
Caloric Restriction Mimetic 2-Deoxyglucose Reduces Inflammatory Signaling in Human Astrocytes: Implications for Therapeutic Strategies Targeting Neurodegenerative Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
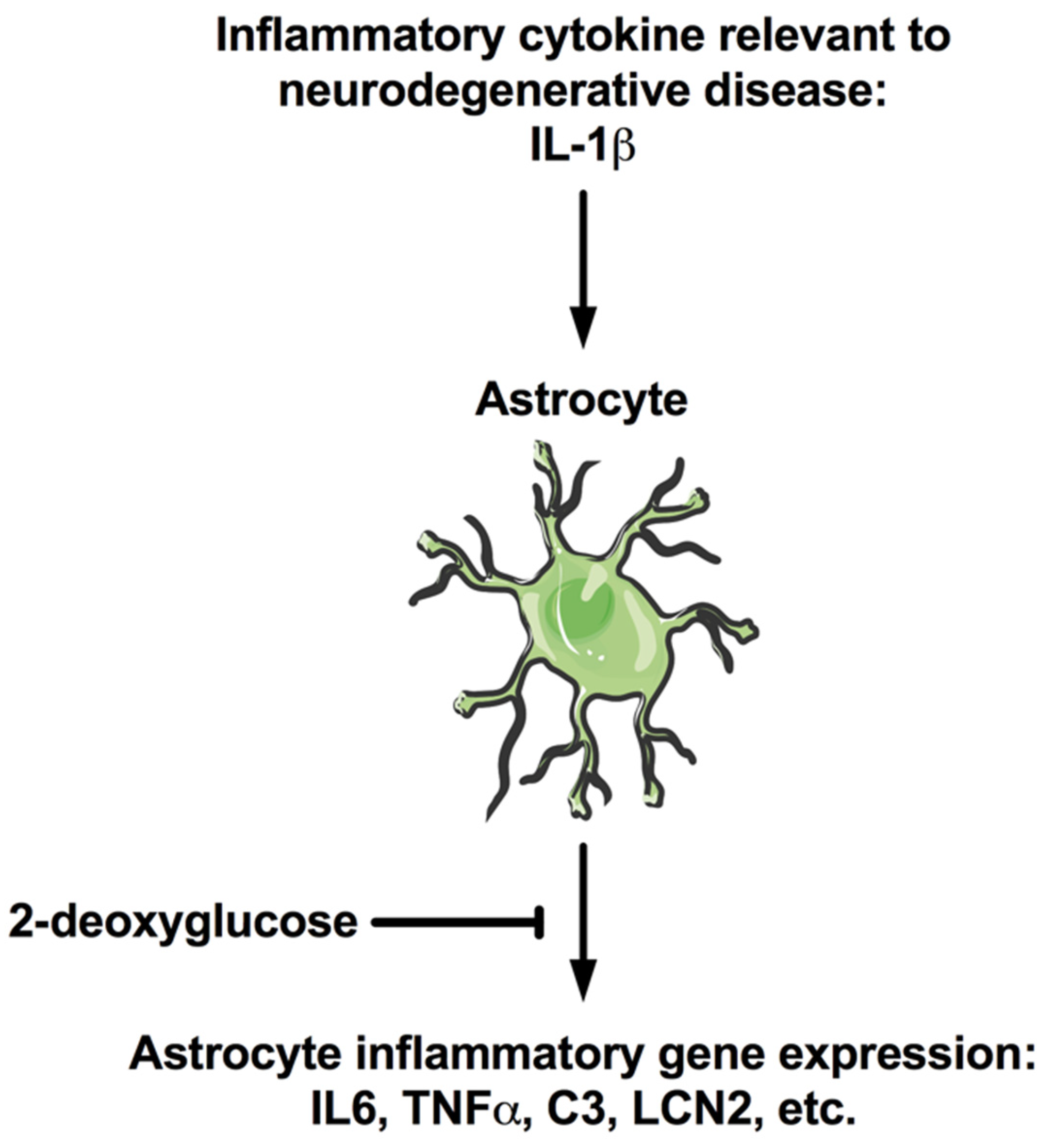
:1. Introduction
2. Materials and Methods
2.1. Generation of Human Astrocytes
2.2. Treatment of Astrocytes
2.3. RNA Isolation and TaqMan® Human Inflammation Array and Real-Time Reverse Transcription Polymerase Chain Reaction (RT2PCR)
2.4. Immunoblot
2.5. Quantification of Astrocyte Nuclei
2.6. Cytotoxicity Assay
3. Results
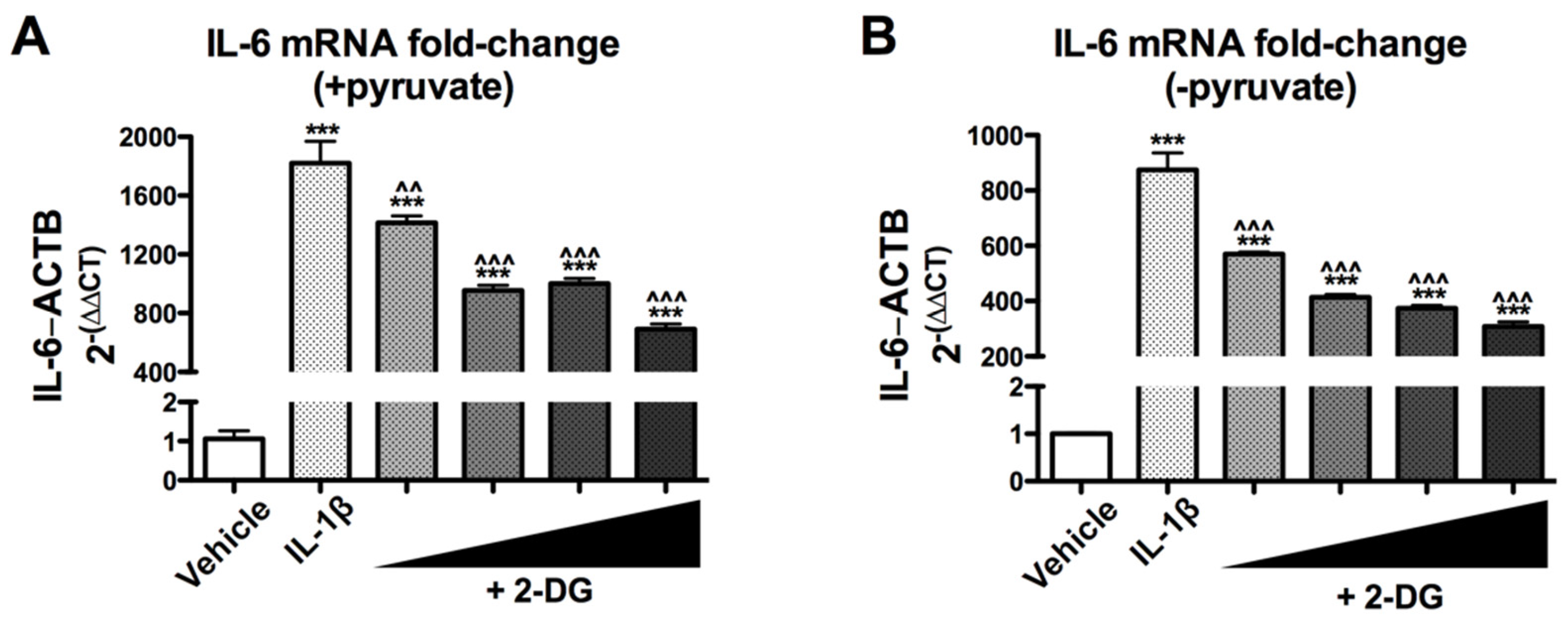
3.1. 2-Deoxyglucose Blocks IL-1β-Induced Inflammatory mRNA, IL6, in a Dose-Dependent Manner
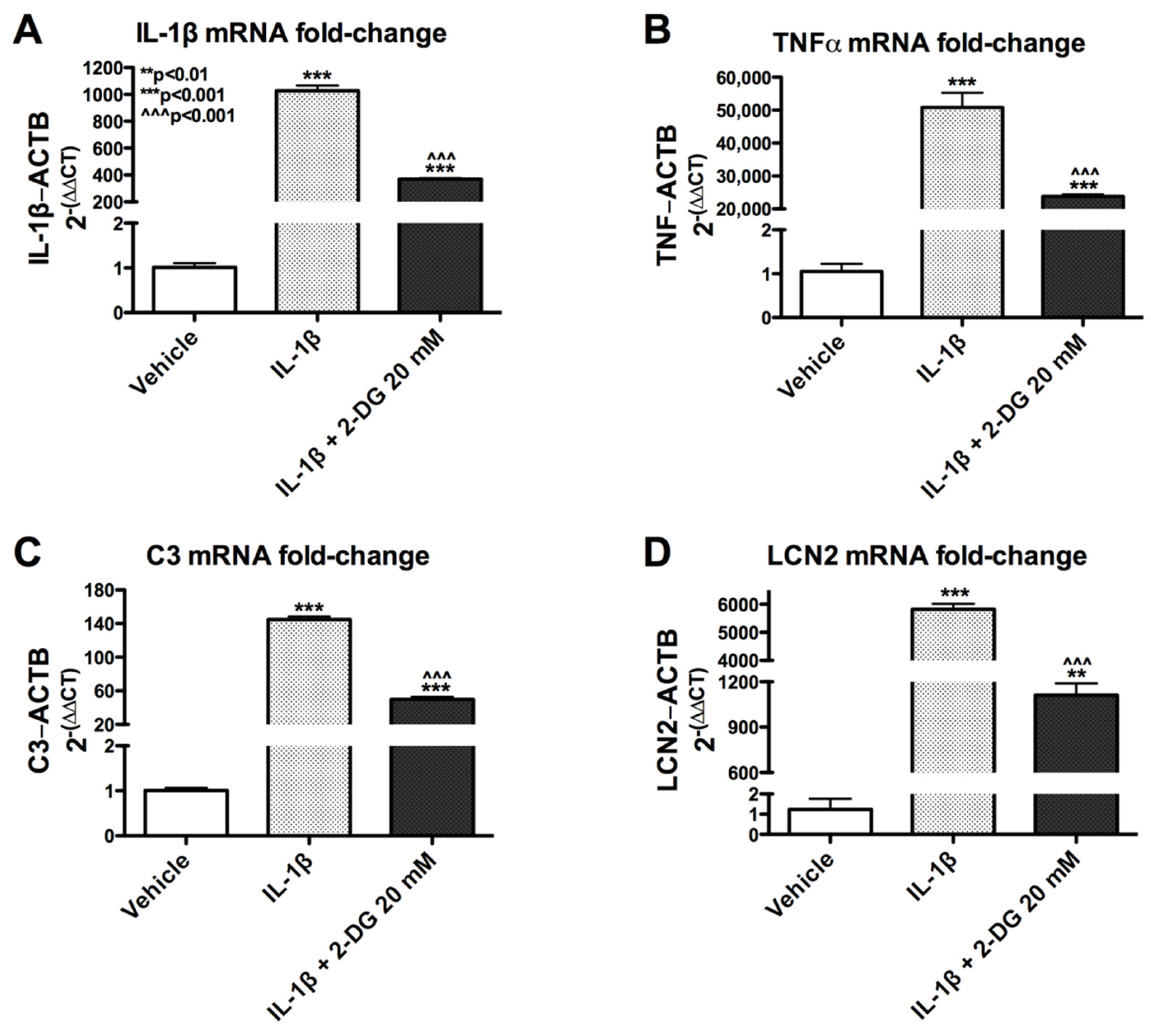
3.2. 2-Deoxyglucose Blocks IL-1β-Induced Inflammatory mRNA in Human Astrocyte
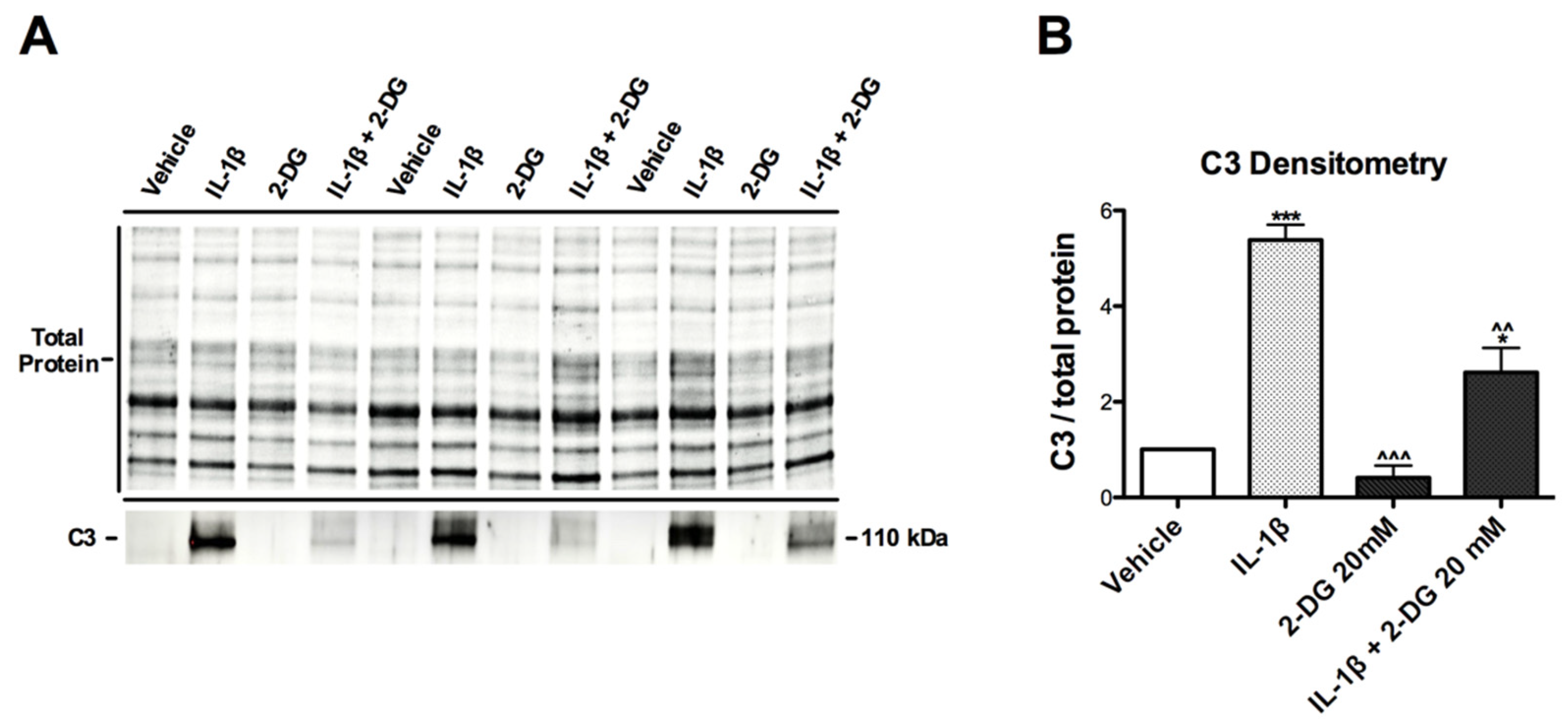
3.3. IL-1β-Induced C3 Protein Production Hindered by 2-DG in Human Astrocytes
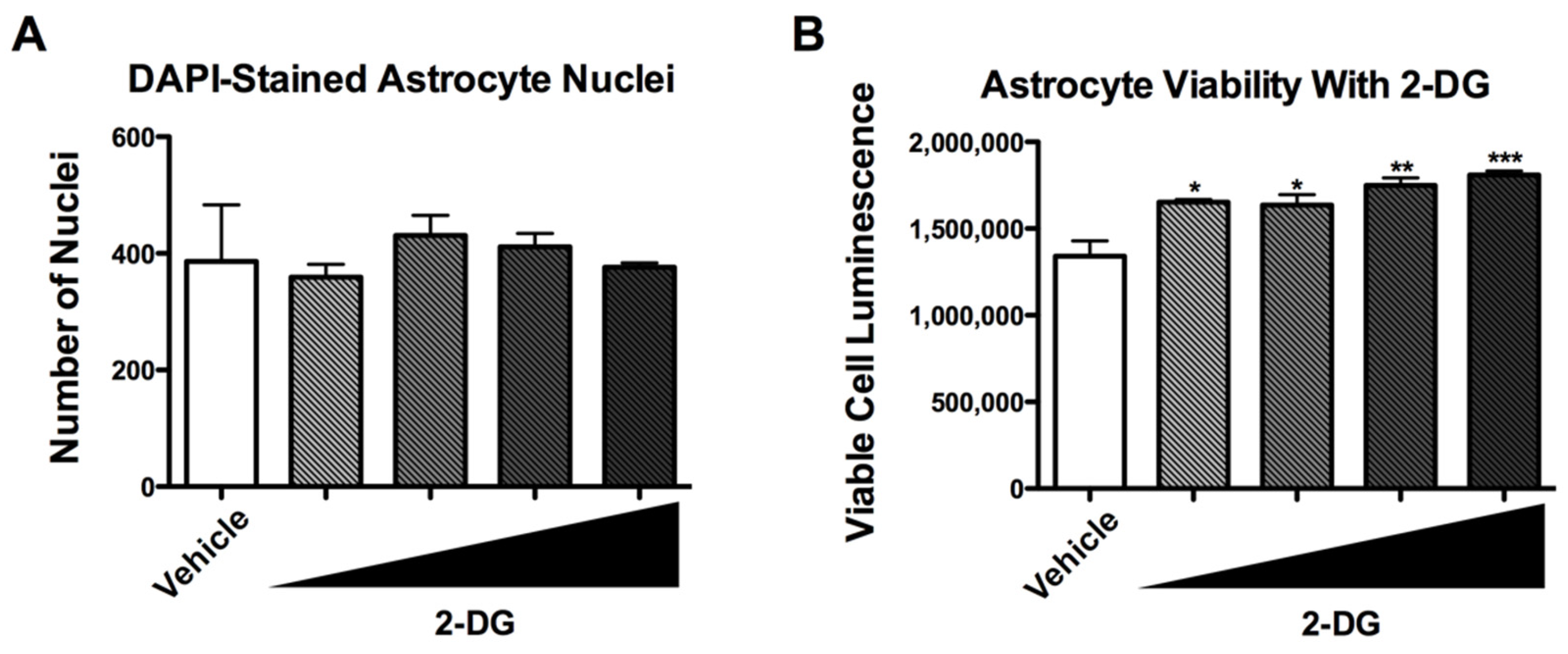
3.4. 2-DG Is Not Cytotoxic to Human Astrocytes
3.5. 2-DG Blocks IL-1β-Induced Gene Expression in Human Astrocytes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Koroshetz, W.J.; Mucke, L. Translational Neuroscience: Toward New Therapies; Nikolich, K., Hyman, S.E., Eds.; MIT Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Muthuraju, S.; Zakaria, R.; Karuppan, M.K.M.; Al-Rahbi, B. The Role of Neuroinflammation in Cellular Damage in Neurodegenerative Diseases. BioMed Res. Int. 2020, 2020, 9231452. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.E.; Liddelow, S.; Chakraborty, C.; Münch, A.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V. Astrocyte failure as a cause of CNS dysfunction. Mol. Psychiatry 2000, 5, 230–232. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [Green Version]
