A Comparison in the Use of the Crystallographic Structure of the Human A1 or the A2A Adenosine Receptors as a Template for the Construction of a Homology Model of the A3 Subtype



Abstract
:Featured Application
Abstract

1. Introduction
2. Materials and Methods
2.1. Computational Facilities
2.2. Protein Preparation
2.3. Sequence Alignment
2.4. Homology Modeling
2.5. Agonists and Antagonists Selection
2.6. Molecular Docking
2.7. Docking Analysis
2.8. Docking-based Homology Models’ Comparison
- one pose is better than the other (x model pose is better than y model pose, blue spots, −+; y model pose is better than x model pose, red spots, +−);
- both the poses are disfavored (++, orange spots);
- both the poses are favored (−−, white spots).
2.9. MMSDocking Video Maker
3. Results and Discussion
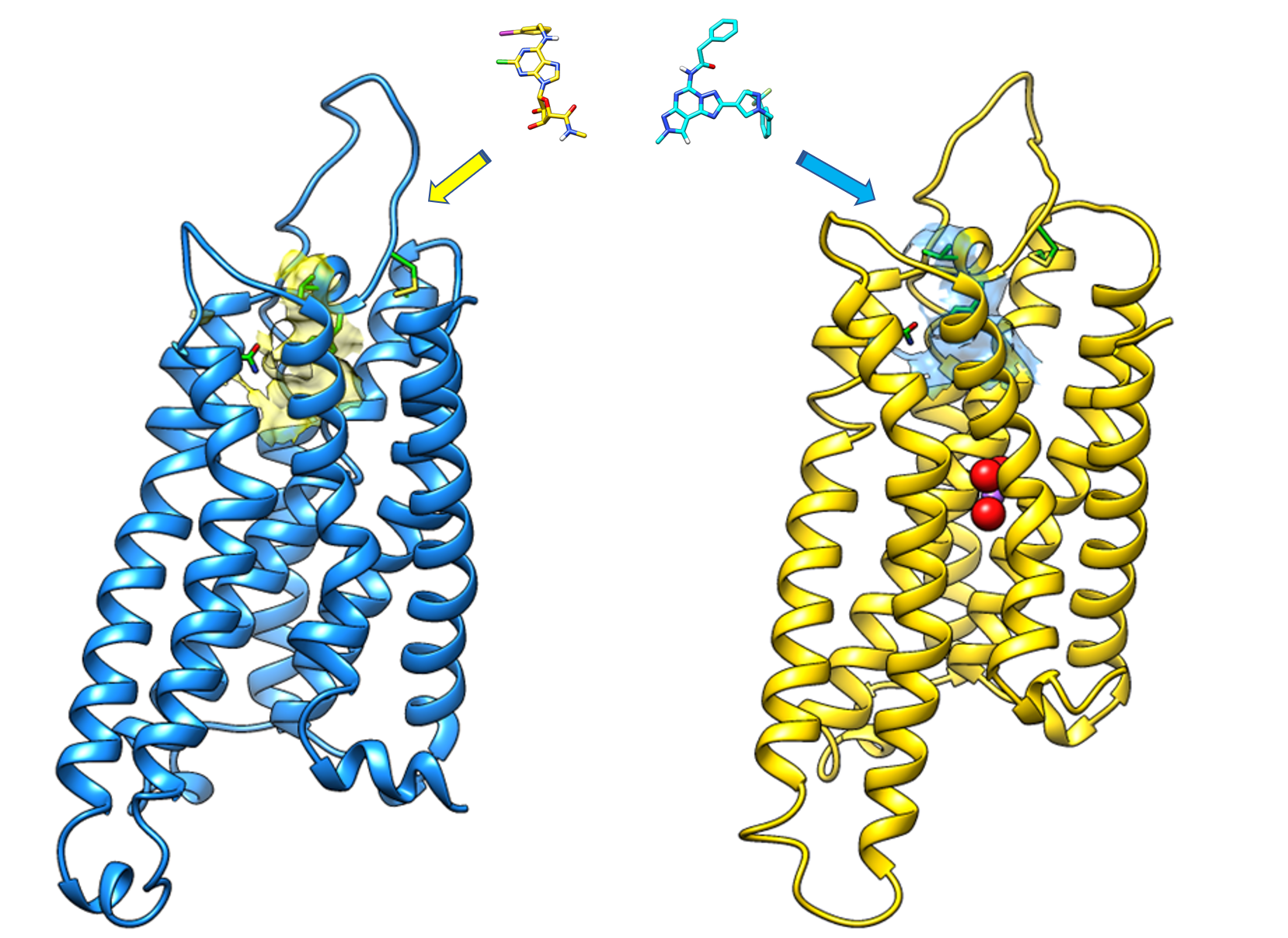
3.1. Sequence Comparison and Homology Modeling
3.2. “Agonist-driven” hA3 AR Models
3.3. “Antagonist-driven” hA3 AR Models
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | adenylate cyclase |
| ADO | adenosine |
| AR | adenosine receptor |
| BW-A522 | 3-(3-iodo-4-aminobenzyl)-8-(4-oxyacetate)-1-propylxanthine |
| CI-IB-MECA | Namodenoson |
| CVL3 | N-{8-methyl-2-[1-(3-trifluoromethyl-benzyl)-1H-pyrazol-4-yl]-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-yl}-phenyl-acetamide |
| DZ20 | 2-furan-2-yl-5-methylamino-[1,2,4]triazolo[1,5-c]pyrimidine-8-carboxylic Acid Ethyl Ester |
| DZ86 | 2-Furan-2-yl-5-methylamino-[1,2,4]triazolo[1,5-c]pyrimidine-8-carboxylic acid 4-trifluoromethyl-benzylamide |
| EL | extracellular loop |
| Ele | electrostatic |
| GPCR | G protein-coupled receptor |
| h | human |
| Hyd | hydrophobic |
| IL | intracellular loop |
| -in | induced fit |
| I125-APNEA | (N6-2-(4-amino-3iodophenyl)ethyladenosine) |
| MRS-1220 | N-[9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazolin-5-yl]benzeneacetamide |
| MRS-1334 | 1,4-Dihydro-2-methyl-6-phenyl-4-(phenylethynyl)-3,5-pyridinedicarboxylic acid 3-ethyl-5-[(3-nitrophenyl)methyl] ester |
| MRS-5127 | (1′R,2′R,3′S,4′R,5′S)-4′-[2-chloro-6-(3-iodobenzylamino)-purine]-2′,3′-O-dihydroxybicyclo-[3.1.0]hexane |
| MRS-5644 | (1S,2R,3S,4R,5S)-2,3-Dihydroxy-N-methyl-4-(6-(methylamino)-2-(phenylethynyl)-9H-purin-9-yl)bicyclo[3.1.0]hexane-1-carboxamide |
| MRS-7110 | (1S,2R,3S,4R,5S)-2,3dihydroxy-N-methyl-4-[6-(methylamino)-2-(4-phenyl-1H-1,2,3-triazol-1-yl)-5,9-dihydro-4H-purin-9-yl]bicyclo[3.1.0]hexane-1-carboxamide |
