Role of NAD+—Modulated Mitochondrial Free Radical Generation in Mechanisms of Acute Brain Injury



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
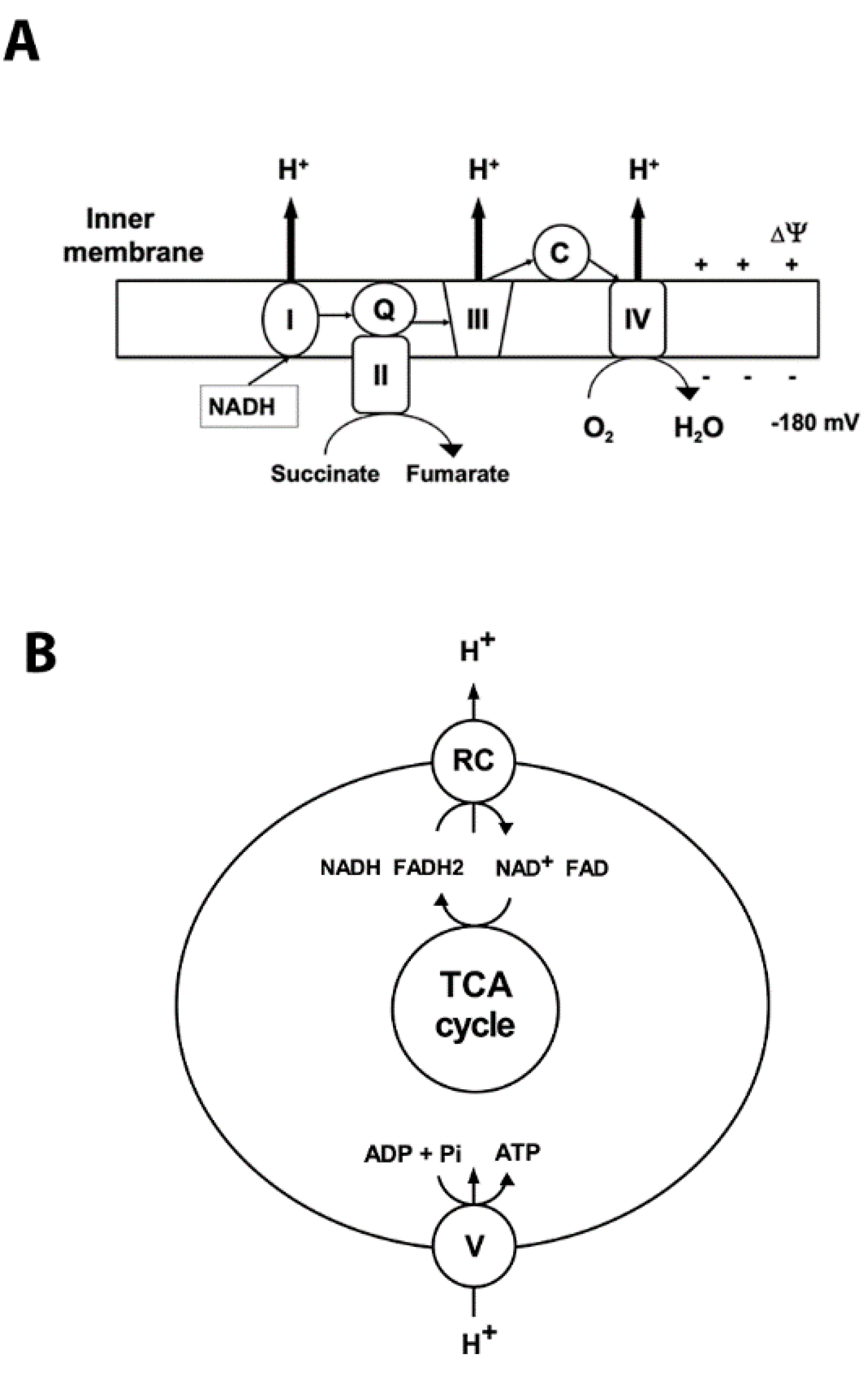
2. Mitochondrial Oxidative Phosphorylation and ROS Production
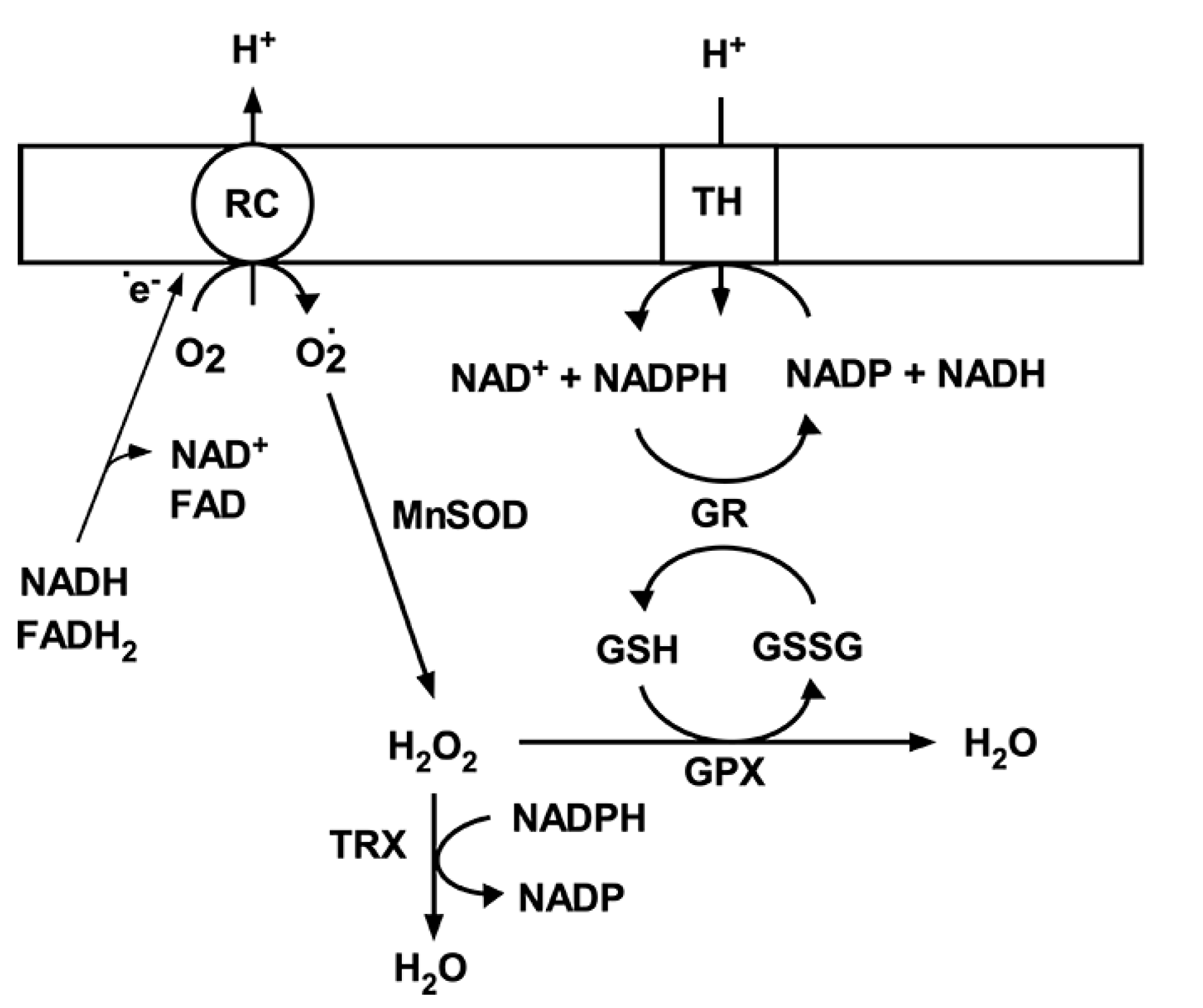
3. Mitochondrial Antioxidant Mechanisms
4. Effect of Ischemia on Mitochondrial Metabolism
4.1. Two Phases of Post-Ischemic Mitochondrial Respiratory Failure
4.2. Mitochondrial Free Radical Production and Ischemic Brain Injury
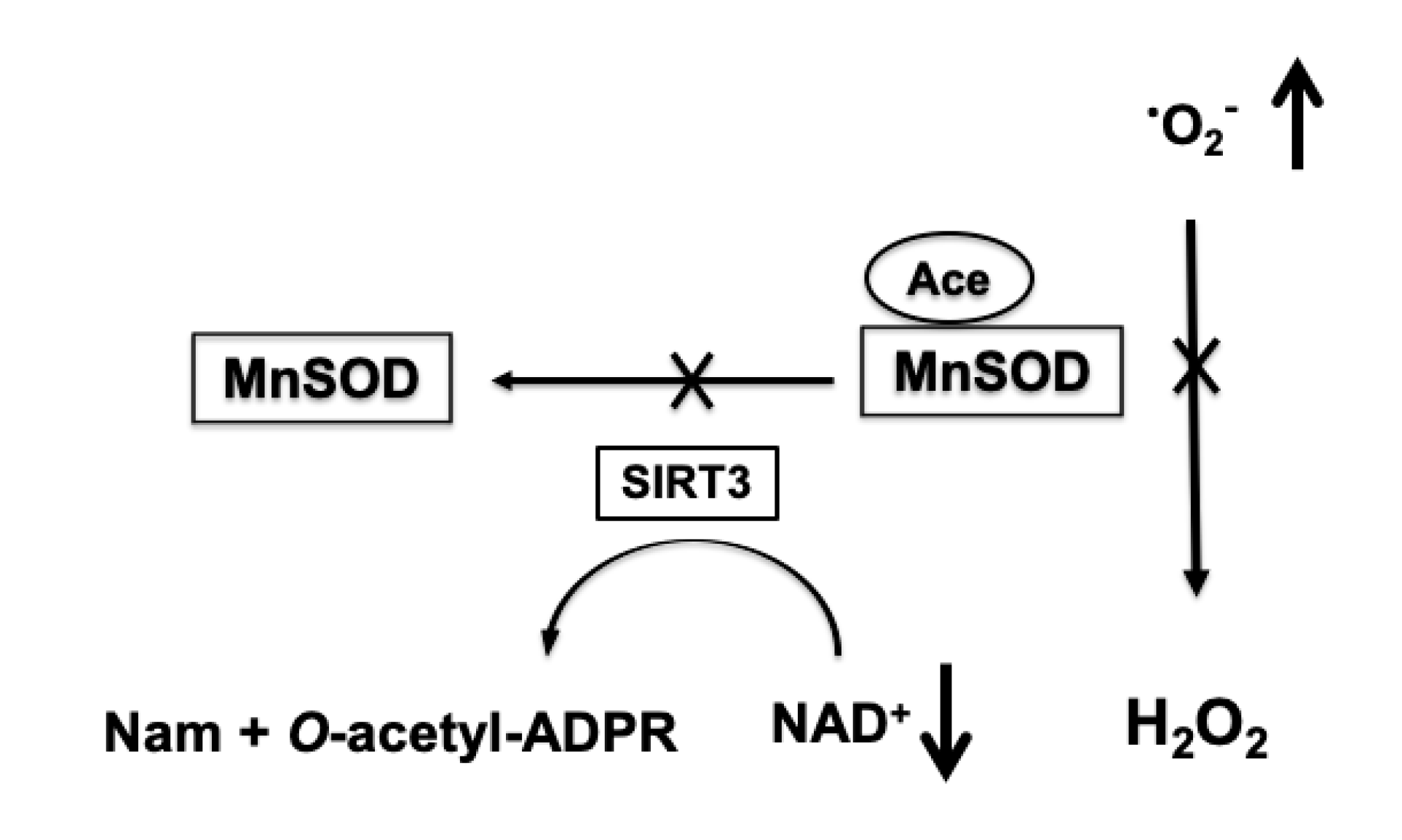
4.3. Role of Protein Acetylation in Mitochondrial ROS Generation
5. Therapeutic Approaches to Reduce Mitochondrially-Generated ROS
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Zweier, J.L. Cardiac mitochondria and reactive oxygen species generation. Circ. Res. 2014, 114, 524–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Tretter, L.; Takacs, K.; Hegedus, V.; Adam-Vizi, V. Characteristics of alpha-glycerophosphate-evoked H2O2 generation in brain mitochondria. J. Neurochem. 2007, 100, 650–663. [Google Scholar] [CrossRef]
- Patole, M.S.; Swaroop, A.; Ramasarma, T. Generation of H2O2 in brain mitochondria. J. Neurochem. 1986, 47, 1–8. (In English) [Google Scholar] [CrossRef]
- Kwong, L.K.; Sohal, R.S. Substrate and site specificity of hydrogen peroxide generation in mouse mitochondria. Arch. Biochem. Biophys. 1998, 350, 118–126. (In English) [Google Scholar] [CrossRef]
- Babady, N.E.; Pang, Y.P.; Elpeleg, O.; Isaya, G. Cryptic proteolytic activity of dihydrolipoamide dehydrogenase. Proc. Natl. Acad. Sci. USA 2007, 104, 6158–6163. (In English) [Google Scholar] [CrossRef] [Green Version]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef] [Green Version]
- Skulachev, V.P. A biochemical approach to the problem of aging: "Megaproject" on membrane-penetrating ions. The first results and prospects. Biochemistry 2007, 72, 1385–1396. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am. J. Physiol. Cell Physiol. 2008, 294, 460–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, A.; Boveris, A. The mitochondrial energy transduction system and the aging process. Am. J. Physiol. Cell Physiol. 2007, 292, 670–686. [Google Scholar] [CrossRef]
- Han, D.; Canali, R.; Rettori, D.; Kaplowitz, N. Effect of glutathione depletion on sites and topology of superoxide and hydrogen peroxide production in mitochondria. Mol. Pharmacol. 2003, . 64, 1136–1144. [Google Scholar] [CrossRef]
- Genova, M.L.; Ventura, B.; Giuliano, G.; Bovina, C.; Formiggini, G.; Castelli, G.P.; Lenaz, G. The site of production of superoxide radical in mitochondrial Complex I is not a bound ubisemiquinone but presumably iron-sulfur cluster N2. FEBS Lett. 2001, 505, 364–368. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264. (In English) [Google Scholar] [CrossRef] [Green Version]
