
Escalation of Tau Accumulation after a Traumatic Brain Injury: Findings from Positron Emission Tomography

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Cognitive Measures
2.3. Cerebrospinal Fluid (CSF) Sample Collection and Analysis
2.4. MRI/PET Image Acquisition
2.5. Data Pre-Processing
2.6. Regions of Interest to Estimate Braak Staging According to [18F]-AV1451 SUVr
2.7. Statistical Analysis
2.7.1. Analysis of the Neuropsychological Measures
2.7.2. Voxel-Based Analysis of the PET Data
3. Results
3.1. Study Subjects, Demographics, and Neuropsychological Data
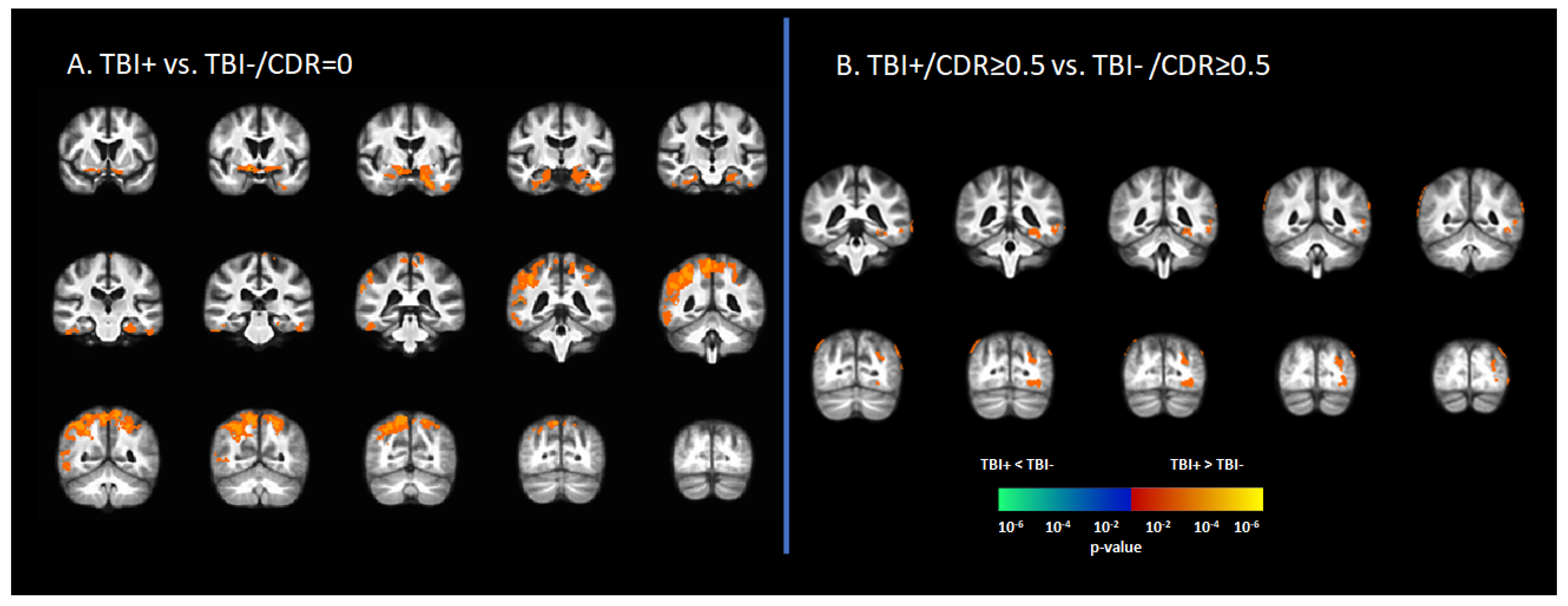
3.2. Tau Deposition Is Increased in the TBI+ Group
3.3. Braak Staging of [18F]-AV1451 SUVr in Subjects with History of TBI
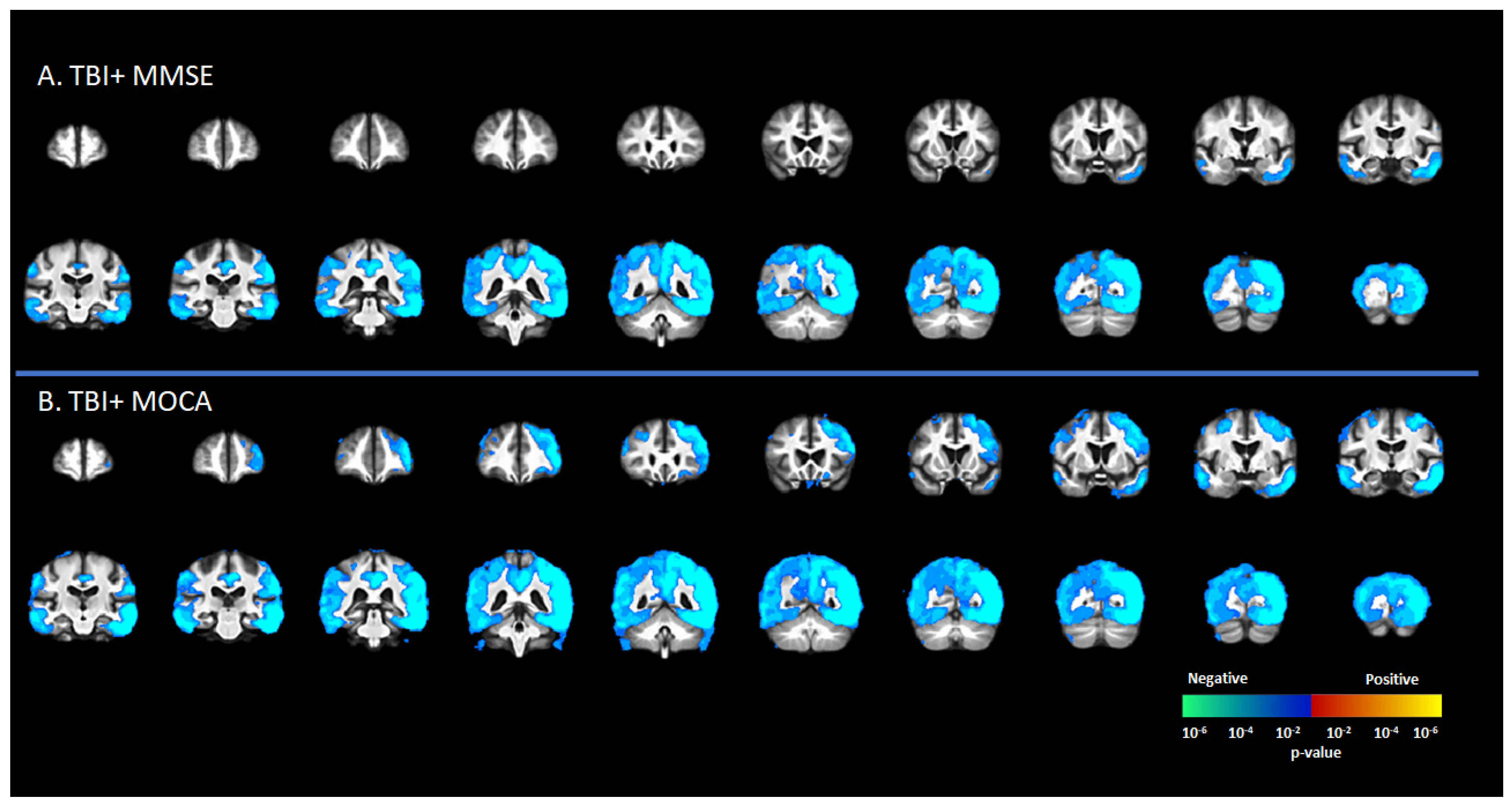
3.4. Correlation [18F]-AV1451 SUVr and Neuropsychological Scores
3.5. Correlation between [18F]-AV1451 SUVr and CSF Levels of Tau and Amyloid
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shen, Q.; Hiebert, J.B.; Hartwell, J.; Thimmesch, A.R.; Pierce, J.D. Systematic Review of Traumatic Brain Injury and the Impact of Antioxidant Therapy on Clinical Outcomes. Worldviews Evid. -Based Nurs. 2016, 13, 380–389. [Google Scholar] [CrossRef]
- Mohamed, A.Z.; Corrigan, F.; Collins-Praino, L.E.; Plummer, S.L.; Soni, N.; Nasrallah, F.A. Evaluating Spatiotemporal Microstructural Alterations Following Diffuse Traumatic Brain Injury. NeuroImage Clin. 2020, 25, 102136. [Google Scholar] [CrossRef]
- Mohamed, A.Z.; Cumming, P.; Nasrallah, F.A. White Matter Alterations Are Associated With Cognitive Dysfunction Decades After Moderate-to-Severe Traumatic Brain Injury and/or Posttraumatic Stress Disorder. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2021, 6, 1100–1109. [Google Scholar] [CrossRef]
- Mohamed, A.Z.; Cumming, P.; Nasrallah, F.A. Traumatic Brain Injury Augurs Ill for Prolonged Deficits in the Brain’s Structural and Functional Integrity Following Controlled Cortical Impact Injury. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- To, X.V.; Mohamed, A.Z.; Cumming, P.; Nasrallah, F.A. Subacute Cytokine Changes after a Traumatic Brain Injury Predict Chronic Brain Microstructural Alterations on Advanced Diffusion Imaging in the Male Rat. Brain. Behav. Immun. 2022. [Google Scholar] [CrossRef]
- Soni, N.; Mohamed, A.Z.; Kurniawan, N.D.; Borges, K.; Nasrallah, F. Diffusion Magnetic Resonance Imaging Unveils the Spatiotemporal Microstructural Gray Matter Changes Following Injury in the Rodent Brain. J. Neurotrauma 2019, 36, 1306–1317. [Google Scholar] [CrossRef]
- Mohamed, A.Z.; Nestor, P.J.; Cumming, P.; Nasrallah, F.A. Traumatic Brain Injury Fast-Forwards Alzheimer’s Pathology: Evidence from Amyloid Positron Emission Tomorgraphy Imaging. J. Neurol. 2022, 269, 873–884. [Google Scholar] [CrossRef]
