Relative Stability of Small Silver, Platinum, and Palladium Doped Gold Cluster Cations


Abstract
:Featured Application
Abstract
1. Introduction
2. Methods
2.1. Photofragmentation Experiments
2.2. Theoretical Calculations
3. Results
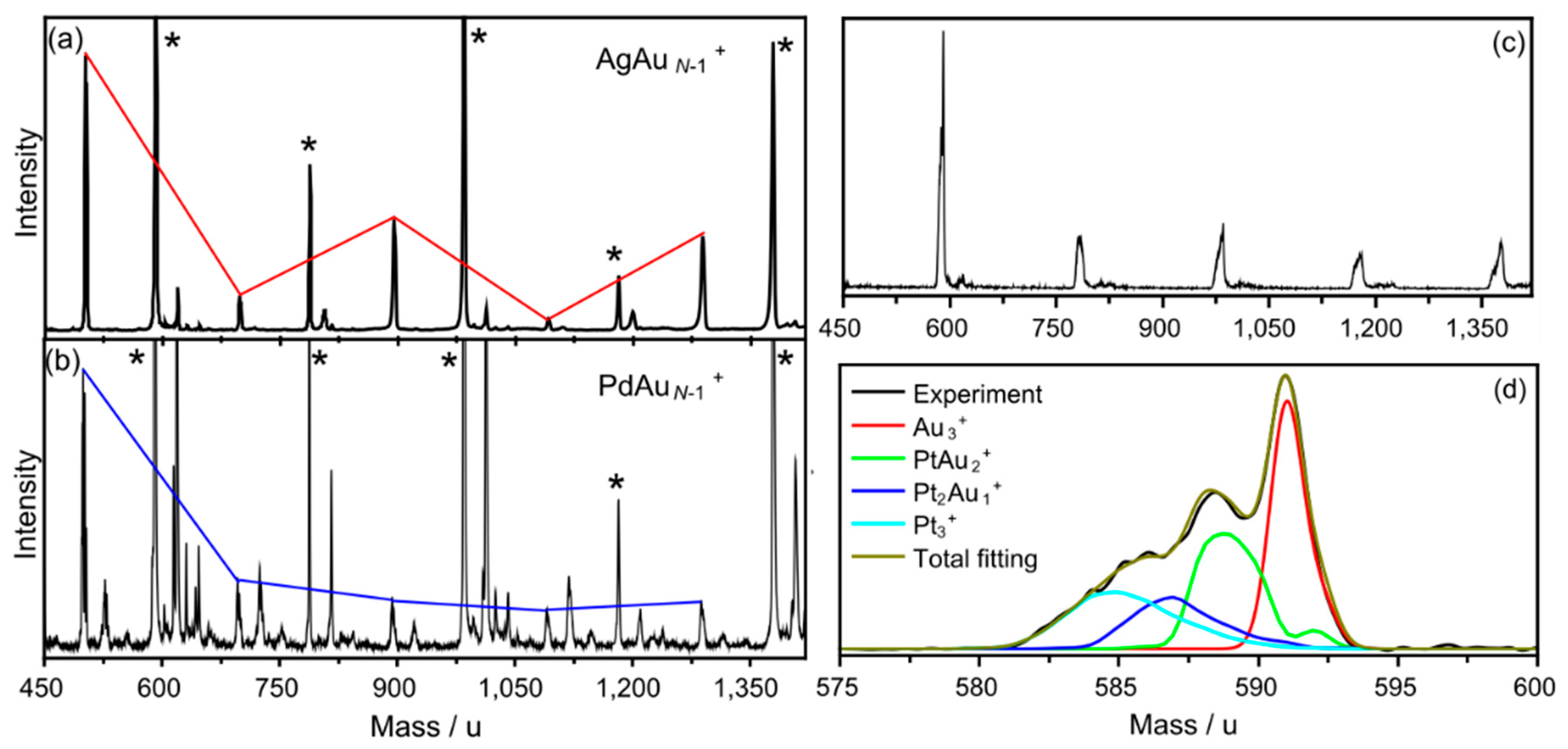
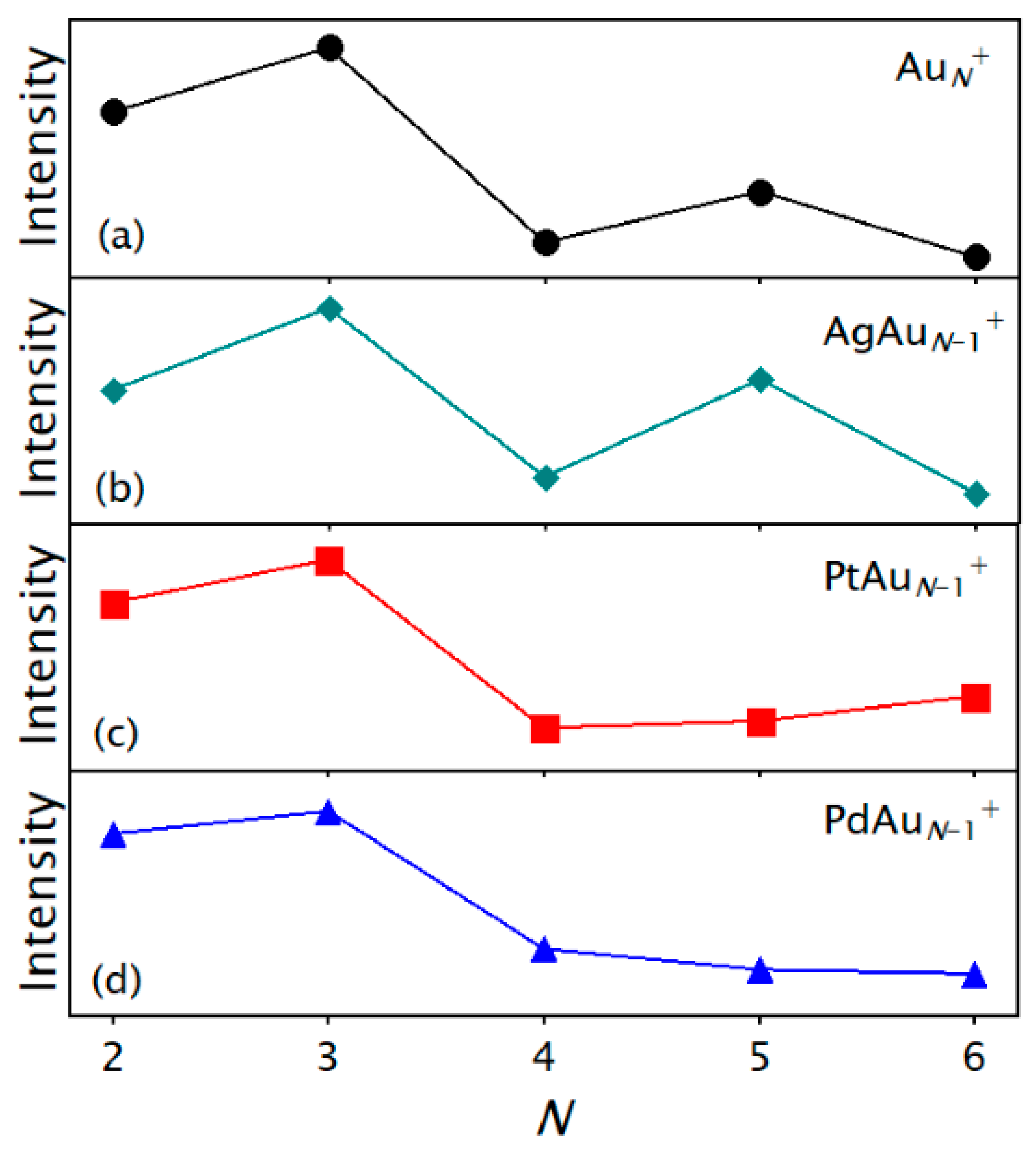
3.1. Abundances of Photofragmented Clusters
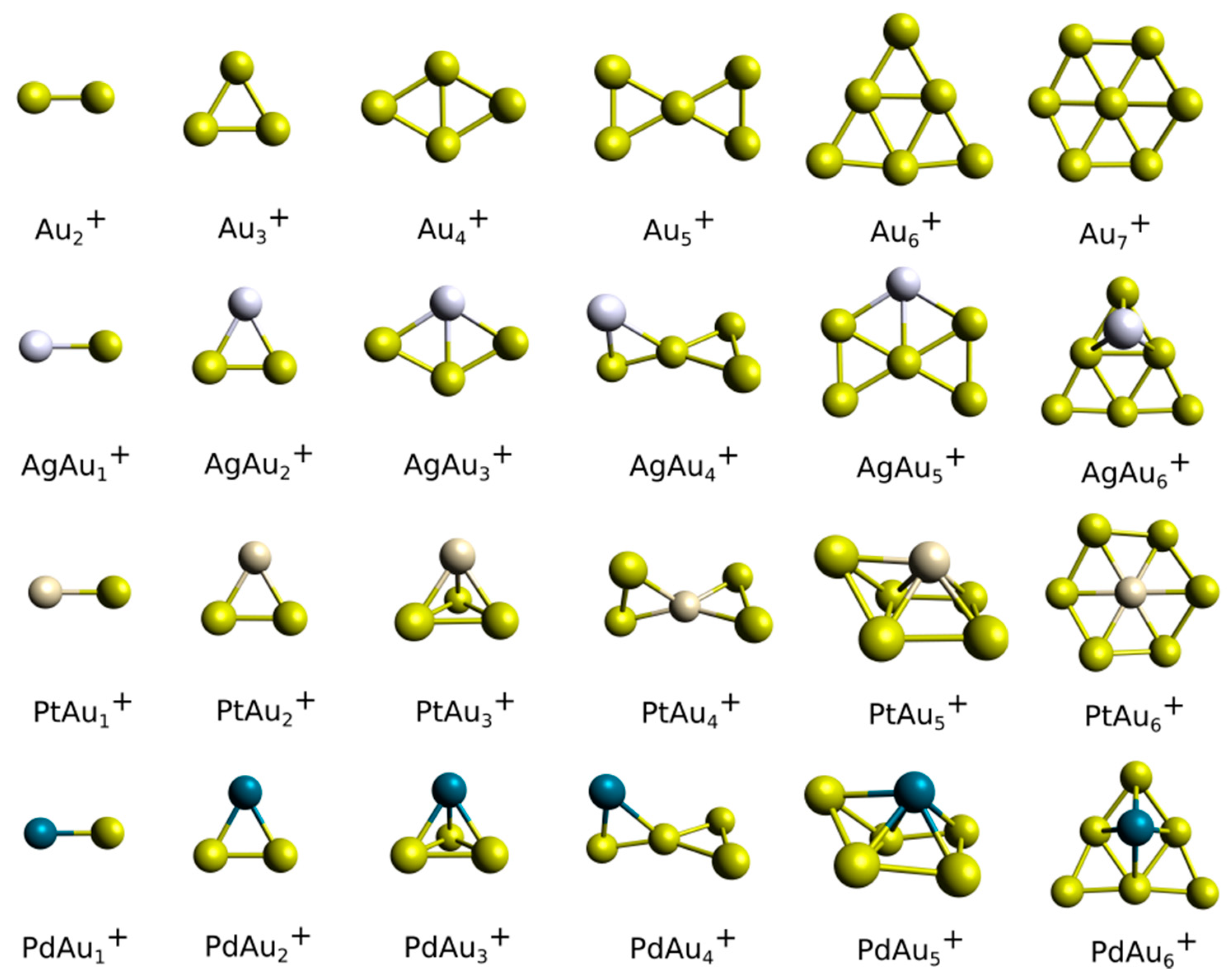
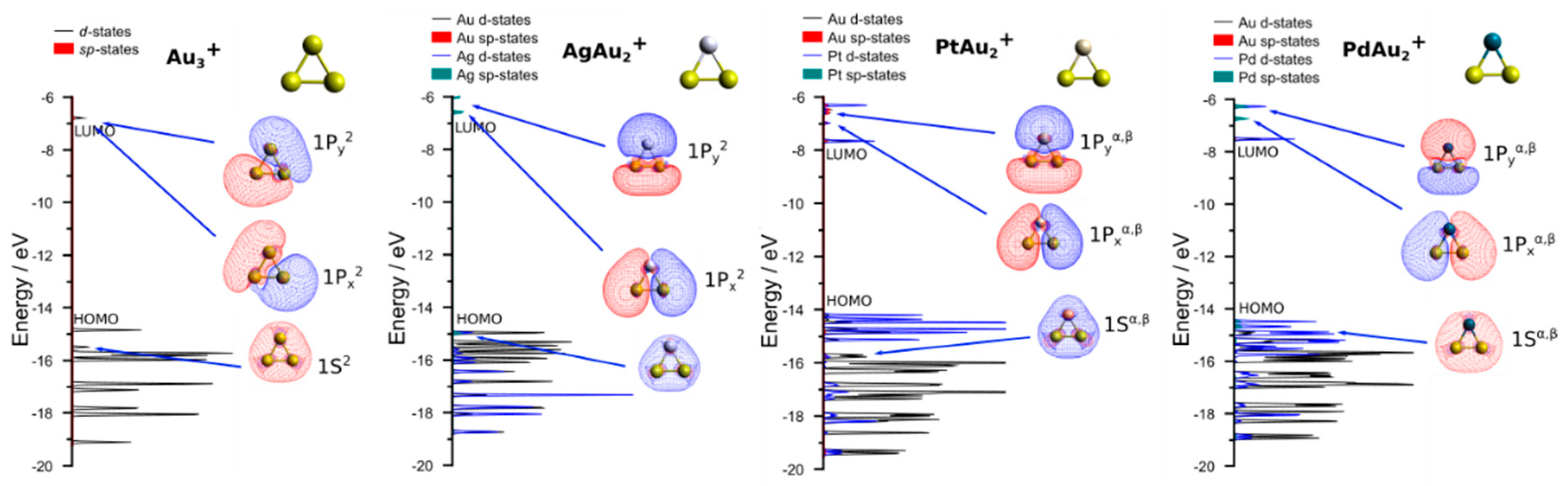
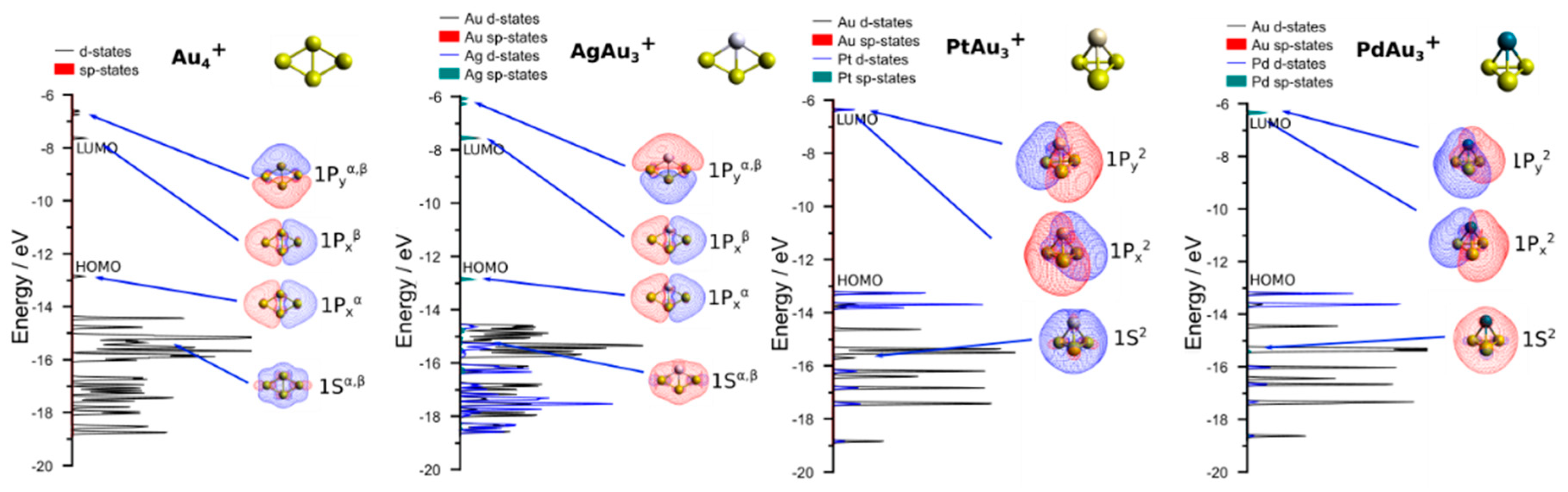
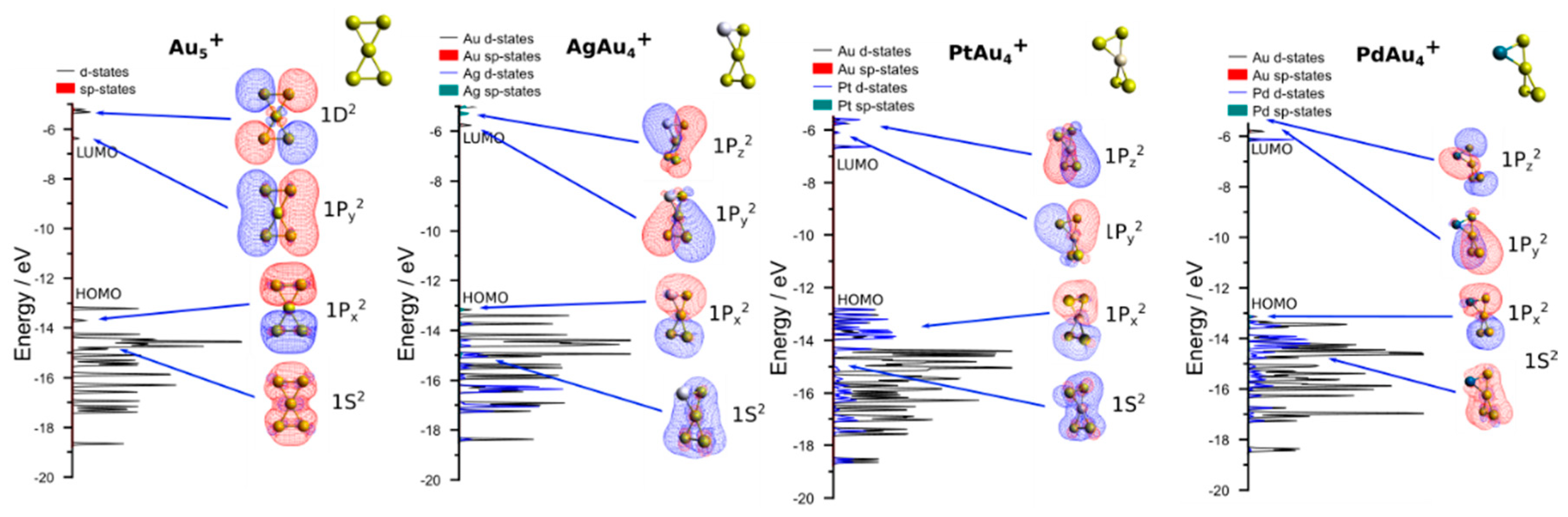
3.2. Theoretical Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liao, T.-W.; Yadav, A.; Hu, K.-J.; van der Tol, J.; Cosentino, S.; D’Acapito, F.; Palmer, R.E.; Lenardi, C.; Ferrando, R.; Grandjean, D.; et al. Unravelling the nucleation mechanism of bimetallic nanoparticles with composition-tunable core–shell arrangemen. Nanoscale 2018, 10, 6684. [Google Scholar] [CrossRef]
- Neukermans, S.; Janssens, E.; Chen, Z.F.; Silverans, R.E.; Schleyer, P.v.R.; Lievens, P. Extremely Stable Metal-Encapsulated AlPb10+ and AlPb12+ Clusters: Mass-Spectrometric Discovery and Density Functional Theory Study. Phys. Rev. Lett. 2004, 92, 163401. [Google Scholar] [CrossRef]
- Zhou, S.; Li, J.; Schlangen, M.; Schwarz, H. Bond Activation by Metal–Carbene Complexes in the Gas Phase. Acc. Chem. Res 2016, 49, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, P.; Molina, L.M.; Kaydashev, V.E.; Alonso, J.A.; Lievens, P.; Janssens, E. Controlling the Adsorption of Carbon Monoxide on Platinum Clusters by Dopant-Induced Electronic Structure Modification. Angew. Chem. Int. Ed. 2016, 55, 11059–11063. [Google Scholar] [CrossRef] [Green Version]
- Philip, R.; Chantharasupawong, P.; Qian, H.; Jin, R.; Thomas, J. Evolution of Nonlinear Optical Properties: From Gold Atomic Clusters to Plasmonic Nanocrystals. Nano Lett. 2012, 12, 4661. [Google Scholar] [CrossRef] [PubMed]
