

Combined Steam and CO2 Reforming of Methane over Ni-Based CeO2-MgO Catalysts: Impacts of Preparation Mode and Pd Addition

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Material
2.2. Methods for Preparing Catalysts
2.3. Methods for Studying the Activity of Catalysts
3. Results and Discussion
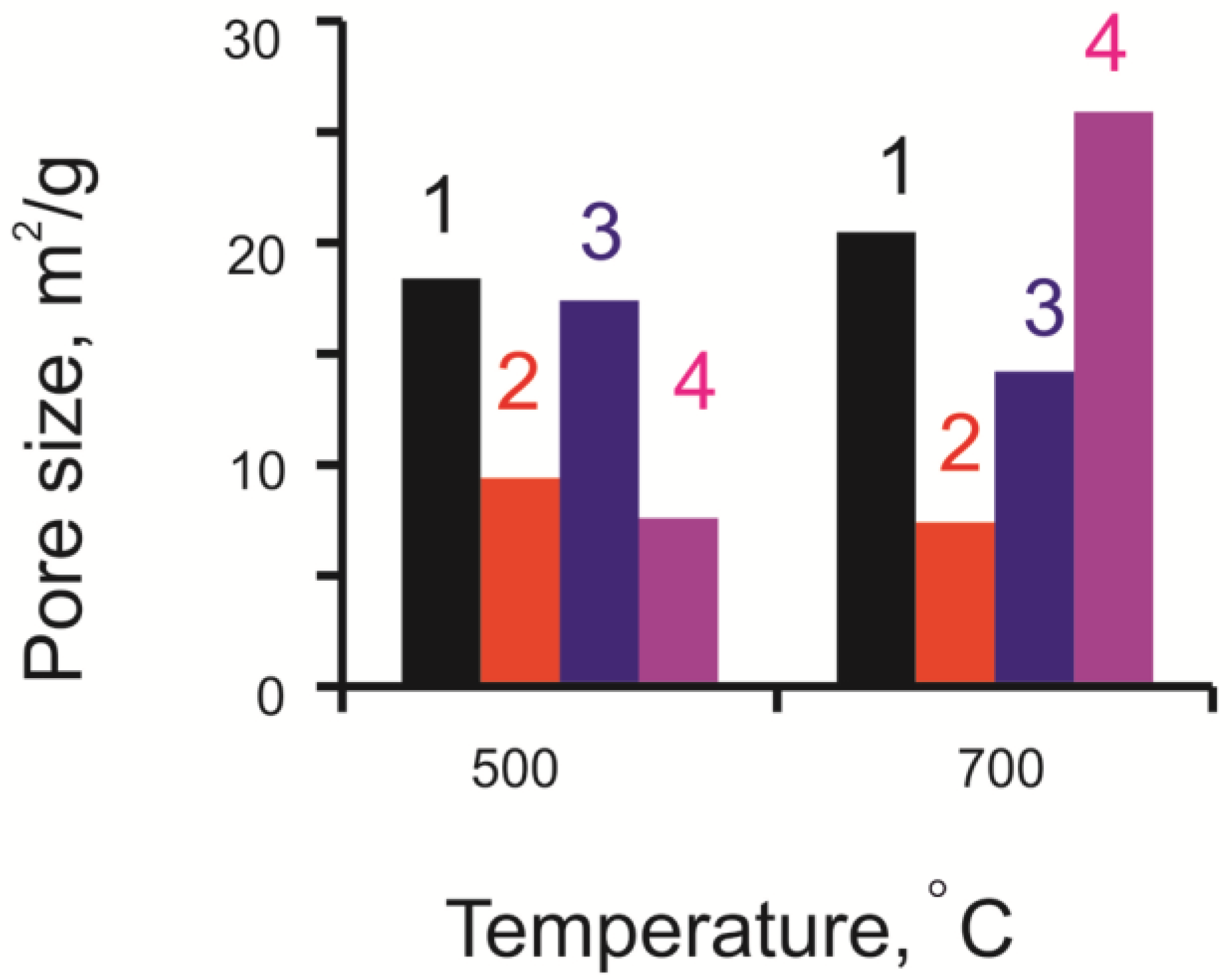
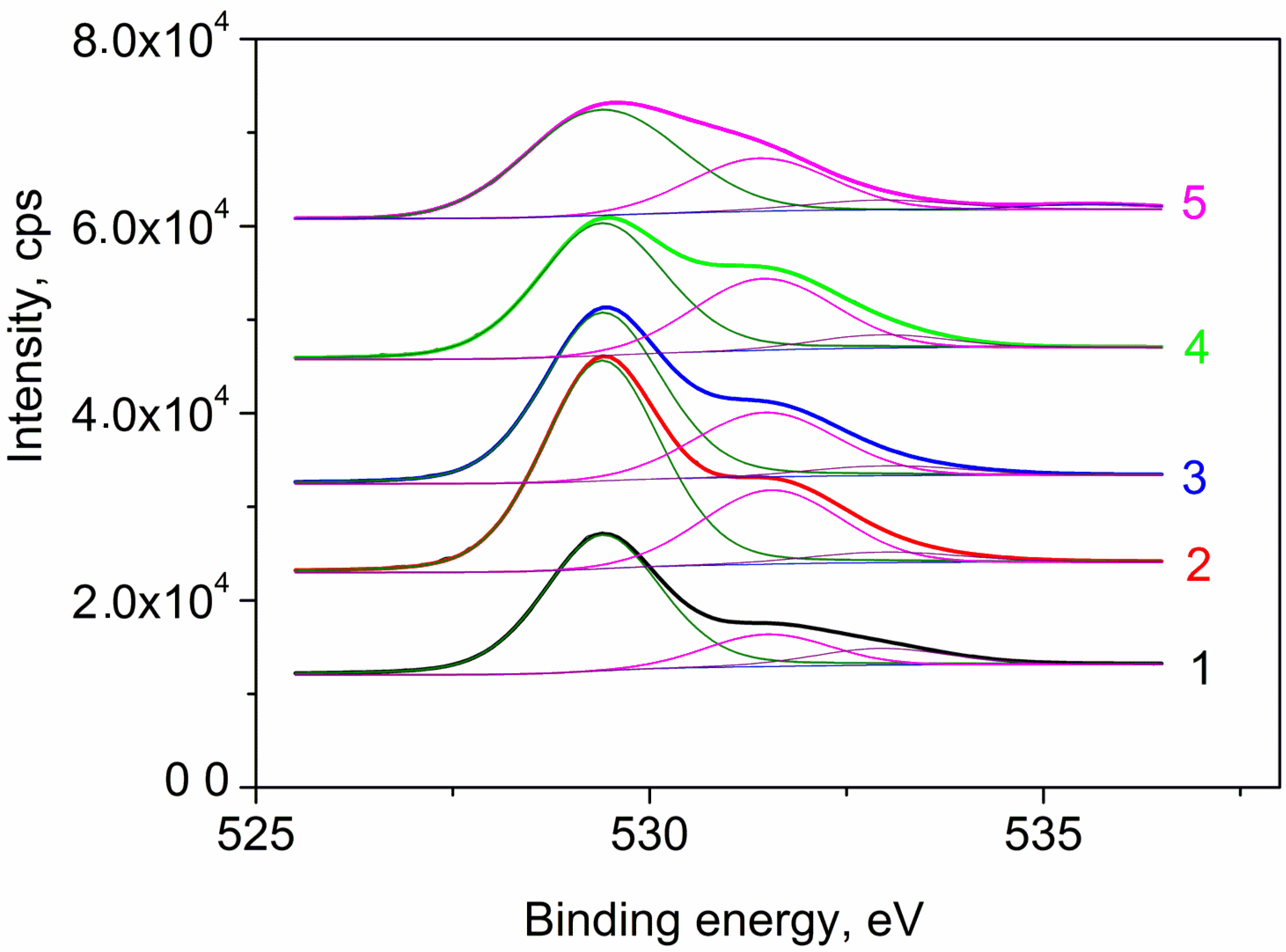
