3221–3222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Belle, J.E.; Orozco, N.M.; Paucar, A.A.; Saxe, J.P.; Mottahedeh, J.; Pyle, A.D.; Wu, H.; Kornblum, H.I. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 2011, 8, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Bigarella, C.L.; Liang, R.; Ghaffari, S. Stem cells and the impact of ROS signaling. Development 2014, 141, 4206–4218. [Google Scholar] [CrossRef] [Green Version]
- Tsatmali, M.; Walcott, E.C.; Makarenkova, H.; Crossin, K.L. Reactive oxygen species modulate the differentiation of neurons in clonal cortical cultures. Mol. Cell Neurosci. 2006, 33, 345–357. [Google Scholar] [CrossRef] [Green Version]
- Vieira, H.L.; Alves, P.M.; Vercelli, A. Modulation of neuronal stem cell differentiation by hypoxia and reactive oxygen species. Prog. Neurobiol. 2011, 93, 444–455. [Google Scholar] [CrossRef]
- Marchetti, L.; Klein, M.; Schlett, K.; Pfizenmaier, K.; Eisel, U.L. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J. Biol. Chem. 2004, 279, 32869–32881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, M.K.; Bisht, B.; Darmawan, D.O.; Chiou, R.; Ha, V.L.; Wallace, W.D.; Chon, A.T.; Hegab, A.E.; Grogan, T.; Elashoff, D.A.; et al. Dynamic changes in intracellular ROS levels regulate airway basal stem cell homeostasis through Nrf2-dependent Notch signaling. Cell Stem Cell 2014, 15, 199–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libert, S.; Cohen, D.; Guarente, L. Neurogenesis directed by Sirt1. Nat. Cell Biol. 2008, 10, 373–374. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Crosstalk between Oxidative Stress and SIRT1: Impact on the Aging Process. Int. J. Mol. Sci. 2013, 14, 3834–3859. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Cao, J.; Huang, K.J.; Zhang, F.; Jiang, T.; Zhu, L.; Qiu, Z.J. Inhibition of STAT3 activity with AG490 decreases the invasion of human pancreatic cancer cells in vitro. Cancer Sci. 2006, 97, 1417–1423. [Google Scholar] [CrossRef]
- Wang, B.; Bai, S.; Wang, J.; Ren, N.; Xie, R.; Cheng, G.; Yu, Y. TPCA-1 negatively regulates inflammation mediated by NF-κB pathway in mouse chronic periodontitis model. Mol. Oral Microbiol. 2021, 36, 192–201. [Google Scholar] [CrossRef]
- Nan, J.; Du, Y.; Chen, X.; Bai, Q.; Wang, Y.; Zhang, X.; Zhu, N.; Zhang, J.; Hou, J.; Wang, Q.; et al. TPCA-1 is a direct dual inhibitor of STAT3 and NF-κB and regresses mutant EGFR-associated human non-small cell lung cancers. Mol. Cancer Ther. 2014, 13, 617–629. [Google Scholar] [CrossRef] [Green Version]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.F.; Perkins, N.D. Nuclear factor-κB, p53, and mitochondria: Regulation of cellular metabolism and the Warburg effect. Trends Biochem. Sci. 2012, 37, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugatto, F.; Fernández-Deudero, Á.; Bailén, Á.; Fernández-Macías, R.; Hervías-Vivancos, B.; Bartha, J.L. Second-trimester amniotic fluid proinflammatory cytokine levels in normal and overweight women. Obstet. Gynecol. 2010, 115, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Atègbo, J.M.; Grissa, O.; Yessoufou, A.; Hichami, A.; Dramane, K.L.; Moutairou, K.; Miled, A.; Grissa, A.; Jerbi, M.; Tabka, Z.; et al. Modulation of adipokines and cytokines in gestational diabetes and macrosomia. J. Clin. Endocrinol. Metab. 2006, 91, 4137–4143. [Google Scholar] [CrossRef] [PubMed]