- Mohamed, A.Z.; Cumming, P.; Srour, H.; Gunasena, T.; Uchida, A.; Haller, C.N.; Nasrallah, F. Amyloid Pathology Fingerprint Differentiates Post-Traumatic Stress Disorder and Traumatic Brain Injury. NeuroImage Clin. 2018, 19, 716–726. [Google Scholar] [CrossRef]
- Mohamed, A.Z.; Cumming, P.; Götz, J.; Nasrallah, F. Tauopathy in Veterans with Long-Term Posttraumatic Stress Disorder and Traumatic Brain Injury. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1139–1151. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Shi, R. Current Advances in Neurotrauma Research: Diagnosis, Neuroprotection, and Neurorepair. Neural Regen. Res. 2014, 9, 1093–1095. [Google Scholar] [CrossRef]
- Nemetz, P.N.; Leibson, C.; Naessens, J.M.; Beard, M.; Kokmen, E.; Annegers, J.F.; Kurland, L.T. Traumatic Brain Injury and Time to Onset of Alzheimer’s Disease: A Population-Based Study. Am. J. Epidemiol. 1999, 149, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.K.; Lin, S.H.; Sung, P.S.; Wu, M.H.; Hung, K.W.; Wang, L.C.; Huang, C.Y.; Lu, K.; Chen, H.J.; Tsai, K.J. Population Based Study on Patients with Traumatic Brain Injury Suggests Increased Risk of Dementia. J. Neurol. Neurosurg. Psychiatry 2012, 83, 1080–1085. [Google Scholar] [CrossRef]
- Weiner, M.W.; Crane, P.K.; Montine, T.J.; Bennett, D.A.; Veitch, D.P. Traumatic Brain Injury May Not Increase the Risk of Alzheimer Disease. Neurology 2017, 89, 1923–1925. [Google Scholar] [CrossRef] [Green Version]
- Sugarman, M.A.; McKee, A.C.; Stein, T.D.; Tripodis, Y.; Besser, L.M.; Martin, B.; Palmisano, J.N.; Steinberg, E.G.; O’Connor, M.K.; Au, R.; et al. Failure to Detect an Association between Self-Reported Traumatic Brain Injury and Alzheimer’s Disease Neuropathology and Dementia. Alzheimer’s Dement. 2019, 15, 686–698. [Google Scholar] [CrossRef]
- Koerte, I.K.; Lin, A.P.; Willems, A.; Muehlmann, M.; Hufschmidt, J.; Coleman, M.J.; Green, I.; Liao, H.; Tate, D.F.; Wilde, E.A.; et al. A Review of Neuroimaging Findings in Repetitive Brain Trauma. Brain Pathol. 2015, 25, 318–349. [Google Scholar] [CrossRef]
- Washington, P.M.; Villapol, S.; Burns, M.P. Polypathology and Dementia after Brain Trauma: Does Brain Injury Trigger Distinct Neurodegenerative Diseases, or Should They Be Classified Together as Traumatic Encephalopathy? Exp. Neurol. 2016, 275, 381–388. [Google Scholar] [CrossRef] [Green Version]
- DeVos, S.L.; Corjuc, B.T.; Oakley, D.H.; Nobuhara, C.K.; Bannon, R.N.; Chase, A.; Commins, C.; Gonzalez, J.A.; Dooley, P.M.; Frosch, M.P.; et al. Synaptic Tau Seeding Precedes Tau Pathology in Human Alzheimer’s Disease Brain. Front. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.E.; McKee, A.C.; Salat, D.H.; Rasmusson, A.M.; Radigan, L.J.; Catana, C.; Milberg, W.P.; McGlinchey, R.E. Positron Emission Tomography of Tau in Iraq and Afghanistan Veterans with Blast Neurotrauma. NeuroImage Clin. 2019, 21, 101651. [Google Scholar] [CrossRef]