- Grivennikova, V.G.; Kozlovsky, V.S.; Vinogradov, A.D. Respiratory complex II: ROS production and the kinetics of ubiquinone reduction. Biochim. Biophys. Acta. Bioenerg. 2017, 1858, 109–117. (In English) [Google Scholar] [CrossRef]
- Siebels, I.; Dröse, S. Q-site inhibitor induced ROS production of mitochondrial complex II is attenuated by TCA cycle dicarboxylates. Biochim. Biophys. Acta. 2013, 1827, 1156–1164. (In English) [Google Scholar] [CrossRef] [Green Version]
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343. (In English) [Google Scholar] [CrossRef]
- Costa, A.D.; Pierre, S.V.; Cohen, M.V.; Downey, J.M.; Garlid, K.D. cGMP signalling in pre- and post-conditioning: The role of mitochondria. Cardiovasc. Res. 2008, 77, 344–352. [Google Scholar] [CrossRef]
- Novalija, E.; Kevin, L.G.; Camara, A.K.; Bosnjak, Z.J.; Kampine, J.P.; Stowe, D.F. Reactive oxygen species precede the epsilon isoform of protein kinase C in the anesthetic preconditioning signaling cascade. Anesthesiology 2003, 99, 421–428. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Antunes, F.; Han, D.; Cadenas, E. Relative contributions of heart mitochondria glutathione peroxidase and catalase to H(2)O(2) detoxification in in vivo conditions. Free Radic. Biol. Med. 2002, 33, 1260–1267. [Google Scholar] [CrossRef]
- Stowe, D.F.; Camara, A.K. Mitochondrial reactive oxygen species production in excitable cells: Modulators of mitochondrial and cell function. Antioxid. Redox Signal. 2009, 11, 1373–1414. [Google Scholar] [CrossRef] [Green Version]
- Inarrea, P.; Moini, H.; Han, D.; Rettori, D.; Aguiló, I.; Alava, M.A.; Iturralde, M.; Cadenas, E. Mitochondrial respiratory chain and thioredoxin reductase regulate intermembrane Cu, Zn-superoxide dismutase activity: Implications for mitochondrial energy metabolism and apoptosis. Biochem. J. 2007, 405, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Holmgren, A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid. Redox Signal. 2000, 2, 811–820. (In English) [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Yoo, Y.H.; Lee, J.H.; Park, J.W. Mitochondrial NADP(+)-dependent isocitrate dehydrogenase knockdown inhibits tumorigenicity of melanoma cells. Biochem. Biophys. Res. Commun. 2014, 451, 246–251. (In English) [Google Scholar] [CrossRef]
- You, K.S. Stereospecificity for nicotinamide nucleotides in enzymatic and chemical hydride transfer reactions. CRC Crit. Rev. Biochem. 1985, 17, 313–451. [Google Scholar] [CrossRef]
