
New Insight in Hyperinsulinism/Hyperammonemia Syndrome by Magnetic Resonance Imaging and Spectroscopy


Abstract
:1. Introduction
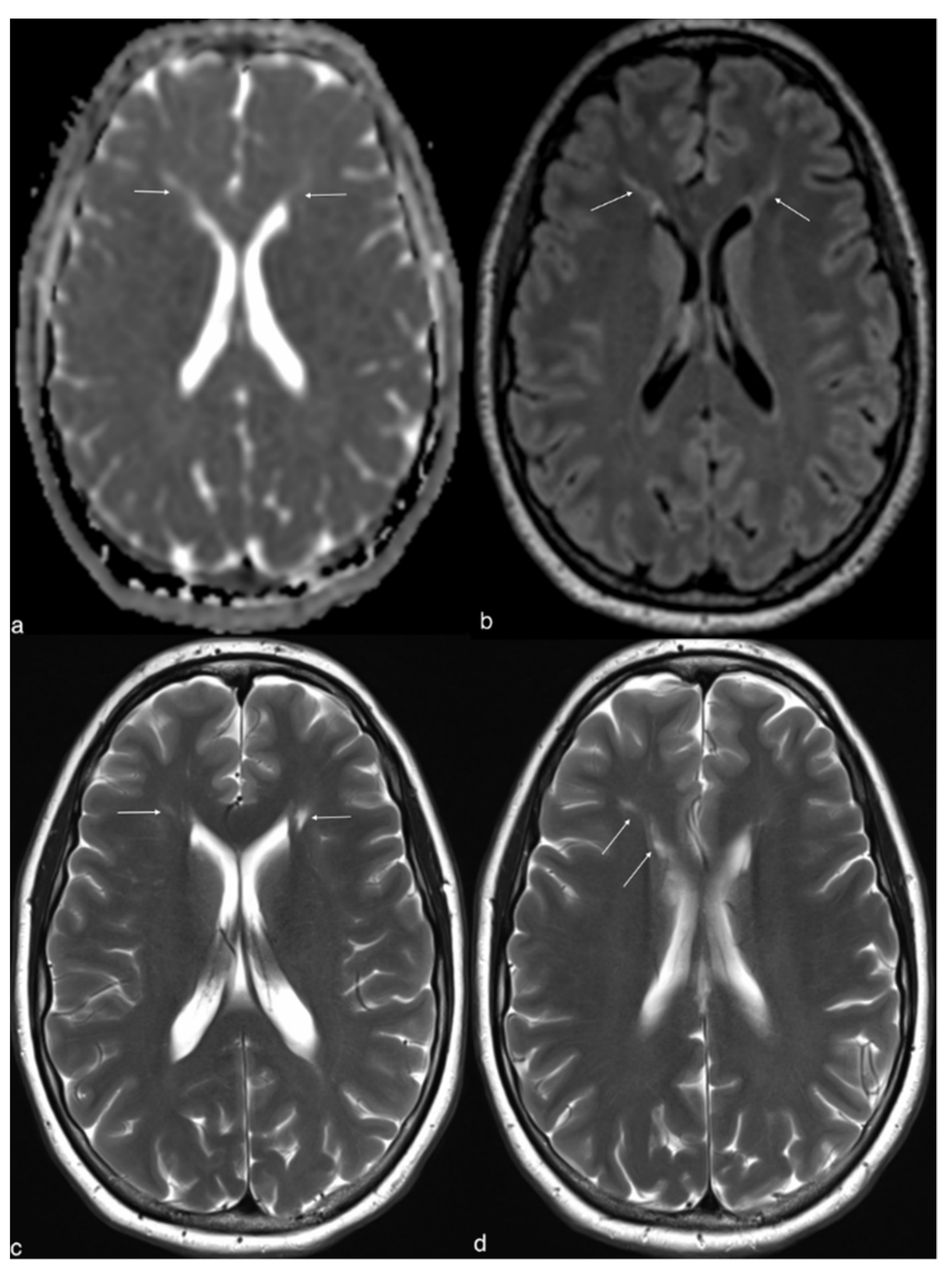
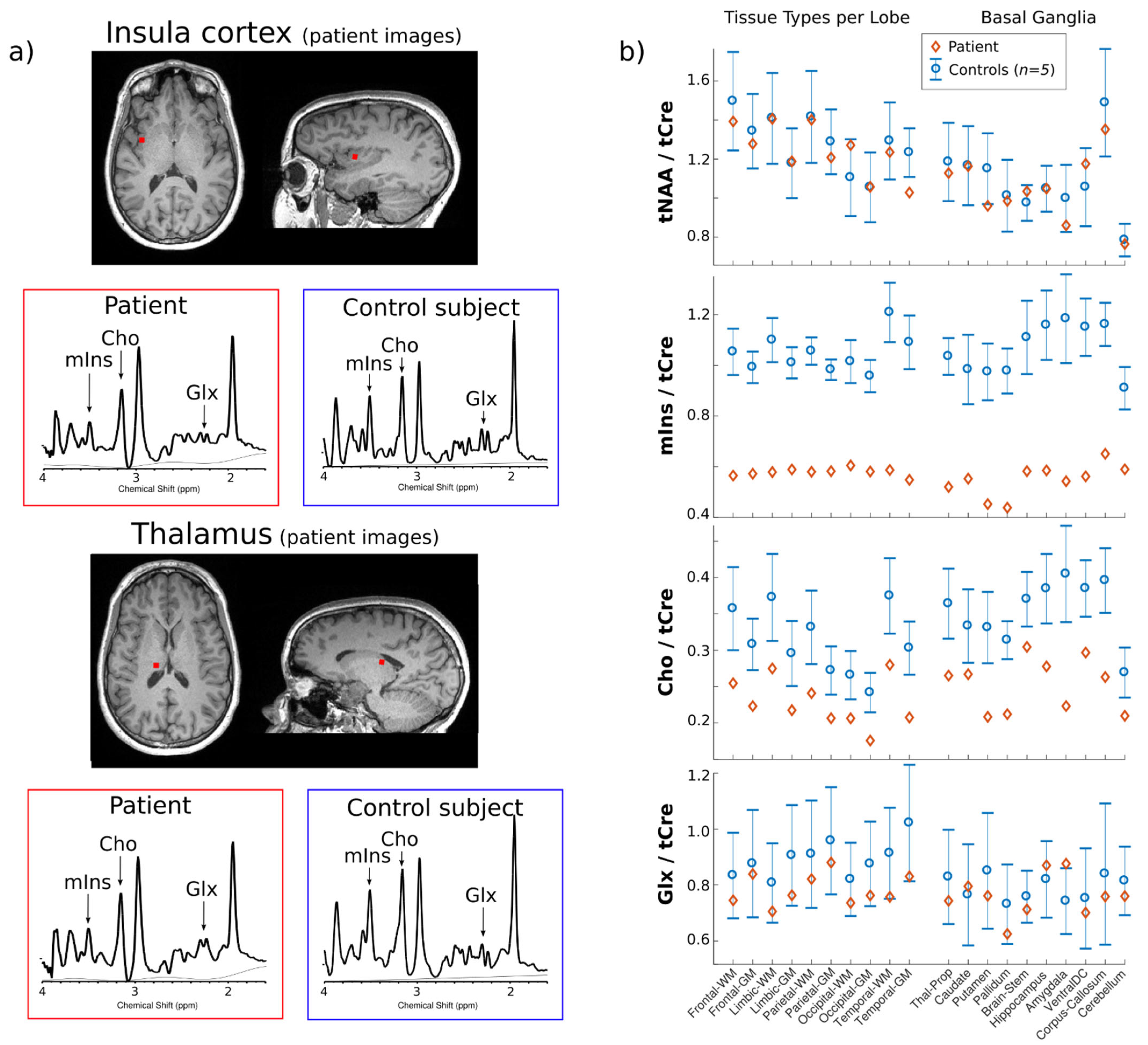
2. Case Report
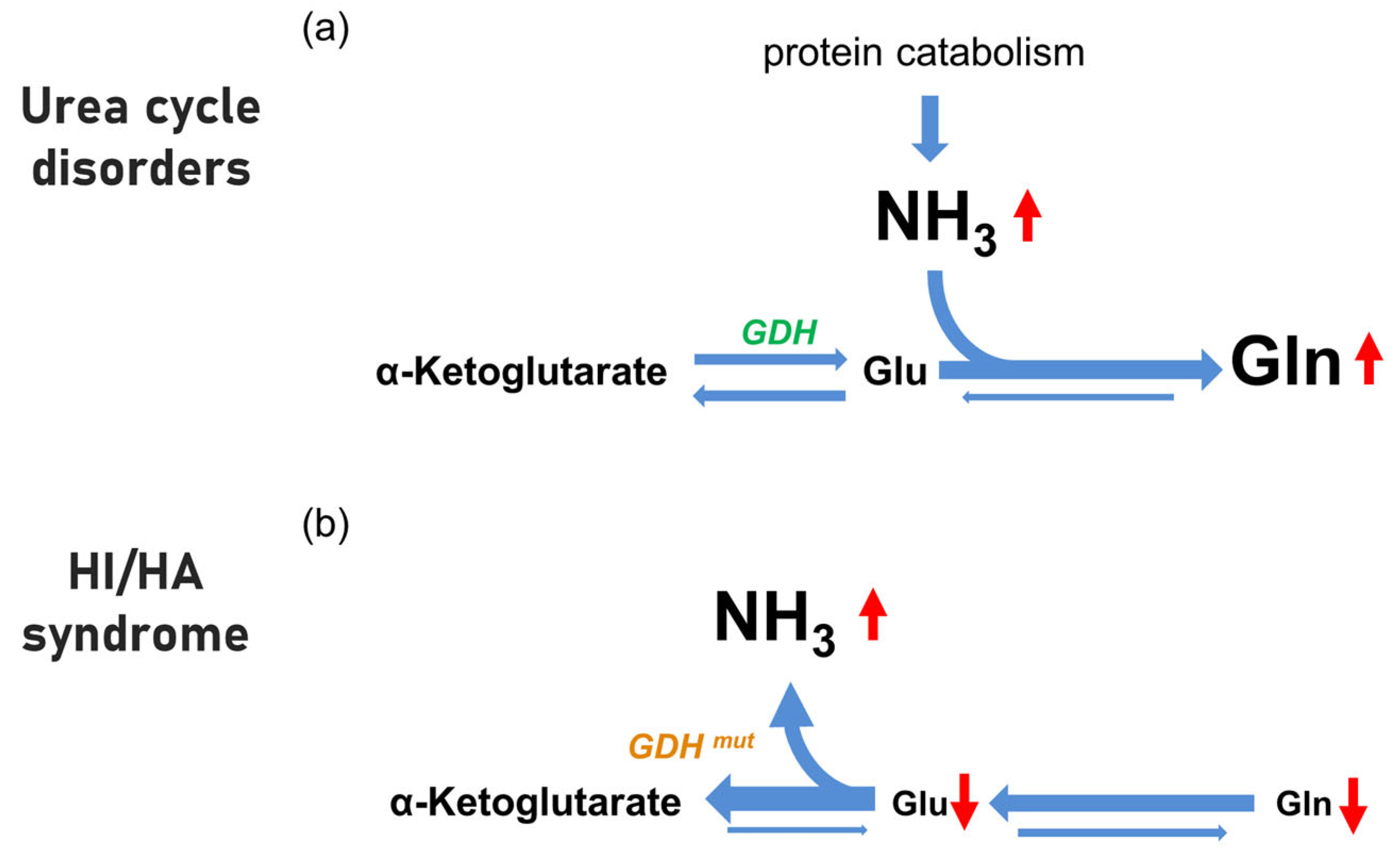
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cochrane, W.A.; Payne, W.W.; Simpkiss, M.J.; Woolf, L.I. Familial hypoglycemia precipitated by amino acids. J. Clin. Investig. 1956, 35, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanley, C.A.; Lieu, Y.K.; Hsu, B.Y.; Burlina, A.B.; Greenberg, C.R.; Hopwood, N.J.; Perlman, K.; Rich, B.H.; Zammarchi, E.; Poncz, M. Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N. Engl. J. Med. 1998, 338, 1352–1357. [Google Scholar] [CrossRef]
- Stanley, C.A. Regulation of glutamate metabolism and insulin secretion by glutamate dehydrogenase in hypoglycemic children. Am. J. Clin. Nutr. 2009, 90, 862S–866S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahi-Buisson, N.; Roze, E.; Dionisi, C.; Escande, F.; Valayannopoulos, V.; Feillet, F.; Heinrichs, C.; Chadefaux-Vekemans, B.; Dan, B.; de Lonlay, P. Neurological aspects of hyperinsulinism-hyperammonaemia syndrome. Dev. Med. Child Neurol. 2008, 50, 945–949. [Google Scholar] [CrossRef]
- Treberg, J.R.; Brosnan, M.E.; Watford, M.; Brosnan, J.T. On the reversibility of glutamate dehydrogenase and the source of hyperammonemia in the hyperinsulinism/hyperammonemia syndrome. Adv. Enzyme Regul. 2010, 50, 34–43. [Google Scholar] [CrossRef] [PubMed]
- De Lonlay, P.; Benelli, C.; Fouque, F.; Ganguly, A.; Aral, B.; Dionisi-Vici, C.; Touati, G.; Heinrichs, C.; Rabier, D.; Kamoun, P.; et al. Hyperinsulinism and hyperammonemia syndrome: Report of twelve unrelated patients. Pediatric Res. 2001, 50, 353–357. [Google Scholar] [CrossRef] [Green Version]
- Adeva, M.M.; Souto, G.; Blanco, N.; Donapetry, C. Ammonium metabolism in humans. Metab. Clin. Exp. 2012, 61, 1495–1511. [Google Scholar] [CrossRef]