| NECA | 5′-N-ethyl-carboxamidoadenosine |
| R | R configuration |
| r | rat |
| (R)-PIA | (R)-(-)-(N6)-1-phenyl-2-propyladenosine |
| RMSD | root mean square deviation |
| S | S configuration |
| SI | sequence identity |
| SS | sequence similarity |
| TM | transmembrane domain |
| vdW | van der Waals |
| VS | virtual screening |
| VUF-5455 | 4-methoxy-N-(7-methyl-3-(2-pyridinyl)-1-isoquinolinyl)benzamide |
| WS51 | (S)-(2-Furan-2-yl-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-yl)-(1-phenylethyl)amine |
| WS60 | Benzhydryl(2-furan-2-yl-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-yl)amine |
| Z82 | N7-Benzyl-2-(furan-2-yl)-N5-isopropyl-[1,2,4]triazolo[1,5-a][1,3,5]triazine-5,7-diamine |
| ZM-241385 | 4-(2-(7-amino-2-(2-furyl)-(1,2,4)triazolo(2,3-a)-(1,3,5)triazin-5-yl-amino)ethyl)phenol |
References
- Kenneth, A.; Jacobson. Adenosine Receptors in Health Disease, Handbook of Experimental Pharmacology; Constance, N., Wilson, S., Mustafa, J., Eds.; Springer: Berlin, Germany, 2009. [Google Scholar]
- Meyerhof, W.; Müller-Brechlin, R.; Richter, D. Molecular cloning of a novel putative G-protein coupled receptor expressed during rat spermiogenesis. FEBS Lett. 1991, 284, 155–160. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.Y.; Li, C.; Olah, M.E.; Johnson, R.A.; Stiles, G.L.; Civelli, O. Molecular cloning and characterization of an adenosine receptor: The A3 adenosine receptor. Proc. Natl. Acad. Sci. USA 1992, 89, 7432–7436. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef] [PubMed]
- Janes, K.; Symons-Liguori, A.M.; Jacobson, K.A.; Salvemini, D. Identification of A3 adenosine receptor agonists as novel non-narcotic analgesics. Br. J. Pharmacol. 2016, 173, 1253–1267. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.; Bar-Yehuda, S.; Liang, B.T.; Jacobson, K.A. Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discov. Today 2012, 17, 359–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.A.; Spina, D.; Page, C.P. Adenosine receptors and asthma. Br. J. Pharmacol. 2008, 153 (Suppl. 1), S446–S456. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Do, C.W.; Avila, M.Y.; Peterson-Yantorno, K.; Stone, R.A.; Gao, Z.