- Challier, J.C.; Basu, S.; Bintein, T.; Minium, J.; Hotmire, K.; Catalano, P.M.; Hauguel-de Mouzon, S. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta 2008, 29, 274–281. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Zhang, F.; Dai, Y.; Han, L.; Chen, S. Serum TNF-α, GTH and MDA of high-fat diet-induced obesity and obesity resistant rats. Saudi Pharm. J. 2016, 24, 333–336. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.; Huang, Y.; Liu, D.; Chen, X.; Wang, D.; Huang, D.; Zhao, L.; Xiao, X. Obesity induced by neonatal overfeeding worsens airway hyperresponsiveness and inflammation. PLoS ONE 2012, 7, e47013. [Google Scholar] [CrossRef] [Green Version]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Whitney, N.P.; Eidem, T.M.; Peng, H.; Huang, Y.; Zheng, J.C. Inflammation mediates varying effects in neurogenesis: Relevance to the pathogenesis of brain injury and neurodegenerative disorders. J. Neurochem. 2009, 108, 1343–1359. [Google Scholar] [CrossRef]
- Yang, R.; Lirussi, D.; Thornton, T.M.; Jelley-Gibbs, D.M.; A Diehl, S.; Case, L.K.; Madesh, M.; Taatjes, D.J.; Teuscher, C.; Haynes, L.; et al. Mitochondrial Ca2⁺ and membrane potential, an alternative pathway for Interleukin 6 to regulate CD4 cell effector function. Elife 2015, 4, e06376. [Google Scholar] [CrossRef] [PubMed]
- Maffezzini, C.; Calvo-Garrido, J.; Wredenberg, A.; Freyer, C. Metabolic regulation of neurodifferentiation in the adult brain. Cell Mol. Life Sci. 2020, 77, 2483–2496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SSripetchwandee, J.; Chattipakorn, N.; Chattipakorn, S.C. Links Between Obesity-Induced Brain Insulin Resistance, Brain Mitochondrial Dysfunction, and Dementia. Front. Endocrinol. 2018, 9, 496. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.M.R.; Isganaitis, E.; Matthews, T.J.; Hughes, K.; Daher, G.; Dreyfuss, J.M.; da Silva, G.A.; Patti, M.E. Maternal obesity programs mitochondrial and lipid metabolism gene expression in infant umbilical vein endothelial cells. Int. J. Obes. 2016, 40, 1627–1634. [Google Scholar] [CrossRef] [Green Version]
- Pantham, P.; Aye, I.L.; Powell, T.L. Inflammation in maternal obesity and gestational diabetes mellitus. Placenta 2015, 36, 709–715. [Google Scholar] [CrossRef] [Green Version]
- Costa, S.M.R.; Isganaitis, E.; Matthews, T.J.; Hughes, K.; Daher, G.; Dreyfuss, J.M. Maternal obesity alters immune cell frequencies and responses in umbilical cord blood samples. Pediatric Allergy Immunol. 2015, 26, 344–351. [Google Scholar]
- Taga, T.; Fukuda, S. Role of IL-6 in the neural stem cell differentiation. Clin. Rev. Allergy Immunol. 2005, 28, 249–256. [Google Scholar] [CrossRef]
- Crampton, S.J.; Collins, L.M.; Toulouse, A.; Nolan, Y.M.; O’Keeffe, G.W. Exposure of foetal neural progenitor cells to IL-1β impairs their proliferation and alters their differentiation—A role for maternal inflammation? J. Neurochem. 2012, 120, 964–973. [Google Scholar] [CrossRef]
- Park, H.R.; Kim, J.Y.; Park, K.Y.; Lee, J. Lipotoxicity of palmitic Acid on neural progenitor cells and hippocampal neurogenesis. Toxicol. Res. 2011, 27, 103–110. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, D.; Zhang, Q.; Wang, J.; Zhan, J.; Xian, X.; Du, Z.; Wang, X.; Hao, A. Palmitic acid affects proliferation and differentiation of neural stem cells in vitro. J. Neurosci. Res. 2014, 92, 574–586. [Google Scholar] [CrossRef]