- Swinton, M.K.; Carson, A.; Telese, F.; Sanchez, A.B.; Soontornniyomkij, B.; Rad, L.; Batki, I.; Quintanilla, B.; Pérez-Santiago, J.; Achim, C.L.; et al. Mitochondrial biogenesis is altered in HIV+ brains exposed to ART: Implications for therapeutic targeting of astroglia. Neurobiol. Dis. 2019, 130, 104502. [Google Scholar] [CrossRef]
- Fields, J.A.; Swinton, M.K.; Carson, A.; Soontornniyomkij, B.; Lindsay, C.; Han, M.M.; Frizzi, K.; Sambhwani, S.; Murphy, A.; Achim, C.L.; et al. Tenofovir disoproxil fumarate induces peripheral neuropathy and alters inflammation and mitochondrial biogenesis in the brains of mice. Sci. Rep. 2019, 9, 17158. [Google Scholar] [CrossRef]
- Natarajaseenivasan, K.; Cotto, B.; Shanmughapriya, S.; Lombardi, A.A.; Datta, P.; Madesh, M.; Elrod, J.; Khalili, K.; Langford, D. Astrocytic metabolic switch is a novel etiology for Cocaine and HIV-1 Tat-mediated neurotoxicity. Cell Death Dis. 2018, 9, 415. [Google Scholar] [CrossRef] [Green Version]
- Cotto, B.; Natarajanseenivasan, K.; Langford, D. HIV-1 infection alters energy metabolism in the brain: Contributions to HIV-associated neurocognitive disorders. Prog. Neurobiol. 2019, 181, 101616. [Google Scholar] [CrossRef]
- Cotto, B.; Natarajaseenivasan, K.; Langford, D. Astrocyte activation and altered metabolism in normal aging, age-related CNS diseases, and HAND. J. Neurovirol. 2019, 25, 722–733. [Google Scholar] [CrossRef]
- Sivalingam, K.; Cirino, T.J.; McLaughlin, J.P.; Samikkannu, T. HIV-Tat and Cocaine Impact Brain Energy Metabolism: Redox Modification and Mitochondrial Biogenesis Influence NRF Transcription-Mediated Neurodegeneration. Mol. Neurobiol. 2020, 58, 490–504. [Google Scholar] [CrossRef]
- Hamby, M.E.; Sofroniew, M.V. Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics 2010, 7, 494–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef] [Green Version]
- Deitmer, J.W.; Theparambil, S.M.; Ruminot, I.; Noor, S.I.; Becker, H.M. Energy Dynamics in the Brain: Contributions of Astrocytes to Metabolism and pH Homeostasis. Front. Neurosci. 2019, 13, 1301. [Google Scholar] [CrossRef] [PubMed]