- Gorgoraptis, N.; Li, L.M.; Whittington, A.; Zimmerman, K.A.; Maclean, L.M.; McLeod, C.; Ross, E.; Heslegrave, A.; Zetterberg, H.; Passchier, J.; et al. In Vivo Detection of Cerebral Tau Pathology in Long-Term Survivors of Traumatic Brain Injury. Sci. Transl. Med. 2019, 11, 1993. [Google Scholar] [CrossRef] [Green Version]
- Blennow, K.; Brody, D.L.; Kochanek, P.M.; Levin, H.; McKee, A.; Ribbers, G.M.; Yaffe, K.; Zetterberg, H. Traumatic Brain Injuries. Nat. Rev. Dis. Prim. 2016, 2, 16084. [Google Scholar] [CrossRef]
- Mendez, M.F. What Is the Relationship of Traumatic Brain Injury to Dementia? J. Alzheimer’s Dis. 2017, 57, 667–681. [Google Scholar] [CrossRef] [PubMed]
- Roberts, G.W.; Gentleman, S.M.; Lynch, A.; Murray, L.; Landon, M.; Graham, D.I. Β3 Amyloid Protein Deposition in the Brain after Severe Head Injury: Implications for the Pathogenesis of Alzheimer’s Disease. J. Neurol. Neurosurg. Psychiatry 1994, 57, 419–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.T.; Veenith, T.; Dewar, D.; Outtrim, J.G.; Tavares, A.; Canales, R.; Mathis, C.A.; Klunk, W.E.; Aigbirhio, F.I.; Coles, J.P.; et al. Amyloid Imaging with Carbon 11–Labeled Pittsburgh Compound B for Traumatic Brain Injury. JAMA Neurol. 2014, 71, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C. The Clinical Dementia Rating (CDR): Current Version and Scoring Rules. Neurology 1993, 43, 2412–2414. [Google Scholar] [CrossRef]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. “Mini-Mental State”. A Practical Method for Grading the Cognitive State of Patients for the Clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Nasreddine, Z.S.; Phillips, N.A.; Bédirian, V.; Charbonneau, S.; Whitehead, V.; Collin, I.; Cummings, J.L.; Chertkow, H. The Montreal Cognitive Assessment, MoCA: A Brief Screening Tool for Mild Cognitive Impairment. J. Am. Geriatr. Soc. 2005, 53, 695–699. [Google Scholar] [CrossRef]
- Skinner, J.; Carvalho, J.O.; Potter, G.G.; Thames, A.; Zelinski, E.; Crane, P.K.; Gibbons, L.E. The Alzheimer’s Disease Assessment Scale-Cognitive-Plus (ADAS-Cog-Plus): An Expansion of the ADAS-Cog to Improve Responsiveness in MCI. Brain Imaging Behav. 2012, 6, 489–501. [Google Scholar] [CrossRef]
- Farias, S.T.; Mungas, D.; Reed, B.R.; Cahn-Weiner, D.; Jagust, W.; Baynes, K.; DeCarli, C. The Measurement of Everyday Cognition (ECog): Scale Development and Psychometric Properties. Neuropsychology 2008, 22, 531–544. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, J.I.; Yesavage, J.A. 9/Geriatric Depression Scale (Gds) Recent Evidence and Development of a Shorter Version. Clin. Gerontol. 1986, 5, 165–173. [Google Scholar] [CrossRef]
- Ito, K.; Hutmacher, M.M.; Corrigan, B.W. Modeling of Functional Assessment Questionnaire (FAQ) as Continuous Bounded Data from the ADNI Database. J. Pharmacokinet. Pharmacodyn. 2012, 39, 601–618. [Google Scholar] [CrossRef]