-G.; Joshi, B.; Besada, P.; Jeong, L.S.; Jacobson, K.A.; et al. Nucleoside-derived antagonists to A3 adenosine receptors lower mouse intraocular pressure and act across species. Exp. Eye Res. 2010, 90, 146–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciancetta, A.; Sabbadin, D.; Federico, S.; Spalluto, G.; Moro, S. Advances in Computational Techniques to Study GPCR-Ligand Recognition. Trends Pharmacol. Sci. 2015, 36, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Deganutti, G.; Cuzzolin, A.; Ciancetta, A.; Moro, S. Understanding allosteric interactions in G protein-coupled receptors using Supervised Molecular Dynamics: A prototype study analysing the human A3 adenosine receptor positive allosteric modulator LUF6000. Bioorg. Med. Chem. 2015, 23, 4065–4071. [Google Scholar] [CrossRef] [PubMed]
- Moro, S.; Morizzo, E.; Jacobson, K.A. Molecular modeling and reengineering of A3 adenosine receptors. In A3 Adenosine Receptors from Cell Biology to Pharmacology and Therapeutics; Borea, P.A., Ed.; Springer: Dordrecht, The Netherlands, 2010; pp. 149–161. [Google Scholar]
- Sabbadin, D.; Moro, S. Supervised molecular dynamics (SuMD) as a helpful tool to depict GPCR-ligand recognition pathway in a nanosecond time scale. J. Chem. Inf. Model. 2014, 54, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Cuzzolin, A.; Sturlese, M.; Deganutti, G.; Salmaso, V.; Sabbadin, D.; Ciancetta, A.; Moro, S. Deciphering the Complexity of Ligand-Protein Recognition Pathways Using Supervised Molecular Dynamics (SuMD) Simulations. J. Chem. Inf. Model. 2016, 56, 687–705. [Google Scholar] [CrossRef] [PubMed]
- Sabbadin, D.; Salmaso, V.; Sturlese, M.; Moro, S. Supervised Molecular Dynamics (SuMD) Approaches in Drug Design. Methods Mol. Biol. 2018, 1824, 287–298. [Google Scholar] [PubMed]
- Draper-Joyce, C.J.; Khoshouei, M.; Thal, D.M.; Liang, Y.-L.; Nguyen, A.T.N.; Furness, S.G.B.; Venugopal, H.; Baltos, J.-A.; Plitzko, J.M.; Danev, R.; et al. Structure of the adenosine-bound human adenosine A1 receptor-Gi complex. Nature 2018, 558, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Glukhova, A.; Thal, D.M.; Nguyen, A.T.; Vecchio, E.A.; Jörg, M.; Scammells, P.J.; May, L.T.; Sexton, P.M.; Christopoulos, A. Structure of the Adenosine A1 Receptor Reveals the Basis for Subtype Selectivity. Cell 2017, 168, 867–877.e13. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group—Citing MOE. Available online: https://www.chemcomp.com/Research-Citing_MOE.htm (accessed on 3 October 2016).