- Muraleedharan, R.; Gawali, M.V.; Tiwari, D.; Sukumaran, A.; Oatman, N.; Anderson, J.; Nardini, D.; Bhuiyan, M.A.N.; Tkáč, I.; Ward, A.L.; et al. AMPK-Regulated Astrocytic Lactate Shuttle Plays a Non-Cell-Autonomous Role in Neuronal Survival. Cell Rep. 2020, 32, 108092. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A. Lack of appropriate stoichiometry: Strong evidence against an energetically important astrocyte-neuron lactate shuttle in brain. J. Neurosci. Res. 2017, 95, 2103–2125. [Google Scholar] [CrossRef] [Green Version]
- Dienel, G.A. The metabolic trinity, glucose-glycogen-lactate, links astrocytes and neurons in brain energetics, signaling, memory, and gene expression. Neurosci. Lett. 2017, 637, 18–25. [Google Scholar] [CrossRef]
- Sada, N.; Inoue, T. Electrical Control in Neurons by the Ketogenic Diet. Front. Cell. Neurosci. 2018, 12, 208. [Google Scholar] [CrossRef] [Green Version]
- Van der Ven, L.T.; Van de Kuil, T.; Verhoef, A.; Verwer, C.M.; Lilienthal, H.; Leonards, P.E.; Schauer, U.M.; Cantón, R.F.; Litens, S.; De Jong, F.H.; et al. Endocrine effects of tetrabromobisphenol-A (TBBPA) in Wistar rats as tested in a one-generation reproduction study and a subacute toxicity study. Toxicology 2008, 245, 76–89. [Google Scholar] [CrossRef]
- Mayengbam, S.; Ellegood, J.; Kesler, M.; Reimer, R.A.; Shearer, J.; Murari, K.; Rho, J.M.; Lerch, J.P.; Cheng, N. A ketogenic diet affects brain volume and metabolome in juvenile mice. NeuroImage 2021, 244, 118542. [Google Scholar] [CrossRef] [PubMed]
- Cervenka, M.; Pascual, J.M.; Rho, J.M.; Thiele, E.; Yellen, G.; Whittemore, V.; Hartman, A.L. Metabolism-based therapies for epilepsy: New directions for future cures. Ann. Clin. Transl. Neurol. 2021, 8, 1730–1737. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Astrogliosis. Cold Spring Harb. Perspect. Biol. 2015, 7, a020420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proulx, J.; Park, I.