- Haberle, J. Clinical and biochemical aspects of primary and secondary hyperammonemic disorders. Arch. Biochem. Biophys. 2013, 536, 101–108. [Google Scholar] [CrossRef]
- Gonzalez Melo, M.; Remacle, N.; Cudre-Cung, H.P.; Roux, C.; Poms, M.; Cudalbu, C.; Barroso, M.; Gersting, S.W.; Feichtinger, R.G.; Mayr, J.A.; et al. The first knock-in rat model for glutaric aciduria type I allows further insights into pathophysiology in brain and periphery. Mol. Genet. Metab. 2021, 133, 157–181. [Google Scholar] [CrossRef]
- Gonzalez Melo, M.; Fontana, A.O.; Viertl, D.; Allenbach, G.; Prior, J.O.; Rotman, S.; Feichtinger, R.G.; Mayr, J.A.; Costanzo, M.; Caterino, M.; et al. A knock-in rat model unravels acute and chronic renal toxicity in glutaric aciduria type I. Mol. Genet. Metab. 2021, 134, 287–300. [Google Scholar] [CrossRef]
- Oz, G.; Alger, J.R.; Barker, P.B.; Bartha, R.; Bizzi, A.; Boesch, C.; Bolan, P.J.; Brindle, K.M.; Cudalbu, C.; Dincer, A.; et al. Clinical proton MR spectroscopy in central nervous system disorders. Radiology 2014, 270, 658–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudkin, T.M.; Arnold, D.L. Proton magnetic resonance spectroscopy for the diagnosis and management of cerebral disorders. Arch. Neurol. 1999, 56, 919–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gropman, A.L.; Sailasuta, N.; Harris, K.C.; Abulseoud, O.; Ross, B.D. Ornithine transcarbamylase deficiency with persistent abnormality in cerebral glutamate metabolism in adults. Radiology 2009, 252, 833–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco-Colon, I.; Fricke, S.; VanMeter, J.; Gropman, A.L. Advances in urea cycle neuroimaging: Proceedings from the 4th International Symposium on urea cycle disorders, Barcelona, Spain, September 2013. Mol. Genet. Metab. 2014, 113, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Haberle, J.; Burlina, A.; Chakrapani, A.; Dixon, M.; Karall, D.; Lindner, M.; Mandel, H.; Martinelli, D.; Pintos-Morell, G.; Santer, R.; et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J. Inherit. Metab. Dis. 2019, 42, 1192–1230. [Google Scholar] [CrossRef]