- Arsenijevic, Y.; Weiss, S.; Schneider, B.; Aebischer, P. Insulin-like growth factor-I is necessary for neural stem cell proliferation and demonstrates distinct actions of epidermal growth factor and fibroblast growth factor-2. J. Neurosci. 2001, 21, 7194–7202. [Google Scholar] [CrossRef] [Green Version]
- Israsena, N.; Hu, M.; Fu, W.; Kan, L.; Kessler, J.A. The presence of FGF2 signaling determines whether beta-catenin exerts effects on proliferation or neuronal differentiation of neural stem cells. Dev. Biol. 2004, 268, 220–231. [Google Scholar] [CrossRef]
- Ito, H.; Nakajima, A.; Nomoto, H.; Furukawa, S. Neurotrophins facilitate neuronal differentiation of cultured neural stem cells via induction of mRNA expression of basic helix-loop-helix transcription factors Mash1 and Math1. J. Neurosci. Res. 2003, 71, 648–658. [Google Scholar] [CrossRef]
- Hara, Y.; McKeehan, N.; Dacks, P.A.; Fillit, H.M. Evaluation of the Neuroprotective Potential of N-Acetylcysteine for Prevention and Treatment of Cognitive Aging and Dementia. J. Prev. Alzheimer’s Dis. 2017, 4, 201–206. [Google Scholar] [CrossRef]
- Chao, M.W.; Chen, C.P.; Yang, Y.H.; Chuang, Y.C.; Chu, T.Y.; Tseng, C.Y. N-acetylcysteine attenuates lipopolysaccharide-induced impairment in lamination of Ctip2-and Tbr1- expressing cortical neurons in the developing rat fetal brain. Sci. Rep. 2016, 6, 32373. [Google Scholar] [CrossRef] [Green Version]
- Halasi, M.; Wang, M.; Chavan, T.S.; Gaponenko, V.; Hay, N.; Gartel, A.L. ROS inhibitor N-acetyl-L-cysteine antagonizes the activity of proteasome inhibitors. Biochem. J. 2013, 454, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, S.F.; Granger, D.N. Cerebral microvascular inflammation in DOCA salt-induced hypertension: Role of angiotensin II and mitochondrial superoxide. J. Cereb. Blood Flow Metab. 2012, 32, 368–375. [Google Scholar] [CrossRef] [Green Version]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Ni, R.; Cao, T.; Xiong, S.; Ma, J.; Fan, G.C.; Lacefield, J.C.; Lu, Y.; le Tissier, S.; Peng, T. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free. Radic. Biol. Med. 2016, 90, 12–23. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desai, M.; Stiles, L.; Torsoni, A.S.; Torsoni, M.A.; Shirihai, O.S.; Ross, M.G. TNFα-Induced Oxidative Stress and Mitochondrial Dysfunction Alter Hypothalamic Neurogenesis and Promote Appetite Versus Satiety Neuropeptide Expression in Mice. Brain Sci. 2022, 12, 900. https://doi.org/10.3390/brainsci12070900
Desai M, Stiles L, Torsoni AS, Torsoni MA, Shirihai OS, Ross MG. TNFα-Induced Oxidative Stress and Mitochondrial Dysfunction Alter Hypothalamic Neurogenesis and Promote Appetite Versus Satiety Neuropeptide Expression in Mice. Brain Sciences. 2022; 12(7):900. https://doi.org/10.3390/brainsci12070900
Chicago/Turabian StyleDesai, Mina, Linsey Stiles, Adriana S. Torsoni, Marcio A. Torsoni, Orian S. Shirihai, and Michael G. Ross. 2022. "TNFα-Induced Oxidative Stress and Mitochondrial Dysfunction Alter Hypothalamic Neurogenesis and Promote Appetite Versus Satiety Neuropeptide Expression in Mice" Brain Sciences 12, no. 7: 900. https://doi.org/10.3390/brainsci12070900