-W.; Borgmann, K. Cal’MAM’ity at the Endoplasmic Reticulum-Mitochondrial Interface: A Potential Therapeutic Target for Neurodegeneration and Human Immunodeficiency Virus-Associated Neurocognitive Disorders. Front. Neurosci. 2021, 15, 715945. [Google Scholar] [CrossRef]
- Fields, J.; Ghorpade, A. C/EBPbeta regulates multiple IL-1beta-induced human astrocyte inflammatory genes. Neuroinflammation 2012, 9, 177. [Google Scholar] [CrossRef] [Green Version]
- Fields, J.A.; Swinton, M.K.; Montilla-Perez, P.; Ricciardelli, E.; Telese, F. The cannabinoid receptor agonist, WIN, suppresses the activation of pro-inflammatory genes induced by interleukin 1 beta in human astrocytes. Cannabis Cannabinoid Res. 2020, 7, 78–92. [Google Scholar] [CrossRef]
- Schousboe, A.; Scafidi, S.; Bak, L.K.; Waagepetersen, H.S.; McKenna, M.C. Glutamate metabolism in the brain focusing on astrocytes. Adv. Neurobiol. 2014, 11, 13–30. [Google Scholar] [CrossRef] [Green Version]
- Haroon, E.; Miller, A.H.; Sanacora, G. Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders. Neuropsychopharmacology 2016, 42, 193–215. [Google Scholar] [CrossRef]
- Wang, Z.; Trillo-Pazos, G.; Kim, S.-Y.; Canki, M.; Morgello, S.; Sharer, L.R.; Gelbard, H.A.; Su, Z.-Z.; Kang, D.-C.; Brooks, A.I.; et al. Effects of human immunodeficiency virus type 1 on astrocyte gene expression and function: Potential role in neuropathogenesis. J. Neurovirol. 2004, 10 (Suppl. S1), 25–32. [Google Scholar] [CrossRef]
- Zhou, B.Y.; Liu, Y.; Kim, B.O.; Xiao, Y.; He, J. Astrocyte activation and dysfunction and neuron death by HIV-1 Tat expression in astrocytes. Mol. Cell. Neurosci. 2004, 27, 296–305. [Google Scholar] [CrossRef]
- Hwangbo, D.-S.; Lee, H.-Y.; Abozaid, L.S.; Min, K.-J. Mechanisms of Lifespan Regulation by Calorie Restriction and Intermittent Fasting in Model Organisms. Nutrients 2020, 12, 1194. [Google Scholar] [CrossRef] [PubMed]