- Luczkowska, K.; Stekelenburg, C.; Sloan-Bena, F.; Ranza, E.; Gastaldi, G.; Schwitzgebel, V.; Maechler, P. Hyperinsulinism associated with GLUD1 mutation: Allosteric regulation and functional characterization of p.G446V glutamate dehydrogenase. Hum. Genom. 2020, 14, 9. [Google Scholar] [CrossRef] [Green Version]
- Klauser, A.; Klauser, P.; Grouiller, F.; Courvoisier, S.; Lazeyras, F. Whole-brain high-resolution metabolite mapping with 3D compressed-sensing SENSE low-rank (1) H FID-MRSI. NMR Biomed. 2022, 35, e4615. [Google Scholar] [CrossRef]
- U-King-Im , J.M.; Yu, E.; Bartlett, E.; Soobrah, R.; Kucharczyk, W. Acute hyperammonemic encephalopathy in adults: Imaging findings. Am. J. Neuroradiol. 2011, 32, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Hershman, M.; Carmody, R.; Udayasankar, U.K. Case 252: Acute Hyperammonemic Encephalopathy Resulting from Late-Onset Ornithine Transcarbamylase Deficiency. Radiology 2018, 287, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Upadhyay, R.; Bleck, T.P.; Busl, K.M. Hyperammonemia: What Urea-lly Need to Know: Case Report of Severe Noncirrhotic Hyperammonemic Encephalopathy and Review of the Literature. Case Rep. Med. 2016, 2016, 8512721. [Google Scholar] [CrossRef] [Green Version]
- Brusilow, W.S.A. Saul Brusilow: Understanding and treating diseases of ammonia toxicity. Anal. Biochem. 2022, 636, 114478. [Google Scholar] [CrossRef] [PubMed]
- Haussinger, D.; Butz, M.; Schnitzler, A.; Gorg, B. Pathomechanisms in hepatic encephalopathy. Biol. Chem. 2021, 402, 1087–1102. [Google Scholar] [CrossRef] [PubMed]