- Reneau, D.D.; Guilbeau, E.J.; Null, R.E. Oxygen dynamics in brain. Microvasc. Res. 1977, 13, 337–344. [Google Scholar] [CrossRef]
- Hertz, L. Bioenergetics of cerebral ischemia: A cellular perspective. Neuropharmacology 2008, 55, 289–309. [Google Scholar] [CrossRef]
- Folbergrova, J.; Ljunggren, B.; Norberg, K.; Siesjo, B.K. Influence of complete ischemia on glycolytic metabolites, citric acid cycle intermediates, and associated amino acids in the rat cerebral cortex. Brain Res. 1974, 80, 265–279. [Google Scholar] [CrossRef]
- Yoshida, S.; Abe, K.; Busto, R.; Watson, B.D.; Kogure, K.; Ginsberg, M.D. Influence of transient ischemia on lipid-soluble antioxidants, free fatty acids and energy metabolites in rat brain. Brain Res. 1982, 245, 307–316. [Google Scholar] [CrossRef]
- Deutsch, J.; Kalderon, B.; Purdon, A.D.; Rapoport, S.I. Evaluation of brain long-chain acylcarnitines during cerebral ischemia. Lipids 2000, 35, 693–696. [Google Scholar] [CrossRef]
- Kristian, T. Metabolic stages, mitochondria and calcium in hypoxic/ischemic brain damage. Cell Calcium 2004, 36, 221–233. [Google Scholar] [CrossRef]
- Galkin, A. Brain Ischemia/Reperfusion Injury and Mitochondrial Complex I Damage. Biochemistry 2019, 84, 1411–1423. [Google Scholar] [CrossRef]
- Siesjo, B.K.; Elmér, E.; Janelidze, S.; Keep, M.; Kristián, T.; Ouyang, Y.B.; Uchino, H. Role and mechanisms of secondary mitochondrial failure. Acta. Neurochir. Suppl. 1999, 73, 7–13. [Google Scholar] [CrossRef]
- Ekholm, A.; Asplund, B.; Siesjo, B.K. Perturbation of cellular energy state in complete ischemia: Relationship to dissipative ion fluxes. Exp. Brain Res. 1992, 90, 47–53. [Google Scholar] [CrossRef]
- Kristian, T.; Siesjo, B.K. Calcium-related damage in ischemia. Life Sci. 1996, 59, 357–367. [Google Scholar] [CrossRef]
- Brittain, M.K.; Brustovetsky, T.; Sheets, P.L.; Brittain, J.M.; Khanna, R.; Cummins, T.R.; Brustovetsky, N. Delayed calcium dysregulation in neurons requires both the NMDA receptor and the reverse Na+/Ca2+ exchanger. Neurobiol. Dis. 2012, 46, 109–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekholm, A.; Katsura, K.; Kristian, T.; Liu, M.; Folbergrova, J.; Siesjo, B.K. Coupling of cellular energy state and ion homeostasis during recovery following brain ischemia. Brain Res. 1993, 604, 185–191. [Google Scholar] [CrossRef]
- Vannucci, R.C.; Towfighi, J.; Vannucci, S.J. Secondary energy failure after cerebral hypoxia-ischemia in the immature rat. J. Cereb. Blood Flow Metab. 2004, 24, 1090–1097. [Google Scholar] [CrossRef] [Green Version]
- Rehncrona, S.; Mela, L.; Siesjo, B.K. Recovery of brain mitochondrial function in the rat after complete and incomplete cerebral ischemia. Stroke 1979, 10, 437–446. [Google Scholar] [CrossRef] [Green Version]