- Shulman, K.I. Clock-Drawing: Is It the Ideal Cognitive Screening Test? Int. J. Geriatr. Psychiatry 2000, 15, 548–561. [Google Scholar] [CrossRef]
- Vakil, E.; Blachstein, H. Rey Auditory-verbal Learning Test: Structure Analysis. J. Clin. Psychol. 1993, 49, 883–890. [Google Scholar] [CrossRef]
- Barr, A.; Brandt, J. Word-List Generation Deficits in Dementia. J. Clin. Exp. Neuropsychol. 1996, 18, 810–822. [Google Scholar] [CrossRef]
- Arnett, J.A.; Labovitz, S.S. Effect of Physical Layout in Performance of the Trail Making Test. Psychol. Assess. 1995, 7, 220–221. [Google Scholar] [CrossRef]
- Williams, B.W.; Mack, W.; Henderson, V.W. Boston Naming Test in Alzheimer’s Disease. Neuropsychologia 1989, 27, 1073–1079. [Google Scholar] [CrossRef]
- McGurn, B.; Starr, J.M.; Topfer, J.A.; Pattie, A.; Whiteman, M.C.; Lemmon, H.A.; Whalley, L.J.; Deary, I.J. Pronunciation of Irregular Words Is Preserved in Dementia, Validating Premorbid IQ Estimation. Neurology 2004, 62, 1184–1186. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, J. A Fast Diffeomorphic Image Registration Algorithm. Neuroimage 2007, 38, 95–113. [Google Scholar] [CrossRef]
- Johnson, K.A.; Schultz, A.; Betensky, R.A.; Becker, J.A.; Sepulcre, J.; Rentz, D.; Mormino, E.; Chhatwal, J.; Amariglio, R.; Papp, K.; et al. Tau Positron Emission Tomographic Imaging in Aging and Early Alzheimer Disease. Ann. Neurol. 2016, 79, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Tredici, K. Staging of Alzheimer Disease-Associated Neurofibrillary Pathology Using Paraffin Sections and Immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, A.J.; Yu, P.; Miller, B.B.; Shcherbinin, S.; Dickson, J.; Navitsky, M.; Joshi, A.D.; Devous, M.D.; Mintun, M.S. Regional Profiles of the Candidate Tau PET Ligand 18F-AV-1451 Recapitulate Key Features of Braak Histopathological Stages. Brain 2016, 139, 1539–1550. [Google Scholar] [CrossRef] [Green Version]
- Marquié, M.; Siao Tick Chong, M.; Antón-Fernández, A.; Verwer, E.E.; Sáez-Calveras, N.; Meltzer, A.C.; Ramanan, P.; Amaral, A.C.; Gonzalez, J.; Normandin, M.D.; et al. [F-18]-AV-1451 Binding Correlates with Postmortem Neurofibrillary Tangle Braak Staging. Acta Neuropathol. 2017, 134, 619–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aschenbrenner, A.J.; Gordon, B.A.; Benzinger, T.L.S.; Morris, J.C.; Hassenstab, J.J. Influence of Tau PET, Amyloid PET, and Hippocampal Volume on Cognition in Alzheimer Disease. Neurology 2018, 91, e859–e866. [Google Scholar] [CrossRef] [PubMed]
- Pontecorvo, M.J.; Devous, M.D.; Navitsky, M.; Lu, M.; Salloway, S.; Schaerf, F.W.; Jennings, D.; Arora, A.K.; McGeehan, A.; Lim, N.C.; et al. Relationships between Flortaucipir PET Tau Binding and Amyloid Burden, Clinical Diagnosis, Age and Cognition. Brain 2017, 140, 748–763. [Google Scholar] [CrossRef]