- Lichter-Konecki, U. Profiling of astrocyte properties in the hyperammonaemic brain: Shedding new light on the pathophysiology of the brain damage in hyperammonaemia. J. Inherit. Metab. Dis. 2008, 31, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O.; Rackayova, V.; Pierzchala, K.; Grosse, J.; McLin, V.A.; Cudalbu, C. Longitudinal neurometabolic changes in the hippocampus of a rat model of chronic hepatic encephalopathy. J. Hepatol. 2019, 71, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Rackayova, V.; Braissant, O.; McLin, V.A.; Berset, C.; Lanz, B.; Cudalbu, C. 1H and 31P magnetic resonance spectroscopy in a rat model of chronic hepatic encephalopathy: In vivo longitudinal measurements of brain energy metabolism. Metab. Brain Dis. 2016, 31, 1303–1314. [Google Scholar] [CrossRef]
- Wong, Y.C.; Au, W.L.; Xu, M.; Ye, J.; Lim, C.C. Magnetic resonance spectroscopy in adult-onset citrullinemia: Elevated glutamine levels in comatose patients. Arch. Neurol. 2007, 64, 1034–1037. [Google Scholar] [CrossRef] [Green Version]
- Balata, S.; Olde Damink, S.W.; Ferguson, K.; Marshall, I.; Hayes, P.C.; Deutz, N.E.; Williams, R.; Wardlaw, J.; Jalan, R. Induced hyperammonemia alters neuropsychology, brain MR spectroscopy and magnetization transfer in cirrhosis. Hepatology 2003, 37, 931–939. [Google Scholar] [CrossRef]
- Shawcross, D.L.; Balata, S.; Olde Damink, S.W.; Hayes, P.C.; Wardlaw, J.; Marshall, I.; Deutz, N.E.; Williams, R.; Jalan, R. Low myo-inositol and high glutamine levels in brain are associated with neuropsychological deterioration after induced hyperammonemia. Am. J. Physiol. Gastrointest Liver Physiol. 2004, 287, G503–G509. [Google Scholar] [CrossRef] [Green Version]
- Sen, K.; Whitehead, M.T.; Gropman, A.L. Multimodal imaging in urea cycle-related neurological disease—What can imaging after hyperammonemia teach us? Transl. Sci. Rare Dis. 2020, 5, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Sen, K.; Anderson, A.A.; Whitehead, M.T.; Gropman, A.L. Review of Multi-Modal Imaging in Urea Cycle Disorders: The Old, the New, the Borrowed, and the Blue. Front. Neurol. 2021, 12, 632307. [Google Scholar] [CrossRef]