- GOLD—The Cambridge Crystallographic Data Centre (CCDC). Available online: https://www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold/ (accessed on 12 March 2017).
- GNUPLOT Homepage. Available online: http://gnuplot.sourceforge.net/ (accessed on 28 March 2018).
- MarvinSketch. Available online: https://docs.chemaxon.com/display/docs/MarvinSketch+Home (accessed on 28 March 2018).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Floris, M.; Sabbadin, D.; Ciancetta, A.; Medda, R.; Cuzzolin, A.; Moro, S. Implementing the “Best Template Searching” tool into Adenosiland platform. In Silico Pharmacol 1; Springer: Berlin, Germany, 2013. [Google Scholar]
- Labute, P. Protonate3D: Assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins 2009, 75, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Labute, P. The generalized Born/volume integral implicit solvent model: Estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem. 2008, 29, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinert, T.; Olieric, N.; Cheng, R.; Brünle, S.; James, D.; Ozerov, D.; Gashi, D.; Vera, L.; Marsh, M.; Jaeger, K.; et al. Serial millisecond crystallography for routine room-temperature structure determination at synchrotrons. Nat. Commun. 2017, 8, 542. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods I. Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database: Its relevance to human molecular medical research. J. Mol. Med. 1997, 75, 312–316. [Google Scholar] [PubMed]
- Fredholm, B.B. Adenosine, adenosine receptors and the actions of caffeine. Pharmacol. Toxicol. 1995, 76, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.O.; Ji, X.D.; Siddiqi, S.M.; Olah, M.E.; Stiles, G.L.; Jacobson, K.A. 2-Substitution of N6-benzyladenosine-5’-uronamides enhances selectivity for A3 adenosine receptors. J. Med. Chem. 1994, 37, 3614–3621. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Park, K.S.; Jiang, J.L.; Kim, Y.C.; Olah, M.E.; Stiles, G.L.; Ji, X.D. Pharmacological characterization of novel A3 adenosine receptor-selective antagonists. Neuropharmacology 1997, 36, 1157–1165. [Google Scholar] [CrossRef] [Green Version]
- Tosh, D.K.; Finley, A.; Paoletta, S.; Moss, S.M.; Gao, Z.-G.; Gizewski, E.T.; Auchampach, J.A.; Salvemini, D.; Jacobson, K.A. In vivo phenotypic screening for treating chronic neuropathic pain: Modification of C2-arylethynyl group of conformationally constrained A3 adenosine receptor agonists. J. Med. Chem. 2014, 57, 9901–9914. [Google Scholar] [CrossRef] [PubMed]
- Tosh, D.K.; Deflorian, F.; Phan, K.; Gao, Z.-G.; Wan, T.C.; Gizewski, E.; Auchampach, J.A.; Jacobson, K.A. Structure-guided design of A(3) adenosine receptor-selective nucleosides: Combination of 2-arylethynyl and bicyclo[3.1.0]hexane substitutions. J. Med. Chem. 2012, 55, 4847–4860. [Google Scholar] [CrossRef] [PubMed]
- Tosh, D.K.; Paoletta, S.; Chen, Z.; Crane, S.; Lloyd, J.; Gao, Z.-G.; Gizewski, E.T.; Auchampach, J.A.; Salvemini, D.; Jacobson, K.A. Structure-Based Design, Synthesis by Click Chemistry and in Vivo Activity of Highly Selective A3 Adenosine Receptor Agonists. MedChemComm 2015, 6, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Auchampach, J.A.; Gizewski, E.T.; Wan, T.C.; de Castro, S.; Brown, G.G.; Jacobson, K.A. Synthesis and pharmacological characterization of [(125)I]MRS5127, a high affinity, selective agonist radioligand for the A3 adenosine receptor. Biochem. Pharmacol. 2010, 79, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Federico, S.; Redenti, S.; Sturlese, M.; Ciancetta, A.; Kachler, S.; Klotz, K.-N.; Cacciari, B.; Moro, S.; Spalluto, G. The Influence of the 1-(3-Trifluoromethyl-Benzyl)-1H-Pyrazole-4-yl Moiety on the Adenosine Receptors Affinity Profile of Pyrazolo[4,3-e][1,2,4]Triazolo[1,5-c]Pyrimidine Derivatives. PLoS ONE 2015, 10, e0143504. [Google Scholar] [CrossRef] [PubMed]