- Gloess, A.N.; Schneider, H.; Weber, J.M.; Kappes, M.M. Electronically excited states and visible region photodissociation spectroscopy of Aum+⋅Arn. J. Chem. Phys. 2008, 128, 114312. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yin, S.; Moro, R.; de Heer, W.A. Magnetic Moments and Adiabatic Magnetization of Free Cobalt Clusters. Phys. Rev. Lett. 2005, 95, 237209. [Google Scholar] [CrossRef] [PubMed]
- Ngan, V.T.; Janssens, E.; Claes, P.; Lyon, J.T.; Fielicke, A.; Nguyen, M.T.; Lievens, P. High Magnetic Moments in Manganese-Doped Silicon Clusters. Chem. Eur. J. 2012, 18, 15788. [Google Scholar] [CrossRef]
- Haruta, M.; Yamada, N.; Kobayashi, T.; Iijima, S. Gold catalysts prepared by coprecipitation for low-temperature oxidation of hydrogen and of carbon monoxide. J. Catal. 1989, 115, 301–309. [Google Scholar] [CrossRef]
- Neumaier, M.; Weigend, F.; Hampe, O.; Kappes, M.M. Binding energies of CO on gold cluster cations Aun+ (n = 1–65): A radiative association kinetics study. J. Chem. Phys. 2005, 122, 104702. [Google Scholar] [CrossRef]
- Janssens, E.; Le, H.T.; Lievens, P. Adsorption of Propene on Neutral Gold Clusters in the Gas Phase. Chem. Eur. J. 2015, 21, 15256–15262. [Google Scholar] [CrossRef]
- Lang, S.M.; Bernhardt, T.M.; Barnett, R.N.; Landman, U. Methane activation and catalytic ethylene formation on free Au2+. Angew. Chem. Int. Ed. 2010, 49, 980–983. [Google Scholar] [CrossRef]
- Hammer, B.; Norskov, J.K. Why gold is the noblest of all the metals. Nature 1995, 376, 238–240. [Google Scholar] [CrossRef]
- Shayeghi, A.; Heard, C.J.; Johnston, R.L.; Schäfer, R. Optical and electronic properties of mixed Ag-Au tetramer cations. J. Chem. Phys. 2014, 140, 054312. [Google Scholar] [CrossRef]