- Julio-Amilpas, A.; Montiel, T.; Soto-Tinoco, E.; Gerónimo-Olvera, C.; Massieu, L. Protection of hypoglycemia-induced neuronal death by beta-hydroxybutyrate involves the preservation of energy levels and decreased production of reactive oxygen species. J. Cereb. Blood Flow Metab. 2015, 35, 851–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabke, P.; Das, A.M. Mechanism of Action of Ketogenic Diet Treatment: Impact of Decanoic Acid and Beta-Hydroxybutyrate on Sirtuins and Energy Metabolism in Hippocampal Murine Neurons. Nutrients 2020, 12, 2379. [Google Scholar] [CrossRef] [PubMed]
- Fields, J.; Gardner-Mercer, J.; Borgmann, K.; Clark, I.; Ghorpade, A. CCAAT/enhancer binding protein beta expression is increased in the brain during HIV-1-infection and contributes to regulation of astrocyte tissue inhibitor of metalloproteinase-1. J. Neurochem. 2011, 118, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhar, A.; Gardner, J.; Borgmann, K.; Wu, L.; Ghorpade, A. Novel role of TGF-beta in differential astrocyte-TIMP-1 regulation: Implications for HIV-1-dementia and neuroinflammation. J. Neurosci. Res. 2006, 83, 1271–1280. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Pajak, B.; Siwiak, E.; Sołtyka, M.; Priebe, A.; Zieliński, R.; Fokt, I.; Ziemniak, M.; Jaśkiewicz, A.; Borowski, R.; Domoradzki, T.; et al. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int. J. Mol. Sci. 2019, 21, 234. [Google Scholar] [CrossRef] [Green Version]
- Devanney, N.; Stewart, A.N.; Gensel, J.C. Microglia and macrophage metabolism in CNS injury and disease: The role of immunometabolism in neurodegeneration and neurotrauma. Exp. Neurol. 2020, 329, 113310. [Google Scholar] [CrossRef]
- Onyango, I.G. Modulation of mitochondrial bioenergetics as a therapeutic strategy in Alzheimer’s disease. Neural Regen. Res. 2018, 13, 19–25. [Google Scholar] [CrossRef]
