Cytokines and Neurodegeneration in Epileptogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Animals
2.2. Induction of Kainic-Acid-Induced Epilepsy Model
2.3. Extraction of RNA and Proteins
2.4. Analysis of Gene Expression at the RNA Level by Real-Time PCR
2.5. Analysis of Gene Expression at the Protein Level by ELISA
2.6. Quantitative Assessment of the Neurodegeneration Level Using the ELISA Method
2.7. Detection of Neurodegeneration Localization Using Fluoro-Jade C Dye
3. Results
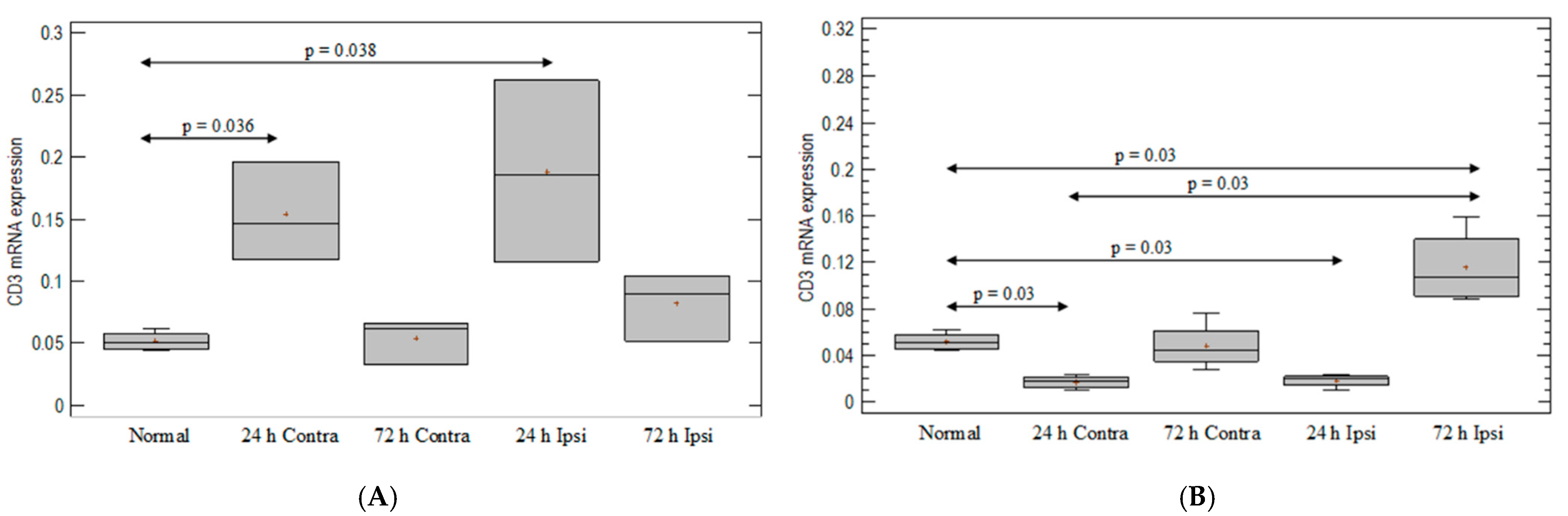
3.1. T-Cell-Marker Expression in the Brain in Animal Model of Epilepsy
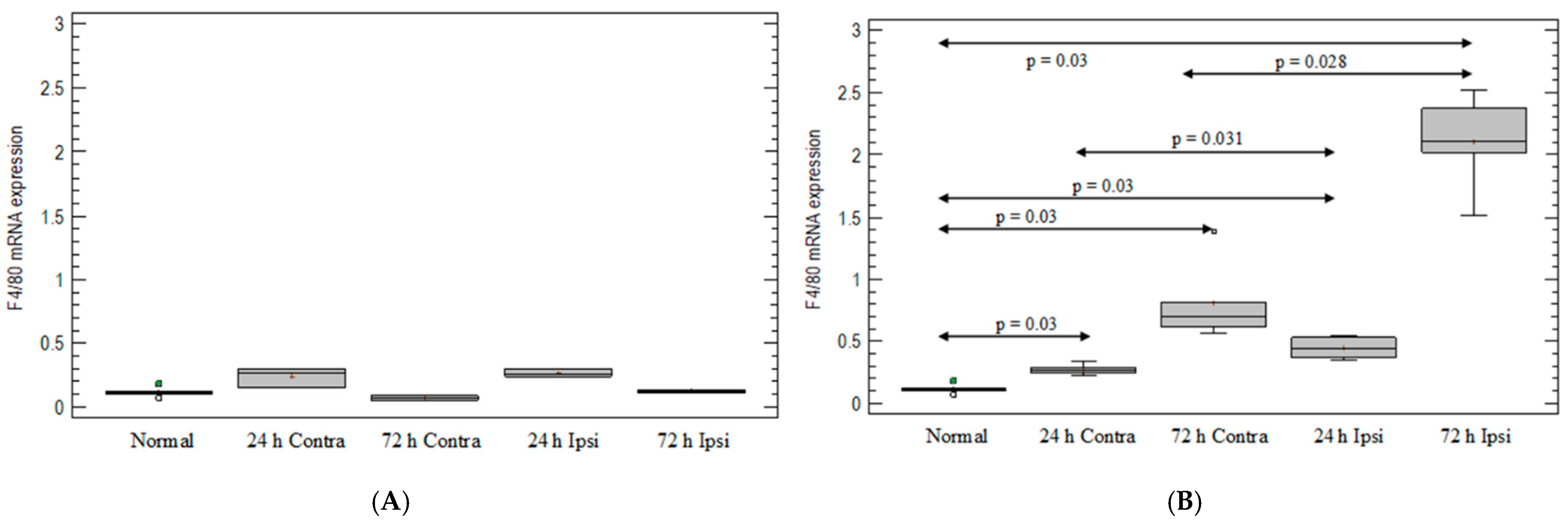
3.2. Monocyte-/Macrophage-Marker Expression in the Brain in Animal Model of Epilepsy
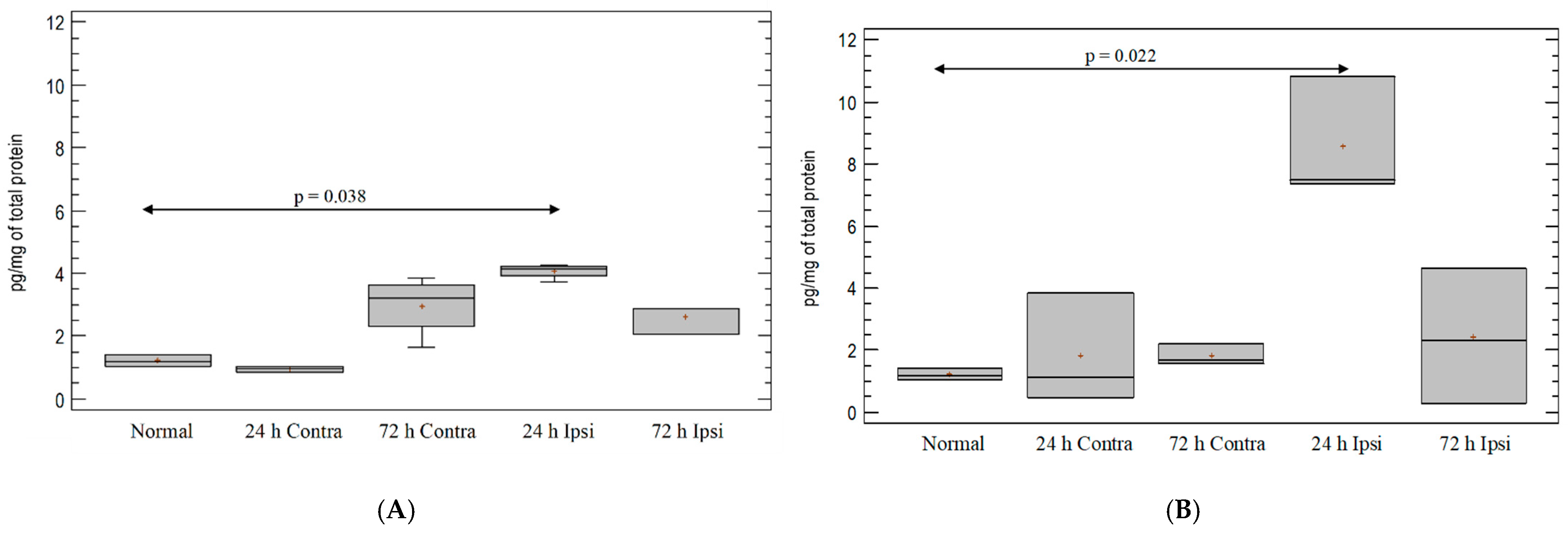
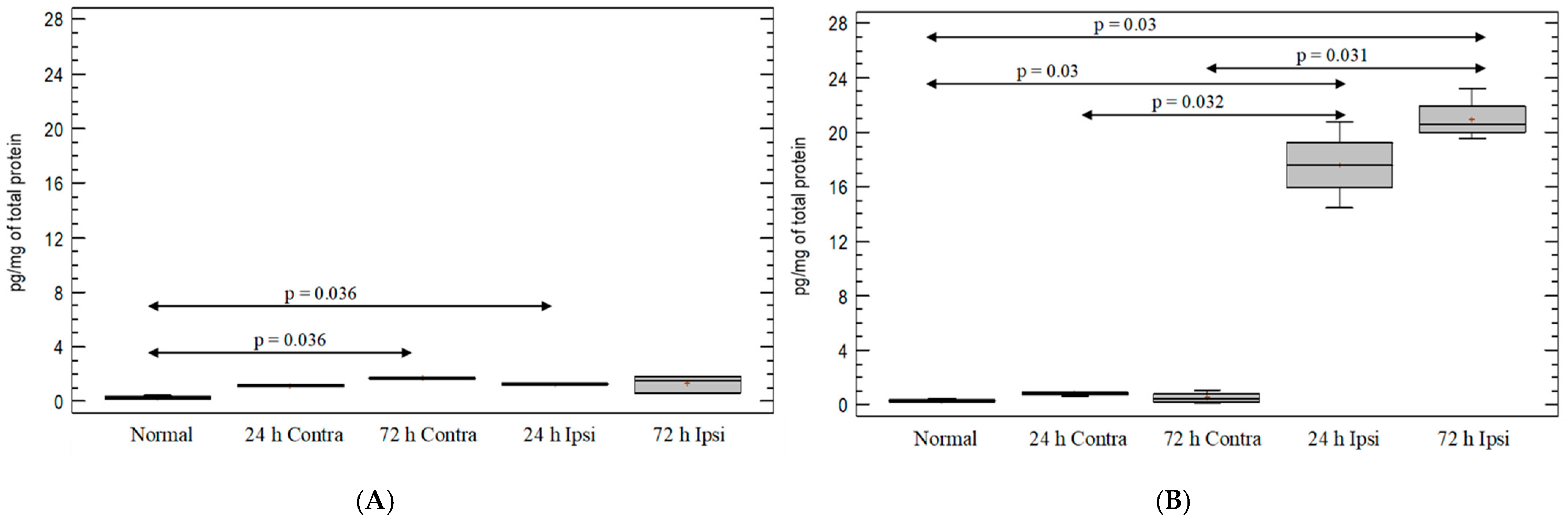
3.3. Cytokine IL-1β Expression in the Brain in Experimental Model of Epilepsy
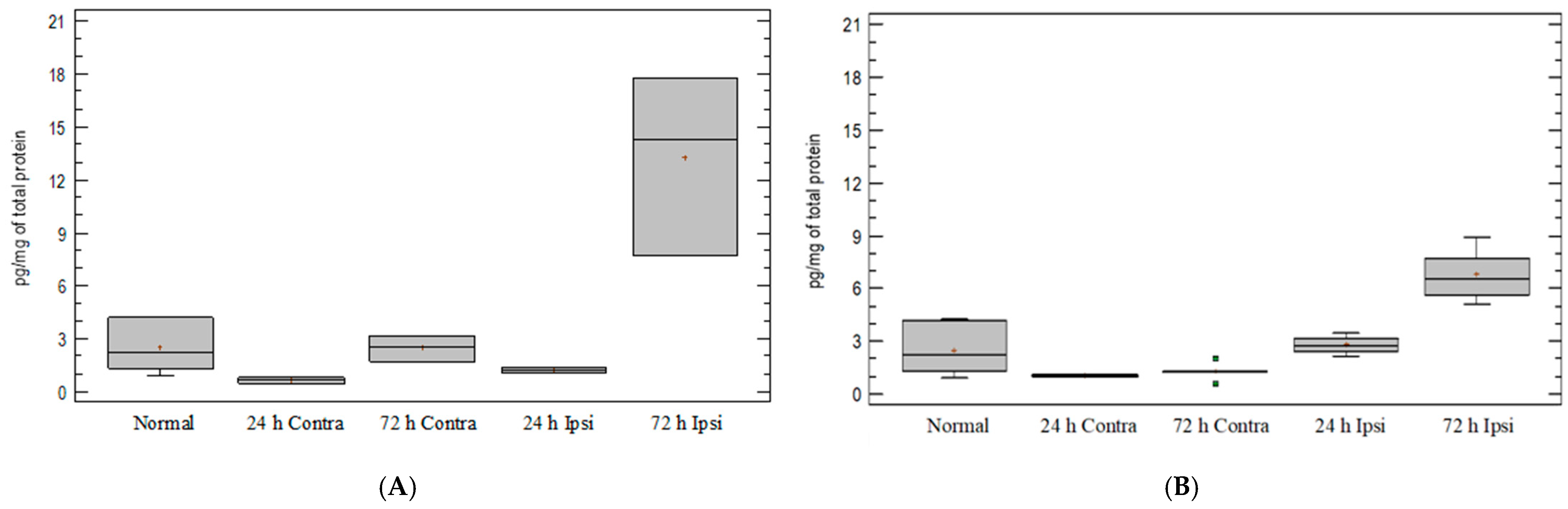
3.4. Expression of Chemokine CCL5 in Experimental Model of Epilepsy
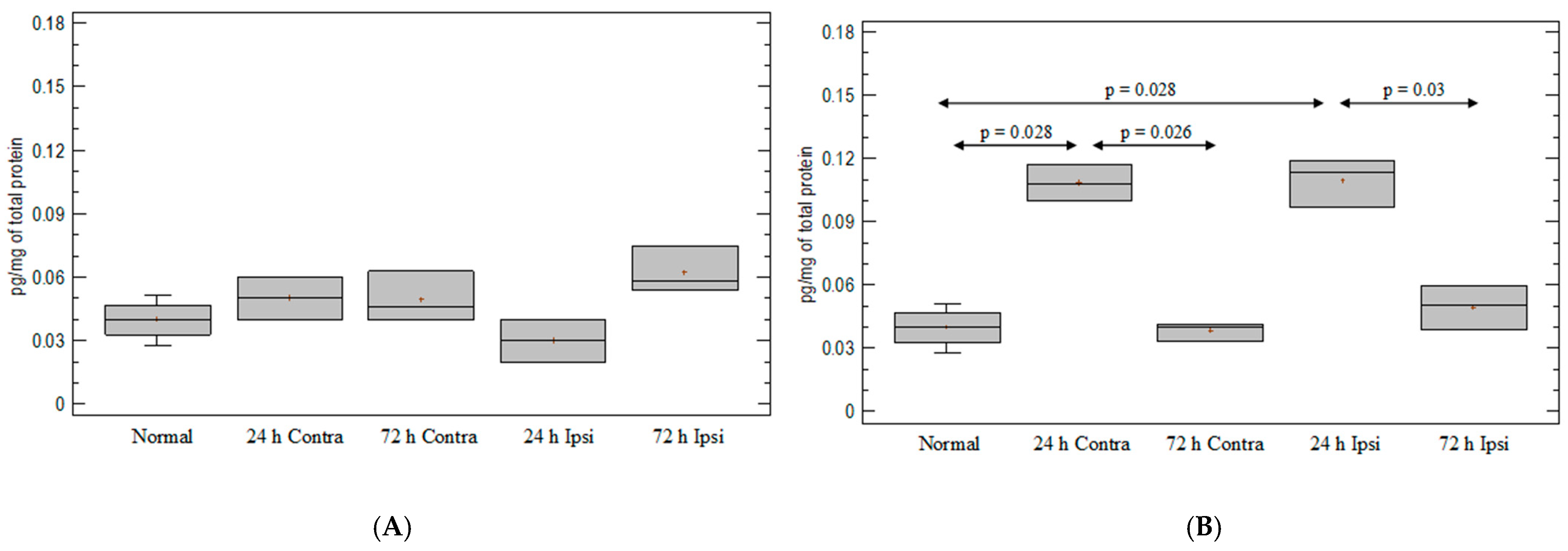
3.5. Expression of Chemokine CXCL2 in Experimental Model of Epilepsy
3.6. Expression of Chemokine CXCL12 in Experimental Model of Epilepsy
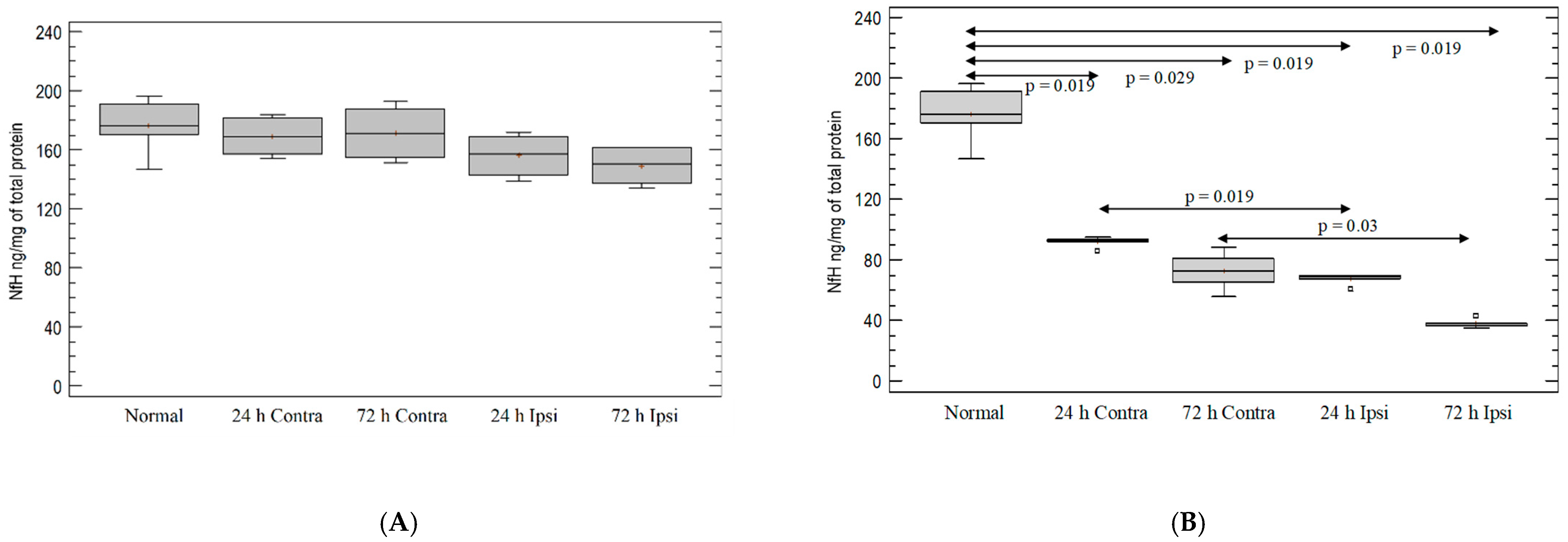
3.7. Analysis of Intensity of Neurodegeneration in KA-Induced Experimental Epilepsy Model
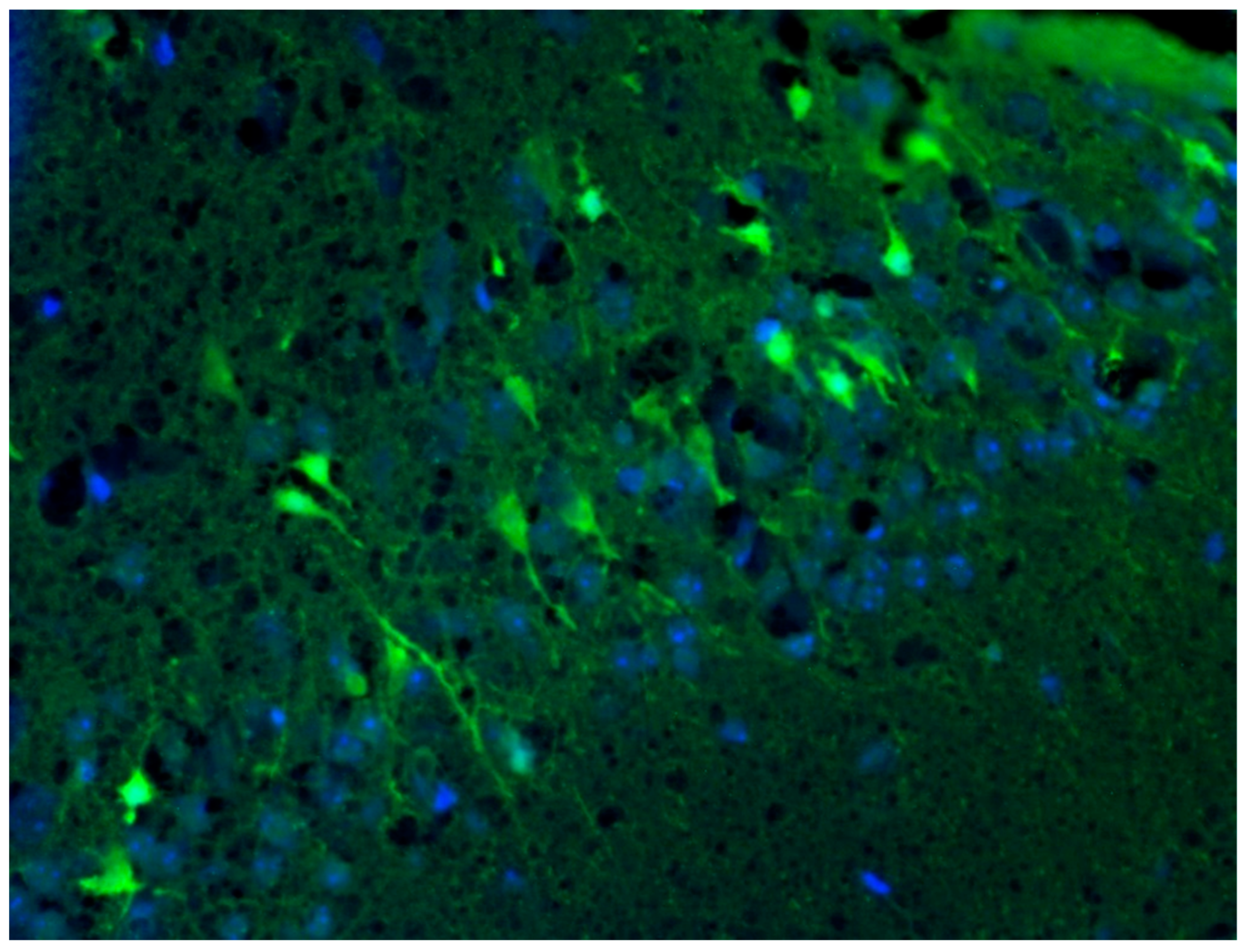
3.8. Analysis of Localization of Neurodegeneration in KA-Induced Experimental Epilepsy Model
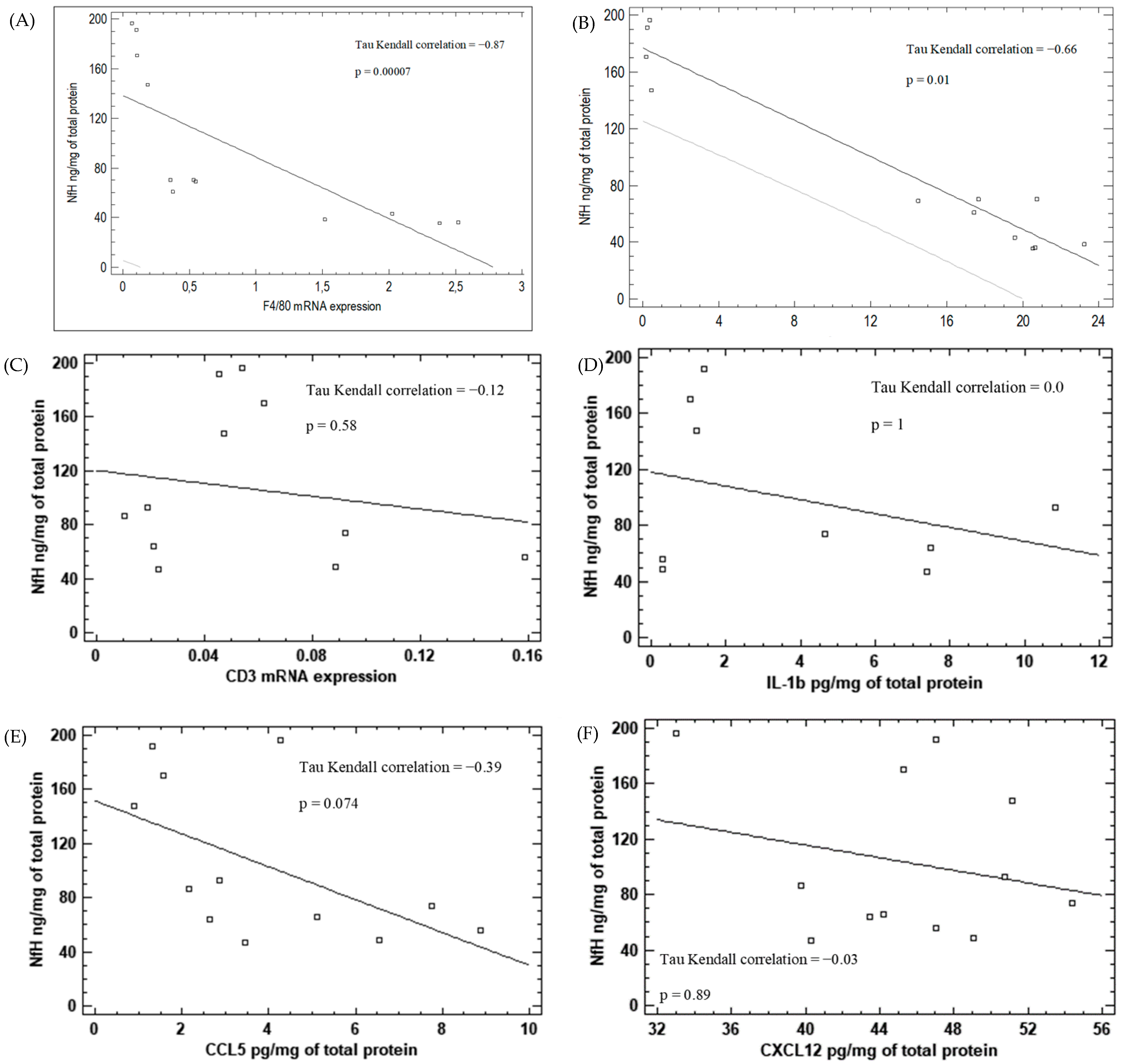
3.9. Correlation between Inflammatory Markers and Neurodegeneration in KA-Induced Epilepsy Model
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Primers 2018, 4, 18024. [Google Scholar] [CrossRef] [PubMed]
- Rakhade, S.N.; Jensen, F.E. Epileptogenesis in the immature brain: Emerging mechanisms. Nat. Rev. Neurol. 2009, 5, 380–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laxer, K.D.; Trinka, E.; Hirsch, L.J.; Cendes, F.; Langfitt, J.; Delanty, N.; Resnick, T.; Benbadis, S.R. The consequences of refractory epilepsy and its treatment. Epilepsy Behav. 2014, 37, 59–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, S.T. Epilepsy after brain insult: Targeting epileptogenesis. Neurology 2002, 59, S21–S26. [Google Scholar] [CrossRef] [PubMed]
- Plata-Salamán, C.R.; Ilyin, S.E.; Turrin, N.P.; Gayle, D.; Flynn, M.C.; Romanovitch, A.E.; Kelly, M.E.; Bureau, Y.; Anisman, H.; McIntyre, D.C. Kindling modulates the IL-1β system, TNF-α, TGF-β1, and neuropeptide mRNAs in specific brain regions. Mol. Brain Res. 2000, 75, 248–258. [Google Scholar] [CrossRef]
- Ravizza, T.; Vezzani, A. Status epilepticus induces time-dependent neuronal and astrocytic expression of interleukin-1 receptor type I in the rat limbic system. Neuroscience 2006, 137, 301–308. [Google Scholar] [CrossRef]