- Federico, S.; Ciancetta, A.; Porta, N.; Redenti, S.; Pastorin, G.; Cacciari, B.; Klotz, K.N.; Moro, S.; Spalluto, G. Scaffold decoration at positions 5 and 8 of 1,2,4-triazolo[1,5-c]pyrimidines to explore the antagonist profiling on adenosine receptors: A preliminary structure-activity relationship study. J. Med. Chem. 2014, 57, 6210–6225. [Google Scholar] [CrossRef] [PubMed]
- Federico, S.; Ciancetta, A.; Porta, N.; Redenti, S.; Pastorin, G.; Cacciari, B.; Klotz, K.N.; Moro, S.; Spalluto, G. 5,7-Disubstituted-[1,2,4]triazolo[1,5-a][1,3,5]triazines as pharmacological tools to explore the antagonist selectivity profiles toward adenosine receptors. Eur. J. Med. Chem. 2016, 108, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Federico, S.; Ciancetta, A.; Sabbadin, D.; Paoletta, S.; Pastorin, G.; Cacciari, B.; Klotz, K.N.; Moro, S.; Spalluto, G. Exploring the directionality of 5-substitutions in a new series of 5-alkylaminopyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine as a strategy to design novel human a(3) adenosine receptor antagonists. J. Med. Chem. 2012, 55, 9654–9668. [Google Scholar] [CrossRef] [PubMed]
- Federico, S.; Margiotta, E.; Salmaso, V.; Pastorin, G.; Kachler, S.; Klotz, K.-N.; Moro, S.; Spalluto, G. [1,2,4]Triazolo[1,5-c]pyrimidines as adenosine receptor antagonists: Modifications at the 8 position to reach selectivity towards A3 adenosine receptor subtype. Eur. J. Med. Chem. 2018, 157, 837–851. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Ji, X.D.; Jacobson, K.A. Derivatives of the triazoloquinazoline adenosine antagonist (CGS15943) are selective for the human A3 receptor subtype. J. Med. Chem. 1996, 39, 4142–4148. [Google Scholar] [CrossRef] [PubMed]
- Li, A.H.; Moro, S.; Melman, N.; Ji, X.D.; Jacobson, K.A. Structure-activity relationships and molecular modeling of 3, 5-diacyl-2,4-dialkylpyridine derivatives as selective A3 adenosine receptor antagonists. J. Med. Chem. 1998, 41, 3186–3201. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.G.; Van Muijlwijk-Koezen, J.E.; Chen, A.; Müller, C.E.; Ijzerman, A.P.; Jacobson, K.A. Allosteric modulation of A(3) adenosine receptors by a series of 3-(2-pyridinyl)isoquinoline derivatives. Mol. Pharmacol. 2001, 60, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Li, A.H.; Ji, X.D.; Melman, N.; Olah, M.E.; Stiles, G.L.; Jacobson, K.A. Selective A(3) adenosine receptor antagonists: Water-soluble 3, 5-diacyl-1,2,4-trialkylpyridinium salts and their oxidative generation from dihydropyridine precursors. J. Med. Chem. 1999, 42, 4232–4238. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Li, A.H.; Jang, S.Y.; Chang, L.; Melman, N.; Moro, S.; Ji, X.; Lobkovsky, E.B.; Clardy, J.C.; Jacobson, K.A. Chiral resolution and stereospecificity of 6-phenyl-4-phenylethynyl- 1,4-dihydropyridines as selective A(3) adenosine receptor antagonists. J. Med. Chem. 1999, 42, 3055–3065. [Google Scholar] [CrossRef] [PubMed]
- Ciancetta, A.; Jacobson, K. Structural Probing and Molecular Modeling of the A3 Adenosine Receptor: A Focus on Agonist Binding. Molecules 2017, 22, 449. [Google Scholar] [CrossRef] [PubMed]
- Ciancetta, A.; Cuzzolin, A.; Moro, S. Alternative quality assessment strategy to compare performances of GPCR-ligand docking protocols: The human adenosine A2A receptor as a case study. J. Chem. Inf. Model. 2014, 54, 2243–2254. [Google Scholar] [CrossRef] [PubMed]
- Cuzzolin, A.; Sturlese, M.; Malvacio, I.; Ciancetta, A.; Moro, S. DockBench: An integrated informatic platform bridging the gap between the robust validation of docking protocols and virtual screening simulations. Molecules 2015, 20, 9977–9993. [Google Scholar] [CrossRef] [PubMed]
- Margiotta, E.; Deganutti, G.; Moro, S. Could the presence of sodium ion influence the accuracy and precision of the ligand-posing in the human A2A adenosine receptor orthosteric binding site using a molecular docking approach? Insights from Dockbench. J. Comput. Aided Mol. Des. 2018, 32, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- RDKit: Cheminformatics and Machine Learning Software. Available online: http://www.rdkit.org (accessed on 28 March 2018).