- Biswas, J.; Gupta, S.; Verma, D.K.; Gupta, P.; Singh, A.; Tiwari, S.; Goswami, P.; Sharma, S.; Singh, S. Involvement of glucose related energy crisis and endoplasmic reticulum stress: Insinuation of streptozotocin induced Alzheimer’s like pathology. Cell. Signal. 2018, 42, 211–226. [Google Scholar] [CrossRef]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Chu, C.T.; Kaufman, B.A. The mitochondrial transcription factor TFAM in neurodegeneration: Emerging evidence and mechanisms. FEBS Lett. 2018, 592, 793–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.P.; Hofman, A.; Breteler, M.M.B. Diabetes mellitus and the risk of dementia. Neurology 1999, 53, 1937. [Google Scholar] [CrossRef] [PubMed]
- Downer, B.; Rote, S.; Markides, K.S.; Snih, S.A. The Comorbid Influence of High Depressive Symptoms and Diabetes on Mortality and Disability in Mexican Americans Aged 75 and Above. Gerontol. Geriatr. Med. 2016, 2. [Google Scholar] [CrossRef]
- Downer, B.; Vickers, B.N.; Snih, S.A.; Raji, M.; Markides, K.S. Effects of Comorbid Depression and Diabetes Mellitus on Cognitive Decline in Older Mexican Americans. J. Am. Geriatr. Soc. 2016, 64, 109–117. [Google Scholar] [CrossRef]
- Bryant, A.K.; Fazeli, P.L.; Letendre, S.L.; Ellis, R.; Potter, M.; Burdo, T.H.; Singh, K.K.; Jeste, D.V.; Grant, I.; Moore, D.J. Complement Component 3 Is Associated with Metabolic Comorbidities in Older HIV-Positive Adults. AIDS Res. Hum. Retrovir. 2016, 32, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Jenner, P.; Dexter, D.; Sian, J.; Schapira, A.; Mardsen, C. Oxidative stress as a cause of nigral cell death in Parkinson’s disease and incidental Lewy body disease. The Royal Kings and Queens Parkinsons’s Disease Research Group. Ann.Neurol. 1992, 32, S82–S87. [Google Scholar] [CrossRef]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neuro-degenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef]