- Gruene, P.; Rayner, D.M.; Redlich, B.; van der Meer, A.F.G.; Lyon, J.T.; Meijer, G.; Fielicke, A. Structures of Neutral Au7, Au19, and Au20 Clusters in the Gas Phase. Science 2008, 321, 674. [Google Scholar] [CrossRef]
- Gilb, S.; Weis, P.; Furche, P.; Ahlrichs, R.; Kappes, M.M. Structures of small gold cluster cations (Aun+, n < 14): Ion mobility measurements versus density functional calculations. J. Chem. Phys. 2002, 116, 4094. [Google Scholar] [CrossRef]
- Furche, F.; Ahlrichs, R.; Weis, P.; Jacob, C.; Gilb, S.; Bierweiler, T.; Kappes, M.M. The structures of small gold cluster anions as determined by a combination of ion mobility measurements and density functional calculations. J. Chem. Phys. 2002, 117, 6982. [Google Scholar] [CrossRef]
- Häkkinen, H.; Yoon, B.; Landman, U.; Li, X.; Zhai, H.-J.; Wang, L.-S. On the Electronic and Atomic Structures of Small AuN− (N = 4−14) Clusters: A Photoelectron Spectroscopy and Density-Functional Study. J. Phys. Chem. A 2003, 107, 6168. [Google Scholar] [CrossRef]
- Xing, X.; Yoon, B.; Landman, U.; Parks, J.H. Structural evolution of Au nanoclusters: From planar to cage to tubular motifs. Phys. Rev. B 2006, 74, 165423. [Google Scholar] [CrossRef]
- Häkkinen, H. Electronic shell structures in bare and protected metal nanoclusters. Adv. Phys. X 2016, 1, 467–491. [Google Scholar] [CrossRef] [Green Version]
- Atkins, P.W.; Friedman, R.S. Molecular Quantum Mechanics, 5th ed.; Oxford University Press Inc.: New York, NY, USA, 2011; ISBN 978-0-19-954142-3. [Google Scholar]
- De Heer, W.A. The physics of simple metal clusters: Experimental aspects and simple models. Rev. Mod. Phys. 1993, 65, 611. [Google Scholar] [CrossRef]