- Hillered, L.; Smith, M.L.; Siesjo, B.K. Lactic acidosis and recovery of mitochondrial function following forebrain ischemia in the rat. J. Cereb. Blood Flow Metab. 1985, 5, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Sims, N.R.; Pulsinelli, W.A. Altered mitochondrial respiration in selectively vulnerable brain subregions following transient forebrain ischemia in the rat. J. Neurochem. 1987, 49, 1367–1374. [Google Scholar] [CrossRef]
- Allen, K.L.; Almeida, A.; Bates, T.E.; Clark, J.B. Changes of respiratory chain activity in mitochondrial and synaptosomal fractions isolated from the gerbil brain after graded ischaemia. J. Neurochem. 1995, 64, 2222–2229. [Google Scholar] [CrossRef]
- Almeida, A.; Allen, K.L.; Bates, T.E.; Clark, J.B. Effect of reperfusion following cerebral ischaemia on the activity of the mitochondrial respiratory chain in the gerbil brain. J. Neurochem. 1995, 65, 1698–1703. [Google Scholar] [CrossRef]
- Hillered, L.; Siesjo, B.K.; Arfors, K.E. Mitochondrial response to transient forebrain ischemia and recirculation in the rat. J. Cereb. Blood Flow Metab. 1984, 4, 438–446. [Google Scholar] [CrossRef] [Green Version]
- Zaidan, E.; Sims, N.R. Selective reductions in the activity of the pyruvate dehydrogenase complex in mitochondria isolated from brain subregions following forebrain ischemia in rats. J. Cereb. Blood Flow Metab. 1993, 13, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Bogaert, Y.E.; Rosenthal, R.E.; Fiskum, G. Postischemic inhibition of cerebral cortex pyruvate dehydrogenase. Free Radic. Biol. Med. 1994, 16, 811–820. [Google Scholar] [CrossRef]
- Sims, N.R. Selective impairment of respiration in mitochondria isolated from brain subregions following transient forebrain ischemia in the rat. J. Neurochem. 1991, 56, 1836–1844. [Google Scholar] [CrossRef] [PubMed]
- Kahl, A.K.A.; Stepanova, A.; Konrad, C.; Anderson, C.; Manfredi, G.; Zhou, P.; Iadecola, C.; Galkin, A. Critical Role of Flavin and Glutathione in Complex I-Mediated Bioenergetic Failure in Brain Ischemia/Reperfusion Injury. Stroke 2018, 49, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, A.; Sosunov, S.; Niatsetskaya, Z.; Konrad, C.; Starkov, A.A.; Manfredi, G.; Wittig, I.; Ten, V.; Galkin, A. Redox-Dependent Loss of Flavin by Mitochondrial Complex I in Brain Ischemia/Reperfusion Injury. Antioxid. Redox Signal. 2019, 31, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niatsetskaya, Z.V.; Sosunov, S.A.; Matsiukevich, D.; Utkina-Sosunova, I.V.; Ratner, V.I.; Starkov, A.A.; Ten, V.S. The oxygen free radicals originating from mitochondrial complex I contribute to oxidative brain injury following hypoxia-ischemia in neonatal mice. J. Neurosci. 