- Mencoder. Available online: http://www.mplayerhq.hu/design7/projects.html (accessed on 28 March 2018).
- Martinelli, A.; Ortore, G. Molecular Modelling of adenosine receptors. Meth. Enzymol. 2013, 522, 37–59. [Google Scholar] [CrossRef] [PubMed]
- Piirainen, H.; Ashok, Y.; Nanekar, R.T.; Jaakola, V.-P. Structural features of adenosine receptors: From crystal to function. Biochim. Biophys. Acta (BBA)-Biomembr. Adenosine Recept. 2011, 1808, 1233–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belardinelli, L.; Pelleg, A. Adenosine and Adenine Nucleotides: From Molecular Biology to Integrative Physiology; Springer Science & Business Media: Berlin, Germany, 1995. [Google Scholar]
- Fiser, A. Template-Based Protein Structure Modeling. Methods Mol. Biol. 2010, 673, 73–94. [Google Scholar] [PubMed] [Green Version]
- Campbell, N.G.; Zhu, C.-B.; Lindler, K.M.; Yaspan, B.L.; Kistner-Griffin, E.; Hewlett, W.A.; Tate, C.G.; Blakely, R.D.; Sutcliffe, J.S. Rare coding variants of the adenosine A3 receptor are increased in autism: On the trail of the serotonin transporter regulome. Mol. Autism 2013, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.-G.; Chen, A.; Barak, D.; Kim, S.-K.; Müller, C.E.; Jacobson, K.A. Identification by Site-directed Mutagenesis of Residues Involved in Ligand Recognition and Activation of the Human A3 Adenosine Receptor. J. Biol. Chem. 2002, 277, 19056–19063. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A3/A1 Sequence Alignment | A3/A2A Sequence Alignment | |
|---|---|---|
| TM1 | 10AAYIGIEVLIALVSVPGNVLVIWAVKV36 | 15AIAVLAILGNVLVCWAVWLNSNLQNVT41 |
| 13VTYITMEIFIGLCAIVGNVLVICVVKL39 | 21FIGLCAIVGNVLVICVVKLNPSLQTTT47 | |
| IL1 | 37NQALRD42 | 42NYFVVS47 |
| 40NPSLQT45 | 48FYFIVS53 | |
| TM2 | 43ATFCFIVSLAVADVAVGALVIPLAILINI71 | 48LAAADIAVGVLAIPFAITISTGFCAACHG76 |
| 46TTFYFIVSLALADIAVGVLVMPLAIVVSL74 | 54LALADIAVGVLVMPLAIVVSLGITIHFYS82 | |
| EL1 | 72GPQTY76 | 77CLFIA81 |
| 75GITIH79 | 83CLFMT87 | |
| TM3 | 77FHTCLMVACPVLILTQSSILALLAIAVDRYLRVK110 | 82CFVLVLTQSSIFSLLAIAIDRYIAIRIPLRYNGL115 |
| 80FYSCLFMTCLLLIFTHASIMSLLAIAVDRYLRVK13 | 88CLLLIFTHASIMSLLAIAVDRYLRVKLTVRYKRV121 | |
| IL2 | 111IPLRYKMVVT120 | 116VTGTRAKGII125 |
| 114LTVRYKRVTT123 | 122TTHRRIWLAL131 | |
| TM4 | 121PRRAAVAIAGCWILSFVVGLTPMF144 | 126AICWVLSFAIGLTPMLGWNNCGQP149 |
| 124HRRIWLALGLCWLVSFLVGLTPMF147 | 132GLCWLVSFLVGLTPMFGWNMKLTS155 | |