- Reber, A.C.; Khanna, S.N. Superatoms: Electronic and Geometric Effects on Reactivity. Acc. Chem. Res. 2017, 50, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Knight, W.D.; Clemenger, K.; de Heer, W.A.; Saunders, W.A.; Chou, M.Y.; Cohen, M.L. Electronic Shell Structure and Abundances of Sodium Clusters. Phys. Rev. Lett. 1984, 53, 510. [Google Scholar] [CrossRef]
- Veldeman, N.; Janssens, E.; Hansen, K.; De Haeck, J.; Silverans, R.E.; Lievens, P. Stability and dissociation pathways of doped AunX+ clusters (X = Y, Er, Nb). Faraday Discuss. 2008, 138, 147–162. [Google Scholar] [CrossRef]
- Hansen, K.; Ferrari, P.; Janssens, E.; Lievens, P. Thermal radiation of gold clusters on microsecond time scales. Phys. Rev. A 2017, 96, 022511. [Google Scholar] [CrossRef] [Green Version]
- Neukermans, S.; Janssens, E.; Tanaka, H.; Silverans, R.E.; Lievens, P. Element- and Size-Dependent Electron Delocalization in AuNX+ Clusters (X = Sc, Ti, V, Cr, Mn, Fe, Co, Ni). Phys. Rev. Lett. 2003, 90, 033401. [Google Scholar] [CrossRef]
- Ferrari, P.; Vanbuel, J.; Janssens, E.; Lievens, P. Tuning the Reactivity of Small Metal Clusters by Heteroatom Doping. Acc. Chem. Res. 2018, 51, 3174. [Google Scholar] [CrossRef] [PubMed]
- Neumaier, M.; Weigend, F.; Hampe, O.; Kappes, M.M. Reactions of mixed silver-gold cluster cations AgmAun+ (m + n = 4, 5, 6) with CO: Radiative association kinetics and density functional theory computations. J. Chem. Phys. 2006, 125, 104308. [Google Scholar] [CrossRef]
- Vanbuel, J.; Fernández, E.M.; Ferrari, P.; Gewinner, S.; Schöllkopf, W.; Balbás, L.C.; Fielicke, A.; Janssens, E. Hydrogen Chemisorption on Singly Vanadium-Doped Aluminum Clusters. Chem. Eur. J. 2017, 23, 15638–15643. [Google Scholar] [CrossRef]
- Medel, V.M.; Reber, A.C.; Chauhan, V.; Sen, P.; Köster, A.M.; Calaminici, P.; Khanna, S.N. Nature of Valence Transition and Spin Moment in AgnV+ Clusters. J. Am. Chem. Soc. 2014, 136, 8229–8236. [Google Scholar] [CrossRef]