- Fields, J.; Dumaop, W.; Elueteri, S.; Campos, S.; Serger, E.; Trejo, M.; Kosberg, K.; Adame, A.; Spencer, B.; Rockenstein, E.; et al. HIV-1 Tat alters neuronal autophagy by modulating autophagosome fusion to the lysosome: Implications for HIV-associated neurocognitive disorders. J. Neurosci. 2015, 35, 1921–1938. [Google Scholar] [CrossRef] [Green Version]
- Fields, J.A.; Dumaop, W.; Crews, L.; Adame, A.; Spencer, B.; Metcalf, J.; He, J.; Rockenstein, E.; Masliah, E. Mechanisms of HIV-1 Tat neurotoxicity via CDK5 translocation and hyper-activation: Role in HIV-associated neurocognitive disorders. Curr. HIV Res. 2015, 13, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Pandhare, J.; Dash, S.; Jones, B.; Villalta, F.; Dash, C. A Novel Role of Proline Oxidase in HIV-1 Envelope Glycoprotein-induced Neuronal Autophagy. J. Biol. Chem. 2015, 290, 25439–25451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, Y.; Shirakabe, A.; Maejima, Y.; Zhai, P.; Sciarretta, S.; Toli, J.; Nomura, M.; Mihara, K.; Egashira, K.; Ohishi, M.; et al. Endogenous Drp1 Mediates Mitochondrial Autophagy and Protects the Heart Against Energy Stress. Circ. Res. 2015, 116, 264–278. [Google Scholar] [CrossRef]
- Saribas, A.S.; Khalili, K.; Sariyer, I.K. Dysregulation of autophagy by HIV-1 Nef in human astrocytes. Cell Cycle 2015, 14, 2899–2904. [Google Scholar] [CrossRef] [Green Version]
- Siman, R.; Cocca, R.; Dong, Y. The mTOR Inhibitor Rapamycin Mitigates Perforant Pathway Neurodegeneration and Synapse Loss in a Mouse Model of Early-Stage Alzheimer-Type Tauopathy. PLoS ONE 2015, 10, e0142340. [Google Scholar] [CrossRef]
- Sultana, R.; Chi, E.Y.; Wood, S.J.; Kendrick, B.S.; Li, C.; Garzon-Rodriguez, W.; Wypych, J.; Randolph, T.W.; Narhi, L.O.; Biere, A.L.; et al. Oxidative dimer formation is the critical rate-limiting step for Parkinson’s disease alpha-synuclein fibrillogenesis. Biochemistry 2003, 42, 829–837. [Google Scholar]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxid. Med. Cell Longev. 2016, 2016, 8910396. [Google Scholar] [CrossRef] [Green Version]