2012, 32, 3235–3244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimova, N.; Fearnow, A.; Long, A.; Kristian, T. NAD(+) precursor modulates post-ischemic mitochondrial fragmentation and reactive oxygen species generation via SIRT3 dependent mechanisms. Exp. Neurol. 2020, 325, 113144. [Google Scholar] [CrossRef] [PubMed]
- Oki, C.; Watanabe, Y.; Yokoyama, H.; Shimoda, T.; Kato, H.; Araki, T. Delayed treatment with arundic acid reduces the MPTP-induced neurotoxicity in mice. Cell Mol. Neurobiol. 2008, 28, 417–430. [Google Scholar] [CrossRef]
- Yokoyama, H.; Kuroiwa, H.; Yano, R.; Araki, T. Targeting reactive oxygen species, reactive nitrogen species and inflammation in MPTP neurotoxicity and Parkinson’s disease. Neurol. Sci. 2008, 29, 293–301. [Google Scholar] [CrossRef]
- Yokoyama, H.; Yano, R.; Aoki, E.; Kato, H.; Araki, T. Comparative pharmacological study of free radical scavenger, nitric oxide synthase inhibitor, nitric oxide synthase activator and cyclooxygenase inhibitor against MPTP neurotoxicity in mice. Metab. Brain Dis. 2008, 23, 335–349. [Google Scholar] [CrossRef]
- Levy, R.J.; Deutschman, C.S. Deficient mitochondrial biogenesis in critical illness: Cause, effect, or epiphenomenon? Crit. Care 2007, 11, 158. [Google Scholar] [CrossRef] [Green Version]
- Ame, J.C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. Bioessays 2004, 26, 882–893. [Google Scholar] [CrossRef]
- Endres, M.; Wang, Z.Q.; Namura, S.; Waeber, C.; Moskowitz, M.A. Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J. Cereb. Blood Flow Metab. 1997, 17, 1143–1151. [Google Scholar] [CrossRef] [Green Version]
- Lo, E.H.; Bosque-Hamilton, P.; Meng, W. Inhibition of poly(ADP-ribose) polymerase: Reduction of ischemic injury and attenuation of N-methyl-D-aspartate-induced neurotransmitter dysregulation. Stroke 1998, 29, 830–836. [Google Scholar] [CrossRef] [Green Version]
- Szabo, C.; Dawson, V.L. Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharmacol. Sci. 1998, 19, 287–298. [Google Scholar] [CrossRef]
- Park, J.H.; Long, A.; Owens, K.; Kristian, T. Nicotinamide mononucleotide inhibits post-ischemic NAD(+) degradation and dramatically ameliorates brain damage following global cerebral ischemia. Neurobiol. Dis. 2016, 95, 102–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, A.; Park, J.H.; Klimova, N.; Fowler, C.; Loane, D.J.; Kristian, T. CD38 Knockout Mice Show Significant Protection Against Ischemic Brain Damage Despite High Level Poly-ADP-Ribosylation. Neurochem. Res. 2017, 42, 283–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; Zhang, X.; Han, Y.Y.; Burke, N.A.; Kochanek, P.M.; Watkins, S.C.; Graham, S.H.; Carcillo, J.A.; Szabó, C.; Clark, R.S.B. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J. Biol. Chem. 2003, 278, 18426–18433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.; Chen, Y.; Watkins, S.C.; Nathaniel, P.D.; Guo, F.; Kochanek, P.M.; Jenkins, L.W.; Szabó, C.; Clark, R.S.B. Identification of poly-ADP-ribosylated mitochondrial proteins after traumatic brain injury. J. Neurochem. 2008, 104, 1700–1711. [Google Scholar] [CrossRef] [PubMed]
- Owens, K.; Park, J.H.; Schuh, R.; Kristian, T. Mitochondrial dysfunction and NAD(+) metabolism alterations in the pathophysiology of acute brain injury. Transl. Stroke Res. 2013, 4, 618–634. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Vassanelli, S.; Veronese, P.; Colonna, R.; Szabo, I.; Zoratti, M. Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J. Biol. Chem. 1992, 267, 2934–2939. [Google Scholar] [PubMed]
- Brustovetsky, N.; Dubinsky, J.M. Limitations of cyclosporin A inhibition of the permeability transition in CNS mitochondria. J. Neurosci. 2000, 20, 8229–8237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristian, T.; Fiskum, G. A fluorescence-based technique for screening compounds that protect against damage to brain mitochondria. Brain Res. Brain Res. Protoc. 2004, 13, 176–182. [Google Scholar] [CrossRef]
- Kristian, T.; Bernardi, P.; Siesjo, B.K. Acidosis promotes the permeability transition in energized mitochondria: Implications for reperfusion injury. J. Neurotrauma. 2001, 18, 1059–1074. [Google Scholar] [CrossRef] [PubMed]
- Kristian, T.; Gertsch, J.; Bates, T.E.; Siesjo, B.K. Characteristics of the calcium-triggered mitochondrial permeability transition in nonsynaptic brain mitochondria: Effect of cyclosporin A and ubiquinone O. J. Neurochem. 2000, 74, 1999–2009. [Google Scholar] [CrossRef] [Green Version]
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef] [Green Version]
- Nakai, A.; Kuroda, S.; Kristian, T.; Siesjo, B.K. The immunosuppressant drug FK506 ameliorates secondary mitochondrial dysfunction following transient focal cerebral ischemia in the rat. Neurobiol. Dis. 1997, 4, 288–300. [Google Scholar] [CrossRef] [Green Version]
- Marmorstein, R.; Trievel, R.C. Histone modifying enzymes: Structures, mechanisms, and specificities. Biochim. Biophys. Acta. 2009, 1789, 58–68. [Google Scholar] [CrossRef] [Green Version]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam. Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Klimova, N.; Long, A.; Kristian, T. Significance of Mitochondrial Protein Post-translational Modifications in Pathophysiology of Brain Injury. Transl. Stroke Res. 2018, 9, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Klimova, N.; Long, A.; Scafidi, S.; Kristian, T. Interplay between NAD(+) and acetylCoA metabolism in ischemia-induced mitochondrial pathophysiology. Biochim. Biophys. Acta. Mol. Basis. Dis. 2019, 1865, 2060–2067. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Kouzarides, T. Acetylation: A regulatory modification to rival phosphorylation? EMBO J. 2000, 19, 1176–1179. [Google Scholar] [CrossRef]
- Norvell, A.; McMahon, S.B. Cell biology. Rise of the rival. Science 2010, 327, 964–965. [Google Scholar] [CrossRef]
- Klimova, N.; Long, A.; Kristian, T. Nicotinamide mononucleotide alters mitochondrial dynamics by SIRT3-dependent mechanism in male mice. J. Neurosci. Res. 2019, 97, 975–990. [Google Scholar] [CrossRef] [Green Version]
- Nogueiras, R.; Habegger, K.M.; Chaudhary, N.; Finan, B.; Banks, A.S.; Dietrich, M.O.; Horvath, T.L.; Sinclair, D.A.; Pfluger, P.T.; Tschöp, M.H. Sirtuin 1 and sirtuin 3: Physiological modulators of metabolism. Physiol. Rev. 2012, 92, 1479–1514. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.; Yang, Y.; Zhou, Y.; Maharana, C.; Lu, D.; Peng, W.; Liu, Y.; Wan, R.; Marosi, K.; Misiak, M.; et al. Mitochondrial SIRT3 Mediates Adaptive Responses of Neurons to Exercise and Metabolic and Excitatory Challenges. Cell Metab. 2016, 23, 128–142. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekaran, K.; Hazelton, J.L.; Wang, Y.; Fiskum, G.; Kristian, T. Neuron-specific conditional expression of a mitochondrially targeted fluorescent protein in mice. J. Neurosci. 2006, 26, 13123–13127. [Google Scholar] [CrossRef] [Green Version]
- Zielonka, J.; Vasquez-Vivar, J.; Kalyanaraman, B. Detection of 2-hydroxyethidium in cellular systems: A unique marker product of superoxide and hydroethidine. Nat. Protoc. 2008, 3, 8–21. [Google Scholar] [CrossRef]
- Owens, K.; Park, J.H.; Gourley, S.; Jones, H.; Kristian, T. Mitochondrial dynamics: Cell-type and hippocampal region specific changes following global cerebral ischemia. J. Bioenerg. Biomembr. 2015, 47, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Bukowski, M.J.; Wider, J.M.; Reynolds, C.A.; Calo, L.; Lepore, B.; Tousignant, R.; Jones, M.; Przyklenk, K.; Sanderson, T.H. Mitochondrial dynamics following global cerebral ischemia. Mol. Cell Neurosci. 2016, 76, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Shen, Y.; Zhou, L.; Sangwung, P.; Fujioka, H.; Zhang, L.; Liao, X. Short-term administration of Nicotinamide Mononucleotide preserves cardiac mitochondrial homeostasis and prevents heart failure. J. Mol. Cell Cardiol. 2017, 112, 64–73. (In English) [Google Scholar] [CrossRef]