- Lehtimäki, K.A.; Keränen, T.; Palmio, J.; Mäkinen, R.; Hurme, M.; Honkaniemi, J.; Peltola, J. Increased plasma levels of cytokines after seizures in localization-related epilepsy. Acta Neurol. Scand. 2007, 116, 226–230. [Google Scholar] [CrossRef]
- Fabene, P.F.; Bramanti, P.; Constantin, G. The emerging role for chemokines in epilepsy. J. Neuroimmunol. 2010, 224, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; Serhan, C.N. Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors. Mol. Asp. Med. 2017, 58, 114–129. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Granata, T. Brain Inflammation in Epilepsy: Experimental and Clinical Evidence. Epilepsia 2005, 46, 1724–1743. [Google Scholar] [CrossRef]
- Musto, A.E.; Gjorstrup, P.; Bazan, N.G. The omega-3 fatty acid-derived neuroprotectin D1 limits hippocampal hyperexcitability and seizure susceptibility in kindling epileptogenesis. Epilepsia 2011, 52, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Musto, A.E.; Rosencrans, R.F.; Walker, C.P.; Bhattacharjee, S.; Raulji, C.M.; Belayev, L.; Fang, Z.; Gordon, W.C.; Bazan, N.G. Dysfunctional epileptic neuronal circuits and dysmorphic dendritic spines are mitigated by platelet-activating factor receptor antagonism. Sci. Rep. 2016, 6, 30298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Nordli, D.R.; Alden, T.D.; DiPatri, A.; Laux, L.; Kelley, K.; Rosenow, J.; Schuele, S.U.; Rajaram, V.; Koh, S. Cellular injury and neuroinflammation in children with chronic intractable epilepsy. J. Neuroinflamm. 2009, 6, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borges, K. Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exp. Neurol. 2003, 182, 21–34. [Google Scholar] [CrossRef]
- Balosso, S.; Ravizza, T.; Perego, C.; Peschon, J.; Campbell, I.L.; de Simoni, M.G.; Vezzani, A. Tumor necrosis factor-α inhibits seizures in mice via p75 receptors. Ann. Neurol. 2005, 57, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, S.C.; Campbell, I.L.; Henriksen, S.J. Site-specific hippocampal pathophysiology due to cerebral overexpression of interleukin-6 in transgenic mice. Brain Res. 1994, 652, 149–153. [Google Scholar] [CrossRef]
- Li, G.; Bauer, S.; Nowak, M.; Norwood, B.; Tackenberg, B.; Rosenow, F.; Knake, S.; Oertel, W.H.; Hamer, H.M. Cytokines and epilepsy. Seizure 2011, 20, 249–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olney, J.W.; Rhee, V.; Ho, O.L. Kainic acid: A powerful neurotoxic analogue of glutamate. Brain Res. 1974, 77, 507–512. [Google Scholar] [CrossRef]
- Coyle, J.T. Kainic Acid: Insights into Excitatory Mechanisms Causing Selective Neuronal Degeneration. In Novartis Foundation Symposia; Wiley: Hoboken, NJ, USA, 2007; pp. 186–203. [Google Scholar] [CrossRef]
- Anzai, T.; Tsuzuki, K.; Yamada, N.; Hayashi, T.; Iwakuma, M.; Inada, K.; Kameyama, K.; Hoka, S.; Saji, M. Overexpression of Ca2+-permeable AMPA receptor promotes delayed cell death of hippocampal CA1 neurons following transient forebrain ischemia. Neurosci. Res. 2003, 46, 41–51. [Google Scholar] [CrossRef]