- Sultana, R.; Boyd-Kimball, D.; Poon, H.F.; Cai, J.; Pierce, W.M.; Klein, J.B.; Markesbery, W.R.; Zhou, X.Z.; Lu, K.P.; Butterfield, D.A. Oxidative modification and down-regulation of Pin1 in Alzheimer’s disease hippocampus: A redox proteomics analysis. Neurobiol. Aging 2006, 27, 918–925. [Google Scholar] [CrossRef]
- McCarty, M.F.; DiNicolantonio, J.J.; O’Keefe, J.H. Ketosis may promote brain macroautophagy by activating Sirt1 and hypoxia-inducible factor-1. Med. Hypotheses 2015, 85, 631–639. [Google Scholar] [CrossRef]
- Kovács, Z.; Brunner, B.; Ari, C. Beneficial Effects of Exogenous Ketogenic Supplements on Aging Processes and Age-Related Neurodegenerative Diseases. Nutrients 2021, 13, 2197. [Google Scholar] [CrossRef] [PubMed]
- Laussel, C.; Sebastien, L. Cellular toxicity of the metabolic inhibitor 2-deoxyglucose and associated resistance mechanisms. Biochem. Pharmacol. 2020, 182, 114213. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallee, K.-A.J.; Fields, J.A. Caloric Restriction Mimetic 2-Deoxyglucose Reduces Inflammatory Signaling in Human Astrocytes: Implications for Therapeutic Strategies Targeting Neurodegenerative Diseases. Brain Sci. 2022, 12, 308. https://doi.org/10.3390/brainsci12030308
Vallee K-AJ, Fields JA. Caloric Restriction Mimetic 2-Deoxyglucose Reduces Inflammatory Signaling in Human Astrocytes: Implications for Therapeutic Strategies Targeting Neurodegenerative Diseases. Brain Sciences. 2022; 12(3):308. https://doi.org/10.3390/brainsci12030308
Chicago/Turabian StyleVallee, Kaylie-Anna Juliette, and Jerel Adam Fields. 2022. "Caloric Restriction Mimetic 2-Deoxyglucose Reduces Inflammatory Signaling in Human Astrocytes: Implications for Therapeutic Strategies Targeting Neurodegenerative Diseases" Brain Sciences 12, no. 3: 308. https://doi.org/10.3390/brainsci12030308