| EL2 | 145GWNNLSAVERAWAANGSMGEPVIKCEFEKVIS176 | 150KEGKNHSQGCGEGQVACLFEDVVP173 |
| 148GWNMKLTSE--YHRNVTF----LSCQFVSVMR173 | 156EYHRNVTFLSCQFVSVMR173------ | |
| TM5 | 177MEYMVYFNFFVWVLPPLLLMVLIYLEVFYLIRKQ210 | 174MNYMVYFNFFACVLVPLLLMLGVYLRIFLAARRQ207 |
| 174MDYMVYFSFLTWIFIPLVVMCAIYLDIFYIIRNK207 | 174MDYMVYFSFLTWIFIPLVVMCAIYLDIFYIIRNK207 | |
| IL3 | 211LNKKVSASSGDPQ223 | 208LKQMESQPLPGERAR222 |
| 208LSLNLS-NSKETG219 | 208LSLNLSN---SKETG219 | |
| TM6 | 224KYYGKELKIAKSLALILFLFALSWLPLHILNCITLF259 | 223STLQKEVHAAKSLAIIVGLFALCWLPLHIINCFTFF258 |
| 220AFYGREFKTAKSLFLVLFLFALSWLPLSIINCIIYF255 | 220AFYGREFKTAKSLFLVLFLFALSWLPLSIINCIIYF255 | |
| EL3 | 260CPSCHKP266 | 259CPDCSHAP266 |
| ----256NGEVP260 | 256N---GEVP260 | |
| TM7 | 267SILTYIAIFLTHGNSAMNPIVYAFR291 | 267LWLMYLAIVLSHTNSVVNPFIYAYR291 |
| 261QLVLYMGILLSHANSMMNPIVYAYK285 | 261QLVLYMGILLSHANSMMNPIVYAYK285 |
| A3/A1 | A3/A2a | |||
|---|---|---|---|---|
| SI(%) | SS(%) | SI(%) | SS(%) | |
| TM1 | 48.1 | 66.7 | 51.9 | 63 |
| IL1 | 33.3 | 66.7 | 66.7 | 83.3 |
| TM2 | 65.5 | 89.7 | 51.7 | 62.1 |
| EL1 | 20 | 40 | 60 | 80 |
| TM3 | 64.7 | 85.3 | 47.1 | 76.5 |
| IL2 | 50 | 70 | 20 | 30 |
| TM4 | 58.3 | 79.2 | 50 | 66.7 |
| EL2 | 25 | 40.6 | 12.5 | 25 |
| TM5 | 52.9 | 79.4 | 44.1 | 67.6 |
| IL3 | 23.1 | 58.3 | 13.3 | 20 |
| TM6 | 69.4 | 80.6 | 50 | 66.7 |
| EL3 | 14.3 | 28.6 | 12.5 | 12.5 |
| TM7 | 52 | 76 | 48 | 80 |
| overall | 48.2 | 68.7 | 31.6 | 47.8 |
| max | TM6 | TM2 | IL1 | IL1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Margiotta, E.; Moro, S. A Comparison in the Use of the Crystallographic Structure of the Human A1 or the A2A Adenosine Receptors as a Template for the Construction of a Homology Model of the A3 Subtype. Appl. Sci. 2019, 9, 821. https://doi.org/10.3390/app9050821
Margiotta E, Moro S. A Comparison in the Use of the Crystallographic Structure of the Human A1 or the A2A Adenosine Receptors as a Template for the Construction of a Homology Model of the A3 Subtype. Applied Sciences. 2019; 9(5):821. https://doi.org/10.3390/app9050821
Chicago/Turabian StyleMargiotta, Enrico, and Stefano Moro. 2019. "A Comparison in the Use of the Crystallographic Structure of the Human A1 or the A2A Adenosine Receptors as a Template for the Construction of a Homology Model of the A3 Subtype" Applied Sciences 9, no. 5: 821. https://doi.org/10.3390/app9050821