- Liu, S.; Lau, L.; Wei, J.; Zhu, D.; Zou, S.; Sun, H.-S.; Fu, Y.; Liu, F.; Lu, Y. Expression of Ca2+-Permeable AMPA Receptor Channels Primes Cell Death in Transient Forebrain Ischemia. Neuron 2004, 43, 43–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, T.-H.; Hwang, H.-M.; Chen, J.-J.; Wu, T.; Li, A.H.; Wang, H.-L. Glutamate transporter function of rat hippocampal astrocytes is impaired following the global ischemia. Neurobiol. Dis. 2005, 18, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Schmued, L.C.; Albertson, C.; Slikker, W. Fluoro-Jade: A novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997, 751, 37–46. [Google Scholar] [CrossRef]
- Tanaka, K.; Jimenez-Mateos, E.M.; Matsushima, S.; Taki, W.; Henshall, D.C. Hippocampal damage after intra-amygdala kainic acid-induced status epilepticus and seizure preconditioning-mediated neuroprotection in SJL mice. Epilepsy Res. 2010, 88, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Glabinski, A.R.; Tani, M.; Tuohy, V.K.; Ransohoff, R.M. [13] Murine experimental autoimmune encephalomyelitis: A model of immune-mediated inflammation and multiple sclerosis. Methods Enzymol. 1997, 288, 182–190. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Vezzani, A.; Lang, B.; Aronica, E. Immunity and Inflammation in Epilepsy. Cold Spring Harb. Perspect. Med. 2016, 6, a022699. [Google Scholar] [CrossRef] [Green Version]
- Vezzani, A.; Maroso, M.; Balosso, S.; Sanchez, M.-A.; Bartfai, T. IL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav. Immun. 2011, 25, 1281–1289. [Google Scholar] [CrossRef]
- Choi, J.; Koh, S. Role of Brain Inflammation in Epileptogenesis. Yonsei Med. J. 2008, 49, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, J.; Becker, A.J.; Elyaman, W.; Peltola, J.; Rüegg, S.; Titulaer, M.J.; Varley, J.A.; Beghi, E. Innate and adaptive immunity in human epilepsies. Epilepsia 2017, 58, 57–68. [Google Scholar] [CrossRef]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, A.; Zurolo, E.; Spliet, W.G.M.; van Rijen, P.C.; Baayen, J.C.; Gorter, J.A.; Aronica, E. Evaluation of the innate and adaptive immunity in type I and type II focal cortical dysplasias. Epilepsia 2010, 51, 1763–1773. [Google Scholar] [CrossRef]
- Arena, A.; Zimmer, T.S.; van Scheppingen, J.; Korotkov, A.; Anink, J.J.; Mühlebner, A.; Jansen, F.E.; van Hecke, W.; Spliet, W.G.; van Rijen, P.C.; et al. Oxidative stress and inflammation in a spectrum of epileptogenic cortical malformations: Molecular insights into their interdependence. Brain Pathol. 2019, 29, 351–365. [Google Scholar] [CrossRef]
- Pitkänen, A.; Löscher, W.; Vezzani, A.; Becker, A.J.; Simonato, M.; Lukasiuk, K.; Gröhn, O.; Bankstahl, J.P.; Friedman, A.; Aronica, E.; et al. Advances in the development of biomarkers for epilepsy. Lancet Neurol. 2016, 15, 843–856. [Google Scholar] [CrossRef]