- Montenigro, P.H.; Baugh, C.M.; Daneshvar, D.H.; Mez, J.; Budson, A.E.; Au, R.; Katz, D.I.; Cantu, R.C.; Stern, R.A. Clinical Subtypes of Chronic Traumatic Encephalopathy: Literature Review and Proposed Research Diagnostic Criteria for Traumatic Encephalopathy Syndrome. Alzheimer’s Res. Ther. 2014, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Morris, H.R.; Neal, J.W.; Lees, A.J.; Hardy, J.; Holton, J.L.; Revesz, T.; Williams, D.D.R. Mixed Pathologies Including Chronic Traumatic Encephalopathy Account for Dementia in Retired Association Football (Soccer) Players. Acta Neuropathol. 2017, 133, 337–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieniek, K.F.; Ross, O.A.; Cormier, K.A.; Walton, R.L.; Soto-Ortolaza, A.; Johnston, A.E.; DeSaro, P.; Boylan, K.B.; Graff-Radford, N.R.; Wszolek, Z.K.; et al. Chronic Traumatic Encephalopathy Pathology in a Neurodegenerative Disorders Brain Bank. Acta Neuropathol. 2015, 130, 877–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquié, M.; Normandin, M.D.; Vanderburg, C.R.; Costantino, I.M.; Bien, E.A.; Rycyna, L.G.; Klunk, W.E.; Mathis, C.A.; Ikonomovic, M.D.; Debnath, M.L.; et al. Validating Novel Tau Positron Emission Tomography Tracer [F-18]-AV-1451 (T807) on Postmortem Brain Tissue. Ann. Neurol. 2015, 78, 787–800. [Google Scholar] [CrossRef] [Green Version]
- Marquié, M.; Normandin, M.D.; Meltzer, A.C.; Siao Tick Chong, M.; Andrea, N.V.; Antón-Fernández, A.; Klunk, W.E.; Mathis, C.A.; Ikonomovic, M.D.; Debnath, M.; et al. Pathological Correlations of [F-18]-AV-1451 Imaging in Non-Alzheimer Tauopathies. Ann. Neurol. 2017, 81, 117–128. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Widespread Tau and Amyloid-Beta Pathology Many Years after a Single Traumatic Brain Injury in Humans. Brain Pathol. 2012, 22, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Zanier, E.R.; Bertani, I.; Sammali, E.; Pischiutta, F.; Chiaravalloti, M.A.; Vegliante, G.; Masone, A.; Corbelli, A.; Smith, D.H.; Menon, D.K.; et al. Induction of a Transmissible Tau Pathology by Traumatic Brain Injury. Brain 2018, 141, 2685–2699. [Google Scholar] [CrossRef]
- Polanco, J.C.; Li, C.; Bodea, L.G.; Martinez-Marmol, R.; Meunier, F.A.; Götz, J. Amyloid-β and Tau Complexity - Towards Improved Biomarkers and Targeted Therapies. Nat. Rev. Neurol. 2018, 14, 22–40. [Google Scholar] [CrossRef]
- Blennow, K.; Hardy, J.; Zetterberg, H. The Neuropathology and Neurobiology of Traumatic Brain Injury. Neuron 2012, 76, 886–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadzadeh, H.; Smith, D.H.; Shenoy, V.B. Mechanical Effects of Dynamic Binding between Tau Proteins on Microtubules during Axonal Injury. Biophys. J. 2015, 109, 2328–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadzadeh, H.; Smith, D.H.; Shenoy, V.B. Viscoelasticity of Tau Proteins Leads to Strain Rate-Dependent Breaking of Microtubules during Axonal Stretch Injury: Predictions from a Mathematical Model. Biophys. J. 2014, 106, 1123–1133. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Cejudo, J.; Wisniewski, T.; Marmar, C.; Zetterberg, H.; Blennow, K.; de Leon, M.J.; Fossati, S. Traumatic Brain Injury and Alzheimer’s Disease: The Cerebrovascular Link. EBioMedicine 2018, 28, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, G.; Zhao, J.; Dash, P.K.; Soto, C.; Moreno-Gonzalez, I. Traumatic Brain Injury Induces Tau Aggregation and Spreading. J. Neurotrauma 2020, 37, 80–92. [Google Scholar] [CrossRef] [Green Version]
- La Joie, R.; Bejanin, A.; Fagan, A.M.; Ayakta, N.; Baker, S.L.; Bourakova, V.; Boxer, A.; Cha, J.; Karydas, A.; Jerome, G.; et al. Associations between [18F] AV1451 Tau PET and CSF Measures of Tau Pathology in a Clinical Sample. Neurology 2018, 90, E282–E290. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a Biological Definition of Alzheimer’s Disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Lowe, V.J.; Curran, G.; Fang, P.; Liesinger, A.M.; Josephs, K.A.; Parisi, J.E.; Kantarci, K.; Boeve, B.F.; Pandey, M.K.; Bruinsma, T.; et al. An Autoradiographic Evaluation of AV-1451 Tau PET in Dementia. Acta Neuropathol. Commun. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Corrigan, J.D.; Bogner, J. Initial Reliability and Validity of the Ohio State University TBI Identification Method. J. Head Trauma Rehabil. 2007, 22, 318–329. [Google Scholar] [CrossRef]
- Vermeiren, C.; Motte, P.; Viot, D.; Mairet-Coello, G.; Courade, J.P.; Citron, M.; Mercier, J.; Hannestad, J.; Gillard, M. The Tau Positron-Emission Tomography Tracer AV-1451 Binds with Similar Affinities to Tau Fibrils and Monoamine Oxidases. Mov. Disord. 2018, 33, 273–281. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sex | APOE | Age | TSI | Number of TBI Incidents | Source | LOC | Braak Stage | Aβ+ | CDR | MOCA | ADAS | ADAS-Cog | MMSE | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | M | 4/4 | 77 | 69 | 1 | 1 | 1 | 0 | NA | 5 | 7 | 29 | ||
| P2 | M | 3/3 | 79 | 28 | 1 | Fall | LOC | 1 | 0 | 0 | 25 | 9 | 13 | 27 |
| P3 | F | 2/3 | 74 | 56 | 1 | LOC | 3 | 1 | 0 | 26 | 8 | 11 | 29 | |
| P4 | M | 2/3 | 68 | 53 | 1 | 4 | 0 | 0 | 26 | 15 | 20 | 29 | ||
| P5 | M | 3/3 | 72 | 9 | 1 | LOC | 0 | 0 | 0 | 27 | 8 | 10 | 29 | |
| P6 | F | 3/4 | 63 | 50 | 1 | 1 | 1 | 0 | 28 | 6 | 9 | 29 | ||
| P7 | F | 3/3 | 83 | 48 | 1 | 0 | 0 | 0 | 27 | 8 | 13 | 28 | ||
| P8 | F | 3/3 | 71 | 9 | 1 | 0 | 0 | 0 | 30 | 9 | 11 | 30 | ||
| P9 | M | 3/4 | 67 | 54 | 1 | Fall | LOC | 0 | 1 | 0 | 26 | 6 | 11 | 29 |
| P10 | F | 3/3 | 83 | 52 | 1 | LOC | 0 | 1 | 0 | 26 | 10 | 15 | 27 | |
| P11 | M | 3/3 | 85 | 73 | 1 | LOC | 0 | 0 | 0.5 | 25 | 12 | 19 | 29 | |
| P12 | M | 3/3 | 75 | 30 | 1 | Accident | 5 | 1 | 1 | 14 | 18 | 27 | 24 | |
| P13 | M | 4/4 | 81 | 7 | 2 | MVA | 2 | 1 | 0.5 | 17 | 15 | 25 | 24 | |
| P14 | M | 3/3 | 80 | 15 | 1 | Fall | LOC | 0 | 1 | 0.5 | 27 | 5 | 8 | 27 |