- Roze, E.; Azuar, C.; Menuel, C.; Haberle, J.; Guillevin, R. Usefulness of magnetic resonance spectroscopy in urea cycle disorders. Pediatric Neurol. 2007, 37, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.G.; Yoo, H.W. Localized proton MR spectroscopy in infants with urea cycle defect. Am. J. Neuroradiol. 2001, 22, 834–837. [Google Scholar] [PubMed]
- Chavarria, L.; Alonso, J.; Garcia-Martinez, R.; Simon-Talero, M.; Ventura-Cots, M.; Ramirez, C.; Torrens, M.; Vargas, V.; Rovira, A.; Cordoba, J. Brain magnetic resonance spectroscopy in episodic hepatic encephalopathy. J. Cereb. Blood Flow Metab. 2013, 33, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Spahr, L.; Vingerhoets, F.; Lazeyras, F.; Delavelle, J.; DuPasquier, R.; Giostra, E.; Mentha, G.; Terrier, F.; Hadengue, A. Magnetic resonance imaging and proton spectroscopic alterations correlate with parkinsonian signs in patients with cirrhosis. Gastroenterology 2000, 119, 774–781. [Google Scholar] [CrossRef]
- Ziyeh, S.; Thiel, T.; Spreer, J.; Klisch, J.; Schumacher, M. Valproate-induced encephalopathy: Assessment with MR imaging and 1H MR spectroscopy. Epilepsia 2002, 43, 1101–1105. [Google Scholar] [CrossRef] [Green Version]
- Gropman, A.L.; Prust, M.; Breeden, A.; Fricke, S.; VanMeter, J. Urea cycle defects and hyperammonemia: Effects on functional imaging. Metab. Brain Dis. 2013, 28, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Gropman, A.L.; Seltzer, R.R.; Yudkoff, M.; Sawyer, A.; VanMeter, J.; Fricke, S.T. 1H MRS allows brain phenotype differentiation in sisters with late onset ornithine transcarbamylase deficiency (OTCD) and discordant clinical presentations. Mol. Genet. Metab. 