- Arisi, G.M. Nervous and immune systems signals and connections: Cytokines in hippocampus physiology and pathology. Epilepsy Behav. 2014, 38, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Kleen, J.K.; Holmes, G.L. Brain inflammation initiates seizures. Nat. Med. 2008, 14, 1309–1310. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-S.; Mane, S.; Eid, T.; Zhao, H.; Lin, A.; Guan, Z.; Kim, J.H.; Schweitzer, J.; King-Stevens, D.; Weber, P.; et al. Gene Expression in Temporal Lobe Epilepsy is Consistent with Increased Release of Glutamate by Astrocytes. Mol. Med. 2007, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gorter, J.A.; van Vliet, E.A.; Aronica, E.; Breit, T.; Rauwerda, H.; Lopes da Silva, F.H.; Wadman, W.J. Potential New Antiepileptogenic Targets Indicated by Microarray Analysis in a Rat Model for Temporal Lobe Epilepsy. J. Neurosci. 2006, 26, 11083–11110. [Google Scholar] [CrossRef]
- Ekdahl, C.T.; Claasen, J.-H.; Bonde, S.; Kokaia, Z.; Lindvall, O. Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13632–13637. [Google Scholar] [CrossRef] [Green Version]
- Bonde, S.; Ekdahl, C.T.; Lindvall, O. Long-term neuronal replacement in adult rat hippocampus after status epilepticus despite chronic inflammation. Eur. J. Neurosci. 2006, 23, 965–974. [Google Scholar] [CrossRef]
- Srivastava, A.; Dixit, A.B.; Banerjee, J.; Tripathi, M.; Sarat Chandra, P. Role of inflammation and its miRNA based regulation in epilepsy: Implications for therapy. Clin. Chim. Acta 2016, 452, 1–9. [Google Scholar] [CrossRef]
- Commins, S.P.; Borish, L.; Steinke, J.W. Immunologic messenger molecules: Cytokines, interferons, and chemokines. J. Allergy Clin. Immunol. 2010, 125, S53–S72. [Google Scholar] [CrossRef]
- Lehtimäki, K.A.; Peltola, J.; Koskikallio, E.; Keränen, T.; Honkaniemi, J. Expression of cytokines and cytokine receptors in the rat brain after kainic acid-induced seizures. Mol. Brain Res. 2003, 110, 253–260. [Google Scholar] [CrossRef]
- Vezzani, A.; Moneta, D.; Richichi, C.; Aliprandi, M.; Burrows, S.J.; Ravizza, T.; Perego, C.; De Simoni, M.G. Functional Role of Inflammatory Cytokines and Antiinflammatory Molecules in Seizures and Epileptogenesis. Epilepsia 2002, 43, 30–35. [Google Scholar] [CrossRef]
- De Simoni, M.G.; Perego, C.; Ravizza, T.; Moneta, D.; Conti, M.; Marchesi, F.; de Luigi, A.; Garattini, S.; Vezzani, A. Inflammatory cytokines and related genes are induced in the rat hippocampus by limbic status epilepticus. Eur. J. Neurosci. 2000, 12, 2623–2633. [Google Scholar] [CrossRef]
- Min, K.; Jou, I.; Joe, E. Plasminogen-induced IL-1β and TNF-α production in microglia is regulated by reactive oxygen species. Biochem. Biophys. Res. Commun. 2003, 312, 969–974. [Google Scholar] [CrossRef]