- Janssens, E.; Neukermans, S.; Wang, X.; Veldeman, N.; Silverans, R.E.; Lievens, P. Stability patterns of transition metal doped silver clusters: Dopant- and size-dependent electron delocalization. Eur. Phys. J. D 2005, 34, 23–27. [Google Scholar] [CrossRef]
- Kaydashev, V.; Ferrari, P.; Heard, C.; Janssens, E.; Johnston, R.L.; Lievens, P. Optical Absorption of Small Palladium-Doped Gold Clusters. Part. Part. Syst. Charact. 2016, 33, 364–372. [Google Scholar] [CrossRef]
- Jia, M.; van der Tol, J.; Li, Y.; Chernyy, V.; Bakker, J.M.; Pham, L.N.; Nguyen, M.T.; Janssens, E. Structures and magnetic properties of small Con+ and Con−1Cr+ (n = 3–5) clusters. J. Phys. Condens. Matter 2018, 30, 474002. [Google Scholar] [CrossRef]
- Ferrari, P.; Hussein, H.A.; Heard, C.J.; Vanbuel, J.; Johnston, R.L.; Lievens, P.; Janssens, E. Effect of palladium doping on the stability and fragmentation patterns of cationic gold clusters. Phys. Rev. A 2018, 97, 052508. [Google Scholar] [CrossRef]
- Ferrari, P.; Vanbuel, J.; Li, Y.; Liao, T.-W.; Janssens, E.; Lievens, P. Modifications of gas aggregation sources: The double laser ablation source approach. In Gas Aggregation Synthesis of Nanoparticles, 1st ed.; Huttel, Y., Ed.; Wiley-VCH: Weinheim, Germany, 2017; pp. 59–78. ISBN 978-3-527-34060-6. [Google Scholar]
- Duncan, M.A. Invited Review Article: Laser vaporization cluster sources. Rev. Sci. Instrum. 2012, 83, 041101. [Google Scholar] [CrossRef] [PubMed]
- Geusic, M.E.; Jarrold, M.F.; McIlrath, T.J.; Freeman, R.R.; Brown, W.L. Photodissociation of carbon cluster cations. J. Chem. Phys. 1987, 86, 3862. [Google Scholar] [CrossRef]
- Hansen, K.; Herlert, A.; Schweikhard, L.; Vogel, M. Dissociation energies of gold clusters AuN+, N = 7–27. Phys. Rev. A 2006, 73, 063202. [Google Scholar] [CrossRef]