| P15 | M | 4/4 | 66 | 52 | 2 | LOC | 5 | 1 | 0.5 | 21 | 13 | 23 | 26 | |
| P16 | M | 3/4 | 74 | 36 | 1 | Fall | 0 | 0 | 0.5 | 22 | 11 | 14 | 29 | |
| P17 | M | 2/3 | 68 | 52 | 3 | Football | LOC | 4 | 0 | 1 | 25 | 16 | 24 | 23 |
| P18 | M | 3/4 | 83 | 74 | 1 | 2 | 1 | 0.5 | 18 | 19 | 28 | 22 | ||
| P19 | M | 3/3 | 89 | 24 | 2 | 0 | 1 | 0.5 | 24 | 11 | 16 | 30 | ||
| P20 | M | 3/4 | 82 | 8 | 1 | 0 | 1 | 1 | 21 | 12 | 16 | 30 |
| TBI-/CDR ≥ 0.5 | TBI-/CDR = 0 | TBI+/CDR ≥ 0.5 | TBI+/CDR = 0 | TBI-/CDR ≥ 0.5 vs. TBI-/CDR = 0 | TBI-/CDR ≥ 0.5 vs. TBI+/CDR ≥ 0.5 | TBI-/CDR = 0 vs. TBI+/CDR = 0 | TBI+/CDR ≥ 0.5 vs. TBI+/CDR = 0 | |
|---|---|---|---|---|---|---|---|---|
| number of participants | N = 50 | N = 50 | N = 10 | N = 10 | ||||
| Gender: | 1 | 0.01 | 1 | 0.065 | ||||
| Female | 24 (48.0%) | 25 (50.0%) | 0 (0.00%) | 5 (50.0%) | ||||
| Male | 26 (52.0%) | 25 (50.0%) | 10 (100%) | 5 (50.0%) | ||||
| APOE4 Status: | 1 | 1 | 1 | 1 | ||||
| Negative | 30 (60.0%) | 32 (64.0%) | 5 (50.0%) | 7 (70.0%) | ||||
| Positive | 20 (40.0%) | 18 (36.0%) | 5 (50.0%) | 3 (30.0%) | ||||
| Age | 75.1 (7.82) | 76.8 (6.30) | 76.6 (7.63) | 75.4 (7.31) | 0.61 | 0.93 | 0.94 | 0.982 |
| TBI Type: | . | . | . | 0.65 | ||||
| Concussion | 7 (70.0%) | 5 (50.0%) | ||||||
| Head Injury | 3 (30.0%) | 5 (50.0%) | ||||||
| TBI Source: | . | . | . | 1 | ||||
| Accident | 1 (20.0%) | 0 (0.00%) | ||||||
| Fall | 2 (40.0%) | 2 (100%) | ||||||
| Football | 1 (20.0%) | 0 (0.00%) | ||||||
| MVA | 1 (20.0%) | 0 (0.00%) | ||||||
| ADAS Total | 10.2 (3.96) | 8.74 (3.68) | 13.5 (4.01) | 8.07 (2.01) | 0.217 | 0.05 * | 0.95 | 0.007 ** |
| ADAS-Cog | 16.0 (6.48) | 12.7 (5.89) | 20.1 (6.34) | 11.9 (3.32) | 0.037 | 0.2 | 0.98 | 0.01 ** |
| MOCA | 23.9 (3.49) | 25.8 (2.92) | 21.5 (4.20) | 26.7 (1.58) | 0.022 | 0.15 | 0.87 | 0.004 ** |
| MMSE | 28.2 (2.06) | 28.9 (1.47) | 26.4 (3.03) | 28.6 (0.97) | 0.182 | 0.03 * | 0.96 | 0.047 * |
| GD Total | 1.84 (2.16) | 0.84 (0.96) | 2.50 (2.12) | 0.78 (0.83) | 0.017 | 0.66 | 1 | 0.12 |
| CDR Memory | 0.53 (0.12) | 0.00 (0.00) | 0.65 (0.58) | 0.00 (0.00) | 0 | 0.25 | 0.84 | <0.001 *** |
| CDR GLOBAL | 0.50 (0.00) | 0.00 (0.00) | 0.50 (0.33) | 0.00 (0.00) | 0 | 1 | 1 | <0.001 *** |
| Every Day Cognitive Assessment | ||||||||
| Memory | 2.35 (0.72) | 1.62 (0.50) | 2.20 (0.72) | 1.71 (0.57) | <0.001 | 0.89 | 0.98 | 0.33 |
| Language | 2.02 (0.70) | 1.45 (0.41) | 1.91 (0.71) | 1.32 (0.31) | <0.001 | 0.95 | 0.92 | 0.12 |
| Visual spatial | 1.49 (0.59) | 1.11 (0.20) | 1.50 (0.56) | 1.06 (0.12) | <0.001 | 1 | 0.99 | 0.13 |
| Planning | 1.57 (0.63) | 1.12 (0.26) | 1.54 (0.57) | 1.11 (0.23) | <0.001 | 0.99 | 1 | 0.21 |
| Organization | 1.75 (0.79) | 1.15 (0.24) | 1.58 (0.50) | 1.24 (0.52) | <0.001 | 0.84 | 0.97 | 0.56 |
| Divided Attention | 2.19 (0.86) | 1.45 (0.56) | 1.82 (0.75) | 1.25 (0.25) | <0.001 | 0.44 | 0.87 | 0.29 |