- Viviani, B.; Bartesaghi, S.; Gardoni, F.; Vezzani, A.; Behrens, M.M.; Bartfai, T.; Binaglia, M.; Corsini, E.; di Luca, M.; Galli, C.L.; et al. Interleukin-1β Enhances NMDA Receptor-Mediated Intracellular Calcium Increase through Activation of the Src Family of Kinases. J. Neurosci. 2003, 23, 8692–8700. [Google Scholar] [CrossRef]
- Wisse, B.E.; Ogimoto, K.; Schwartz, M.W. Role of hypothalamic interleukin-1β (IL-1β) in regulation of energy homeostasis by melanocortins. Peptides 2006, 27, 265–273. [Google Scholar] [CrossRef]
- Alyu, F.; Dikmen, M. Inflammatory aspects of epileptogenesis: Contribution of molecular inflammatory mechanisms. Acta Neuropsychiatr. 2017, 29, 1–16. [Google Scholar] [CrossRef]
- Han, T.; Qin, Y.; Mou, C.; Wang, M.; Jiang, M.; Liu, B. Seizure induced synaptic plasticity alteration in hippocampus is mediated by IL-1β receptor through PI3K/Akt pathway. Am. J. Transl. Res. 2016, 8, 4499–4509. [Google Scholar]
- Kothur, K.; Bandodkar, S.; Wienholt, L.; Chu, S.; Pope, A.; Gill, D.; Dale, R.C. Etiology is the key determinant of neuroinflammation in epilepsy: Elevation of cerebrospinal fluid cytokines and chemokines in febrile infection-related epilepsy syndrome and febrile status epilepticus. Epilepsia 2019, 60, 1678–1688. [Google Scholar] [CrossRef]
- Patterson, K.P.; Brennan, G.P.; Curran, M.; Kinney-Lang, E.; Dubé, C.; Rashid, F.; Ly, C.; Obenaus, A.; Baram, T. Rapid, Coordinate Inflammatory Responses after Experimental Febrile Status Epilepticus: Implications for Epileptogenesis. eNeuro 2015, 2, ENEURO.0034-15.2015. [Google Scholar] [CrossRef] [Green Version]
- Vezzani, A.; Conti, M.; de Luigi, A.; Ravizza, T.; Moneta, D.; Marchesi, F.; de Simoni, M.G. Interleukin-1β Immunoreactivity and Microglia Are Enhanced in the Rat Hippocampus by Focal Kainate Application: Functional Evidence for Enhancement of Electrographic Seizures. J. Neurosci. 1999, 19, 5054–5065. [Google Scholar] [CrossRef] [Green Version]
- Marchi, N.; Fan, Q.; Ghosh, C.; Fazio, V.; Bertolini, F.; Betto, G.; Batra, A.; Carlton, E.; Najm, I.; Granata, T.; et al. Antagonism of peripheral inflammation reduces the severity of status epilepticus. Neurobiol. Dis. 2009, 33, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Noe, F.M.; Polascheck, N.; Frigerio, F.; Bankstahl, M.; Ravizza, T.; Marchini, S.; Beltrame, L.; Banderó, C.R.; Löscher, W.; Vezzani, A. Pharmacological blockade of IL-1β/IL-1 receptor type 1 axis during epileptogenesis provides neuroprotection in two rat models of temporal lobe epilepsy. Neurobiol. Dis. 2013, 59, 183–193. [Google Scholar] [CrossRef]
- Terrone, G.; Salamone, A.; Vezzani, A. Inflammation and Epilepsy: Preclinical Findings and Potential Clinical Translation. Curr. Pharm. Des. 2018, 23, 5569–5576. [Google Scholar] [CrossRef]
- Ravizza, T.; Noé, F.; Zardoni, D.; Vaghi, V.; Sifringer, M.; Vezzani, A. Interleukin Converting Enzyme inhibition impairs kindling epileptogenesis in rats by blocking astrocytic IL-1β production. Neurobiol. Dis. 2008, 31, 327–333. [Google Scholar] [CrossRef]