- Ferrari, P.; Hansen, K.; Lievens, P.; Janssens, E. Stability of small cationic platinum clusters. Phys. Chem. Chem. Phys. 2018, 20, 29085–29090. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Lu, P.; Kuang, X.Y.; Mao, A.J.; Wang, Z.H.; Zhao, Y.R. Structural and electronic properties of silver-doped gold clusters AunAgv (2 ≤ n ≤ 10; v = 0, ±1): Comparison with pure gold clusters. Mol. Phys. 2011, 109, 2057–2068. [Google Scholar] [CrossRef]
- Weis, P.; Welz, O.; Vollmer, E.; Kappes, M.M. Structures of mixed gold-silver cluster cations (AgmAun+, m + n < 6): Ion mobility measurements and density-functional calculations. J. Chem. Phys. 2004, 120, 677. [Google Scholar] [CrossRef]
- Shayeghi, A.; Johnston, R.L.; Rayner, D.M.; Schfer, R.; Fielicke, A. The Nature of Bonding between Argon and Mixed Gold–Silver Trimers. Angew. Chem. Int. Ed. 2015, 54, 10675. [Google Scholar] [CrossRef] [PubMed]
- Shayeghi, A.; Schäfer, R.; Rayner, D.M.; Johnston, R.L.; Fielicke, A. Charge-induced dipole vs. relativistically enhanced covalent interactions in Ar-tagged Au-Ag tetramers and pentamers. J. Chem. Phys. 2015, 143, 024310. [Google Scholar] [CrossRef]
- Weigenda, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Pyykko, P. Relativistic effects in structural chemistry. Chem. Rev. 1988, 88, 563–594. [Google Scholar] [CrossRef]
- Van Wüllen, C. Molecular density functional calculations in the regular relativistic approximation: Method, application to coinage metal diatomics, hydrides, fluorides and chlorides, and comparison with first-order relativistic calculations. J. Chem. Phys. 1998, 109, 392. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Chen, X.-Y.; Landis, C.R.; Neese, N. All-Electron Scalar Relativistic Basis Sets for Third-Row Transition Metal Atoms. J. Chem. Theory Comput. 2008, 4, 908–919. [Google Scholar] [CrossRef]