2008, 94, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Karaca, M.; Martin-Levilain, J.; Grimaldi, M.; Li, L.; Dizin, E.; Emre, Y.; Maechler, P. Liver Glutamate Dehydrogenase Controls Whole-Body Energy Partitioning Through Amino Acid-Derived Gluconeogenesis and Ammonia Homeostasis. Diabetes 2018, 67, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Stanley, C.A. Hyperinsulinism/hyperammonemia syndrome: Insights into the regulatory role of glutamate dehydrogenase in ammonia metabolism. Mol. Genet. Metab. 2004, 81 (Suppl. 1), S45–S51. [Google Scholar] [CrossRef]
- Palladino, A.A.; Stanley, C.A. The hyperinsulinism/hyperammonemia syndrome. Rev. Endocr. Metab. Disord. 2010, 11, 171–178. [Google Scholar] [CrossRef]
- El-Gharbawy, A.H. Hyperinsulinism/Hyperammonemia Syndrome: A synopsis. Mol. Genet. Metab. 2005, 84, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.; Jeitner, T.M. Central Role of Glutamate Metabolism in the Maintenance of Nitrogen Homeostasis in Normal and Hyperammonemic Brain. Biomolecules 2016, 6, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brusilow, S.W.; Koehler, R.C.; Traystman, R.J.; Cooper, A.J. Astrocyte glutamine synthetase: Importance in hyperammonemic syndromes and potential target for therapy. Neurotherapeutics 2010, 7, 452–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Disease | MRI Findings | MRS Findings | Ref. |
|---|---|---|---|
| OTC | NA | Elevated glutamine in posterior cingulate gray matter, parietal and frontal WM. Reduction in myoinositol and choline in parietal and frontal white matter, thalamus and posterior cingulate gray matter | [36] |
| OTC | Increased signal on T2-weighted and diffusion-weighted images in the basal ganglia, claustrum, frontoparietal WM, pontine tegmentum, and left brachium pontis | Elevated glutamine. Reduction in myoinositol and choline | [29] |
| OTC | No structural abnormalities in gray or white matter | Elevated glutamine and glutamate. Reduction in myoinositol and choline | [37] |