- Ravizza, T.; Gagliardi, B.; Noé, F.; Boer, K.; Aronica, E.; Vezzani, A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: Evidence from experimental models and human temporal lobe epilepsy. Neurobiol. Dis. 2008, 29, 142–160. [Google Scholar] [CrossRef]
- Vezzani, A.; Balosso, S.; Ravizza, T. The role of cytokines in the pathophysiology of epilepsy. Brain Behav. Immun. 2008, 22, 797–803. [Google Scholar] [CrossRef]
- Vezzani, A.; Moneta, D.; Conti, M.; Richichi, C.; Ravizza, T.; de Luigi, A.; de Simoni, M.G.; Sperk, G.; Andell-Jonsson, S.; Lundkvist, J.; et al. Powerful anticonvulsant action of IL-1 receptor antagonist on intracerebral injection and astrocytic overexpression in mice. Proc. Natl. Acad. Sci. USA 2000, 97, 11534–11539. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, M.; Hino, H.; Suzuki, Y.; Takahashi, H.; Morimoto, T.; Ishii, E. Postnatal interleukin-1β enhances adulthood seizure susceptibility and neuronal cell death after prolonged experimental febrile seizures in infantile rats. Acta Neurol. Belg. 2014, 114, 179–185. [Google Scholar] [CrossRef]
- Ichiyama, T.; Nishikawa, M.; Yoshitomi, T.; Hayashi, T.; Furukawa, S. Tumor necrosis factor-a, interleukin-l, and interleukin-6 in cerebrospinal fluid from children with prolonged febrile seizures Comparison with acute encephalitis/encephalopathy. Neurology 1998, 50, 407–411. [Google Scholar] [CrossRef]
- Shi, L.; Chen, R.; Zhang, H.; Jiang, C.; Gong, J. Cerebrospinal fluid neuron specific enolase, interleukin-1β and erythropoietin concentrations in children after seizures. Child’s Nerv. Syst. 2017, 33, 805–811. [Google Scholar] [CrossRef]
- Fiala, M.; Avagyan, H.; Merino, J.J.; Bernas, M.; Valdivia, J.; Espinosa-Jeffrey, A.; Witte, M.; Weinand, M. Chemotactic and mitogenic stimuli of neuronal apoptosis in patients with medically intractable temporal lobe epilepsy. Pathophysiology 2013, 20, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Omran, A.; Peng, J.; Zhang, C.; Xiang, Q.-L.; Xue, J.; Gan, N.; Kong, H.; Yin, F. Interleukin-1β and microRNA-146a in an immature rat model and children with mesial temporal lobe epilepsy. Epilepsia 2012, 53, 1215–1224. [Google Scholar] [CrossRef]
- Lachos, J.; Zattoni, M.; Wieser, H.-G.; Fritschy, J.-M.; Langmann, T.; Schmitz, G.; Errede, M.; Virgintino, D.; Yonekawa, Y.; Frei, K. Characterization of the Gene Expression Profile of Human Hippocampus in Mesial Temporal Lobe Epilepsy with Hippocampal Sclerosis. Epilepsy Res. Treat. 2011, 2011, 758407. [Google Scholar] [CrossRef]
- Peltola, J.; Palmio, J.; Korhonen, L.; Suhonen, J.; Miettinen, A.; Hurme, M.; Lindholm, D.; Keränen, T. Interleukin-6 and Interleukin-1 receptor antagonist in cerebrospinal fluid from patients with recent tonic-clonic seizures. Epilepsy Res. 2000, 41, 205–211. [Google Scholar] [CrossRef]