- Häkkinen, H.; Landman, U. Gold clusters (AuN, 2<~N<~10) and their anions. Phys. Rev. B 2000, 62, R2287. [Google Scholar] [CrossRef]
- Massen, C.; Mortimer-Jones, T.V.; Johnston, R.L. Geometries and segregation properties of platinum–palladium nanoalloy clusters. J. Chem. Soc. Dalton Trans. 2002, 0, 4375–4388. [Google Scholar] [CrossRef]
- Martínez, J.I.; Alonso, J.A. An improved descriptor of cluster stability: Application to small carbon clusters. Phys. Chem. Chem. Phys. 2018, 20, 27368–27374. [Google Scholar] [CrossRef]
- Koyasu, K.; Mitsui, M.; Nakajima, A.; Kaya, K. Photoelectron spectroscopy of palladium-doped gold cluster anions; AunPd− (n = 1–4). Chem. Phys. Lett. 2002, 358, 224–230. [Google Scholar] [CrossRef]
- Janssens, E.; Tanaka, H.; Neukermans, S.; Silverans, R.E.; Lievens, P. Two-dimensional magic numbers in mass abundances of photofragmented bimetallic clusters. New J. Phys. 2003, 5, 46.1–46.10. [Google Scholar] [CrossRef]
- Ferrighi, L.; Hammer, B.; Madsen, G.K.H. 2D−3D Transition for Cationic and Anionic Gold Clusters: A Kinetic Energy Density Functional Study. J. Am. Chem. Soc. 2009, 131, 10605–10609. [Google Scholar] [CrossRef] [PubMed]
- Van der Tol, J.; Jia, D.; Li, Y.; Chernyy, V.; Bakker, J.M.; Nguyen, M.T.; Lievens, P.; Janssens, E. Structural assignment of small cationic silver clusters by far-infrared spectroscopy and DFT calculations. Phys. Chem. Chem. Phys. 2017, 19, 19360–19368. [Google Scholar] [CrossRef] [PubMed]
- Harding, D.J.; Kerpal, C.; Rayner, D.M.; Fielicke, A. Communication: The structures of small cationic gas-phase platinum clusters. J. Chem. Phys. 2012, 136, 211103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, X.; Hermann, A.; Kuang, X.; Ju, M.; Lu, C.; Jin, Y.; Xia, X.; Maroulis, G. Insights into the