| Type II citrullinemia | Bilateral, non-enhancing abnormalities of the globus pallidus, insular cortex, and cingulate gyrus on T2-weighted and diffusion-weighted MRI | Elevated glutamine and glutamate. Reduction in myoinositol and choline | [26] |
| Hepatic encephalopathy | Elevated apparent diffusion coefficient values in the corticospinal tract and parietal white matter | Elevated glutamine, reduced myoinositol and choline and non-significant difference in glutamate and N-acetylaspartate | [33] |
| Hepatic encephalopathy | NA | Elevated glutamine and glutamate. Reduction in myoinositol and choline | [27] |
| Hepatic encephalopathy | Occipital white matter and basal ganglia had significant hyperintensity | Elevated glutamine and glutamate. Reduction in myoinositol and choline | [34] |
| Valproate-induced encephalopathy | Metabolic-toxic lesion pattern with bilateral T2-hyperintense lesion in the cerebellar white matter and in the globus pallidus | Elevated glutamine and glutamate. Reduction in myoinositol and choline | [35] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gariani, K.; Klauser, A.; Vargas, M.I.; Lazeyras, F.; Tran, C. New Insight in Hyperinsulinism/Hyperammonemia Syndrome by Magnetic Resonance Imaging and Spectroscopy. Brain Sci. 2022, 12, 389. https://doi.org/10.3390/brainsci12030389
Gariani K, Klauser A, Vargas MI, Lazeyras F, Tran C. New Insight in Hyperinsulinism/Hyperammonemia Syndrome by Magnetic Resonance Imaging and Spectroscopy. Brain Sciences. 2022; 12(3):389. https://doi.org/10.3390/brainsci12030389
Chicago/Turabian StyleGariani, Karim, Antoine Klauser, Maria Isabel Vargas, François Lazeyras, and Christel Tran. 2022. "New Insight in Hyperinsulinism/Hyperammonemia Syndrome by Magnetic Resonance Imaging and Spectroscopy" Brain Sciences 12, no. 3: 389. https://doi.org/10.3390/brainsci12030389