| Total | 1.90 (0.58) | 1.32 (0.26) | 1.76 (0.49) | 1.28 (0.28) | <0.001 | 0.81 | 0.99 | 0.09 |
| FAQ Total | 2.52 (3.80) | 0.14 (0.50) | 5.90 (5.99) | 0.25 (0.46) | 0.001 | 0.009 ** | 1 | 0.001 *** |
| Clock drawing tests | 4.52 (1.01) | 4.78 (0.51) | 4.30 (1.06) | 4.60 (0.70) | 0.394 | 0.86 | 0.92 | 0.84 |
| Clock copy test | 4.86 (0.50) | 4.82 (0.48) | 4.70 (0.67) | 4.50 (1.58) | 0.992 | 0.90 | 0.50 | 0.91 |
| Logic memory—Story | 11.3 (4.74) | 15.2 (3.81) | 10.1 (4.04) | 15.3 (2.54) | <0.001 | 0.83 | 1 | 0.03 |
| Logic memory- Delayed Recall | 9.40 (4.88) | 14.0 (3.37) | 6.80 (3.94) | 15.1 (3.51) | <0.001 | 0.27 | 0.88 | <0.001 *** |
| Category Fluency Test | 18.4 (5.19) | 21.7 (4.56) | 15.6 (5.08) | 20.2 (4.37) | 0.005 | 0.36 | 0.80 | 0.16 |
| Trail Making Test | ||||||||
| Part A—Time to Complete (sec) | 39.1 (25.3) | 31.0 (8.16) | 40.1 (12.1) | 31.1 (9.48) | 0.113 | 1 | 1 | 0.69 |
| Part A—Errors of Commission | 0.12 (0.39) | 0.12 (0.39) | 0.11 (0.33) | 0.00 (0.00) | 1 | 1 | 0.77 | 0.91 |
| Part A—Errors of Omission | 0.26 (1.84) | 0.00 (0.00) | 0.00 (0.00) | 0.00 (0.00) | 0.707 | 0.93 | 1 | 1 |
| Part B—Time to Complete (sec) | 103 (66.7) | 68.6 (27.8) | 155 (90.0) | 74.4 (27.4) | 0.011 | 0.04 * | 0.99 | 0.008 ** |
| Part B—Errors of Commission | 0.69 (1.06) | 0.37 (0.64) | 1.89 (1.76) | 0.20 (0.42) | 0.32 | 0.004 ** | 0.96 | 0.001 ** |
| Part B—Errors of Omission | 0.35 (1.55) | 0.02 (0.14) | 1.40 (2.32) | 0.00 (0.00) | 0.536 | 0.06 | 1 | 0.05 * |
| Trail Making Test | ||||||||
| Forgetting | 3.47 (5.75) | 3.88 (2.85) | 5.33 (2.50) | 4.29 (3.45) | 0.975 | 0.76 | 1 | 0.97 |
| Immediate | 36.6 (11.0) | 44.6 (10.5) | 28.2 (6.59) | 48.6 (7.52) | 0.005 | 0.26 | 0.79 | 0.004 *** |
| Learning | 5.18 (3.41) | 5.81 (2.26) | 2.83 (1.94) | 6.29 (1.80) | 0.737 | 0.22 | 0.97 | 0.11 |
| Percent Forgetting | 41.6 (80.8) | 36.5 (33.4) | 79.0 (23.6) | 36.6 (31.1) | 0.978 | 0.45 | 1 | 0.54 |
| Recognition Score | 11.2 (2.61) | 12.6 (3.12) | 9.40 (4.35) | 13.7 (0.87) | 0.072 | 0.29 | 0.77 | 0.01 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamed, A.Z.; Cumming, P.; Nasrallah, F.A.; Alzheimer’s Disease Neuroimaging Initiative. Escalation of Tau Accumulation after a Traumatic Brain Injury: Findings from Positron Emission Tomography. Brain Sci. 2022, 12, 876. https://doi.org/10.3390/brainsci12070876
Mohamed AZ, Cumming P, Nasrallah FA, Alzheimer’s Disease Neuroimaging Initiative. Escalation of Tau Accumulation after a Traumatic Brain Injury: Findings from Positron Emission Tomography. Brain Sciences. 2022; 12(7):876. https://doi.org/10.3390/brainsci12070876
Chicago/Turabian StyleMohamed, Abdalla Z., Paul Cumming, Fatima A. Nasrallah, and Alzheimer’s Disease Neuroimaging Initiative. 2022. "Escalation of Tau Accumulation after a Traumatic Brain Injury: Findings from Positron Emission Tomography" Brain Sciences 12, no. 7: 876. https://doi.org/10.3390/brainsci12070876