- Hosseini, L.; Vafaee, M.S.; Badalzadeh, R. Melatonin and Nicotinamide Mononucleotide Attenuate Myocardial Ischemia/Reperfusion Injury via Modulation of Mitochondrial Function and Hemodynamic Parameters in Aged Rats. J. Cardiovasc. Pharmacol. Ther. 2020, 25, 240–250. (In English) [Google Scholar] [CrossRef]
- Wang, X.; Hu, X.; Zhang, L.; Xu, X.; Sakurai, T. Nicotinamide mononucleotide administration after sever hypoglycemia improves neuronal survival and cognitive function in rats. Brain Res. Bull. 2020, 160, 98–106. (In English) [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Shen, X.; Zhao, Y.; Zhao, R.; He, X.; Yin, J.; Su, J.; Li, Q.; Liu, J. Mechanisms of transformation of nicotinamide mononucleotides to cerebral infarction hemorrhage based on MCAO model. Saudi J. Biol. Sci. 2020, 27, 899–904. (In English) [Google Scholar] [CrossRef]
- Jou, M.J. Pathophysiological and pharmacological implications of mitochondria-targeted reactive oxygen species generation in astrocytes. Adv. Drug Deliv. Rev. 2008, 60, 1512–1526. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Horváth, B.; Zsengellėr, Z.; Bátkai, S.; Cao, Z.; Kechrid, M.; Holovac, E.; Erdėlyi, K.; Tanchian, G.; Liaudet, L.; et al. Mitochondrial reactive oxygen species generation triggers inflammatory response and tissue injury associated with hepatic ischemia-reperfusion: Therapeutic potential of mitochondrially targeted antioxidants. Free Radic. Biol. Med. 2012, 53, 1123–1138. [Google Scholar] [CrossRef] [Green Version]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.J.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005, 19, 1088–1095. [Google Scholar] [CrossRef]
- Walters, A.M.; Porter, G.A.J.; Brookes, P.S. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circ. Res. 2012, 111, 1222–1236. [Google Scholar] [CrossRef]
- Plotnikov, E.Y.; Zorov, D.B. Pros and Cons of Use of Mitochondria-Targeted Antioxidants. Antioxidants 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klimova, N.; Fearnow, A.; Kristian, T. Role of NAD+—Modulated Mitochondrial Free Radical Generation in Mechanisms of Acute Brain Injury. Brain Sci. 2020, 10, 449. https://doi.org/10.3390/brainsci10070449
Klimova N, Fearnow A, Kristian T. Role of NAD+—Modulated Mitochondrial Free Radical Generation in Mechanisms of Acute Brain Injury. Brain Sciences. 2020; 10(7):449. https://doi.org/10.3390/brainsci10070449
Chicago/Turabian StyleKlimova, Nina, Adam Fearnow, and Tibor Kristian. 2020. "Role of NAD+—Modulated Mitochondrial Free Radical Generation in Mechanisms of Acute Brain Injury" Brain Sciences 10, no. 7: 449. https://doi.org/10.3390/brainsci10070449