geometries, electronic and magnetic properties of neutral and charged palladium clusters. Sci. Rep. 2016, 6, 19656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, M.; Hansen, K.; Herlert, A.; Schweikhard, L. Decay pathways of small gold clusters. Eur. Phys. J. D 2001, 16, 73–76. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dissociation Channel | DN/eV | |
|---|---|---|
| Au3+ | → Au1+ + Au2 | 4.14 |
| Au4+ | → Au3+ + Au1 | 2.25 |
| Au5+ | → Au3+ + Au2 | 2.58 |
| Au6+ | → Au5+ + Au1 | 2.71 |
| Au7+ | → Au5+ + Au2 | 3.20 |
| AgAu2+ | → Au1+ + AgAu1 | 4.69 |
| AgAu3+ | → AgAu2+ + Au1 | 2.18 |
| AgAu4+ | → AgAu2+ + Au2 | 2.44 |
| AgAu5+ | → AgAu4+ + Au1 | 2.51 |
| AgAu6+ | → AgAu4+ + Au2 | 3.41 |
| PtAu2+ | → Pt1+ + Au2 | 3.97 |
| PtAu3+ | → PtAu2+ + Au1 | 3.36 |
| PtAu4+ | → PtAu3+ + Au1 | 2.69 |
| → PtAu2+ + Au2 | 2.89 | |
| → Au3+ + PtAu1 | 2.91 | |
| PtAu5+ | → PtAu3+ + Au2 | 2.62 |
| PtAu6+ | → PtAu4+ + Au2 | 3.61 |
| → PtAu5+ + Au1 | 3.67 | |
| PdAu2+ | → Pd1+ + Au2 | 2.83 |
| PdAu3+ | → PdAu2+ + Au1 | 3.19 |
| PdAu4+ | → PdAu3+ + Au1 | 2.51 |
| → PdAu2+ + Au2 | 2.54 | |
| PdAu5+ | → PdAu3+ + Au2 | 2.46 |
| PdAu6+ | → PdAu4+ + Au2 | 3.32 |
| → PdAu5+ + Au1 | 3.37 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrari, P.; Janssens, E. Relative Stability of Small Silver, Platinum, and Palladium Doped Gold Cluster Cations. Appl. Sci. 2019, 9, 1666. https://doi.org/10.3390/app9081666
Ferrari P, Janssens E. Relative Stability of Small Silver, Platinum, and Palladium Doped Gold Cluster Cations. Applied Sciences. 2019; 9(8):1666. https://doi.org/10.3390/app9081666
Chicago/Turabian StyleFerrari, Piero, and Ewald Janssens. 2019. "Relative Stability of Small Silver, Platinum, and Palladium Doped Gold Cluster Cations" Applied Sciences 9, no. 8: 1666. https://doi.org/10.3390/app9081666