- Hulkkonen, J.; Koskikallio, E.; Rainesalo, S.; Keränen, T.; Hurme, M.; Peltola, J. The balance of inhibitory and excitatory cytokines is differently regulated in vivo and in vitro among therapy resistant epilepsy patients. Epilepsy Res. 2004, 59, 199–205. [Google Scholar] [CrossRef]
- Lauro, C.; di Angelantonio, S.; Cipriani, R.; Sobrero, F.; Antonilli, L.; Brusadin, V.; Ragozzino, D.; Limatola, C. Activity of Adenosine Receptors Type 1 Is Required for CX3 CL1-Mediated Neuroprotection and Neuromodulation in Hippocampal Neurons. J. Immunol. 2008, 180, 7590–7596. [Google Scholar] [CrossRef] [Green Version]
- Guyon, A.; Nahon, J.-L. Multiple actions of the chemokine stromal cell-derived factor-1α on neuronal activity. J. Mol. Endocrinol. 2007, 38, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Louboutin, J.; Chekmasova, A.; Marusich, E.; Agrawal, L.; Strayer, D.S. Role of CCR5 and its ligands in the control of vascular inflammation and leukocyte recruitment required for acute excitotoxic seizure induction and neural damage. FASEB J. 2011, 25, 737–753. [Google Scholar] [CrossRef] [Green Version]
- Valerio, A.; Ferrario, M.; Martinez, F.O.; Locati, M.; Ghisi, V.; Bresciani, L.G.; Mantovani, A.; Spano, P. Gene expression profile activated by the chemokine CCL5/RANTES in human neuronal cells. J. Neurosci. Res. 2004, 78, 371–382. [Google Scholar] [CrossRef]
- Pujol, F.; Kitabgi, P.; Boudin, H. The chemokine SDF-1 differentially regulates axonal elongation and branching in hippocampal neurons. J. Cell Sci. 2005, 118, 1071–1080. [Google Scholar] [CrossRef] [Green Version]
- Mennicken, F.; Chabot, J.-G.; Quirion, R. Systemic administration of kainic acid in adult rat stimulates expression of the chemokine receptor CCR5 in the forebrain. Glia 2002, 37, 124–138. [Google Scholar] [CrossRef]
- Kan, A.A.; van der Hel, W.S.; Kolk, S.M.; Bos, I.W.M.; Verlinde, S.A.M.W.; van Nieuwenhuizen, O.; de Graan, P.N.E. Prolonged increase in rat hippocampal chemokine signalling after status epilepticus. J. Neuroimmunol. 2012, 245, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Louboutin, J.-P.; Reyes, B.A.S.; Agrawal, L.; Maxwell, C.R.; van Bockstaele, E.J.; Strayer, D.S. Blood–brain barrier abnormalities caused by exposure to HIV-1 gp120—Protection by gene delivery of antioxidant enzymes. Neurobiol. Dis. 2010, 38, 313–325. [Google Scholar] [CrossRef]
- Laing, J.M.; Aurelian, L. DeltaRR vaccination protects from KA-induced seizures and neuronal loss through ICP10PK-mediated modulation of the neuronal-microglial axis. Genet. Vaccines Ther. 2008, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Louboutin, J.-P.; Agrawal, L.; Reyes, B.A.S.; van Bockstaele, E.J.; Strayer, D.S. HIV-1 gp120 neurotoxicity proximally and at a distance from the point of exposure: Protection by rSV40 delivery of antioxidant enzymes. Neurobiol. Dis. 2009, 34, 462–476. [Google Scholar] [CrossRef]
- Kan, A.A.; de Jager, W.; de Wit, M.; Heijnen, C.; van Zuiden, M.; Ferrier, C.; van Rijen, P.; Gosselaar, P.; Hessel, E.; van Nieuwenhuizen, O.; et al. Protein expression profiling of inflammatory mediators in human temporal lobe epilepsy reveals co-activation of multiple chemokines and cytokines. J. Neuroinflamm. 2012, 9, 207. [Google Scholar] [CrossRef] [Green Version]
- Krumbholz, M.; Theil, D.; Cepok, S.; Hemmer, B.; Kivisäkk, P.; Ransohoff, R.M.; Hofbauer, M.; Farina, C.; Derfuss, T.; Hartle, C.; et al. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain 2006, 129, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Xu, W.; Zhang, X.; Wang, S.; Zhu, G.; Xiao, T.; Zhao, M.; Zhao, C. CXCR4 Antagonist AMD3100 Suppresses the Long-Term Abnormal Structural Changes of Newborn Neurons in the Intraventricular Kainic Acid Model of Epilepsy. Mol. Neurobiol. 2016, 53, 1518–1532. [Google Scholar] [CrossRef]
- Jung, K.-H.; Chu, K.; Lee, S.-T.; Kim, J.-H.; Kang, K.-M.; Song, E.-C.; Kim, S.-J.; Park, H.-K.; Kim, M.; Lee, S.K.; et al. Region-specific plasticity in the epileptic rat brain: A hippocampal and extrahippocampal analysis. Epilepsia 2009, 50, 537–549. [Google Scholar] [CrossRef]
- Janigro, D. Are you in or out? Leukocyte, ion, and neurotransmitter permeability across the epileptic blood-brain barrier. Epilepsia 2012, 53, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Fabene, P.F.; Mora, G.N.; Martinello, M.; Rossi, B.; Merigo, F.; Ottoboni, L.; Bach, S.; Angiari, S.; Benati, D.; Chakir, A.; et al. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat. Med. 2008, 14, 1377–1383. [Google Scholar] [CrossRef]
- Zattoni, M.; Mura, M.L.; Deprez, F.; Schwendener, R.A.; Engelhardt, B.; Frei, K.; Fritschy, J.-M. Brain Infiltration of Leukocytes Contributes to the Pathophysiology of Temporal Lobe Epilepsy. J. Neurosci. 2011, 31, 4037–4050. [Google Scholar] [CrossRef] [Green Version]
- Petzold, A.; Altintas, A.; Andreoni, L.; Bartos, A.; Berthele, A.; Blankenstein, M.A.; Buee, L.; Castellazzi, M.; Cepok, S.; Comabella, M.; et al. Neurofilament ELISA validation. J. Immunol. Methods 2010, 352, 23–31. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wolinski, P.; Ksiazek-Winiarek, D.; Glabinski, A. Cytokines and Neurodegeneration in Epileptogenesis. Brain Sci. 2022, 12, 380. https://doi.org/10.3390/brainsci12030380
Wolinski P, Ksiazek-Winiarek D, Glabinski A. Cytokines and Neurodegeneration in Epileptogenesis. Brain Sciences. 2022; 12(3):380. https://doi.org/10.3390/brainsci12030380
Chicago/Turabian StyleWolinski, Pawel, Dominika Ksiazek-Winiarek, and Andrzej Glabinski. 2022. "Cytokines and Neurodegeneration in Epileptogenesis" Brain Sciences 12, no. 3: 380. https://doi.org/10.3390/brainsci12030380