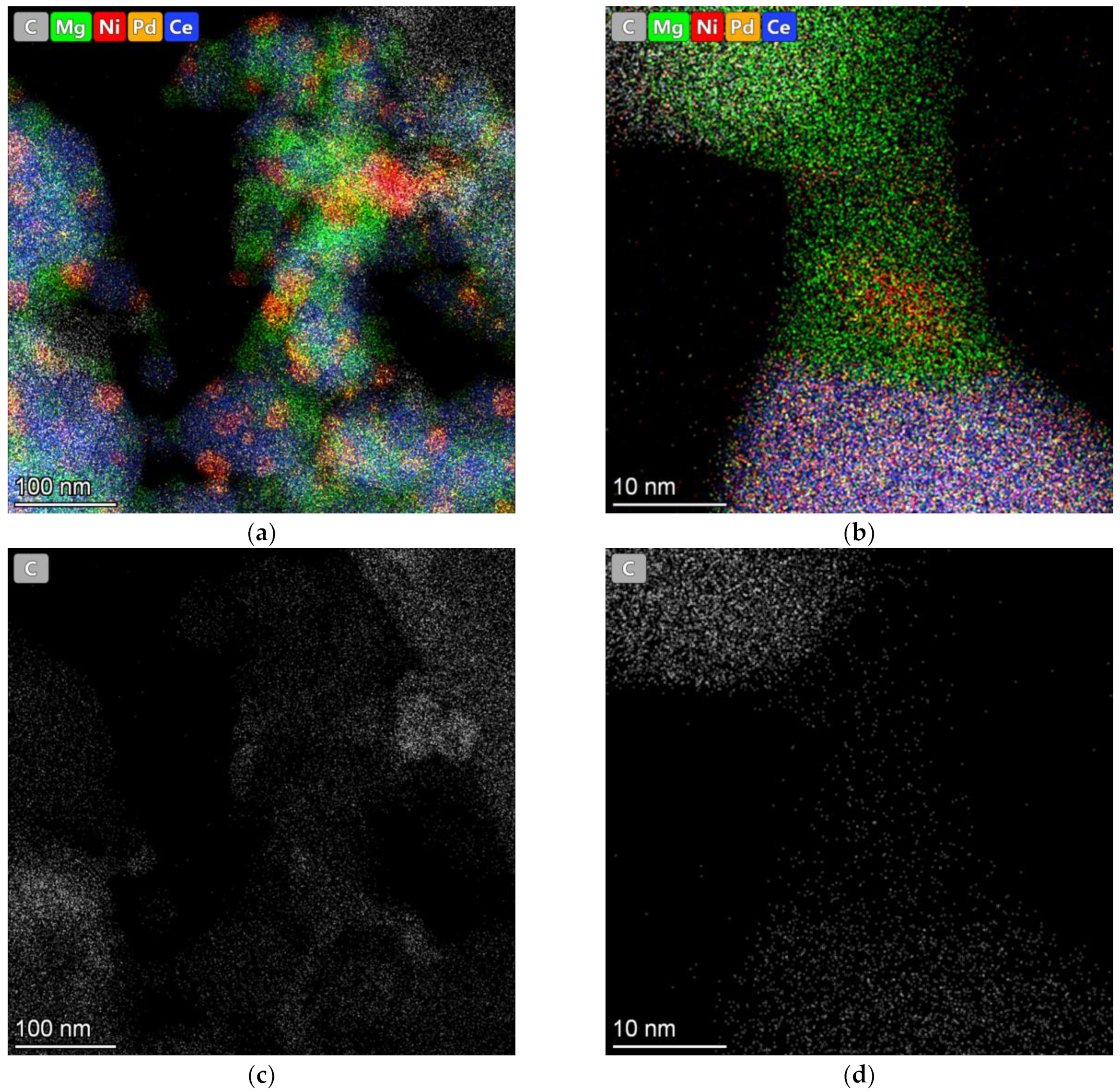
3.1. Physicochemical Research of Fresh Catalysts
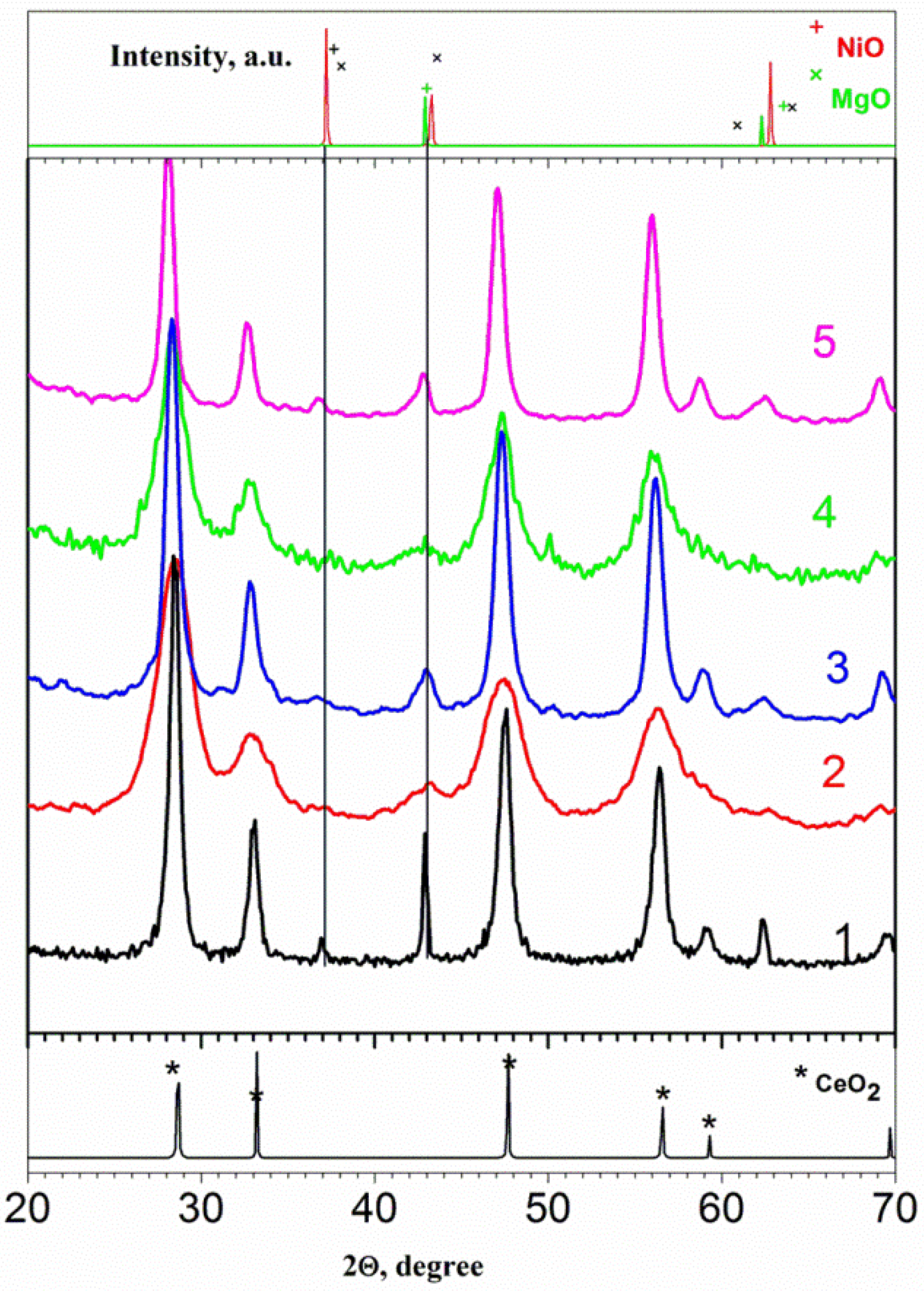
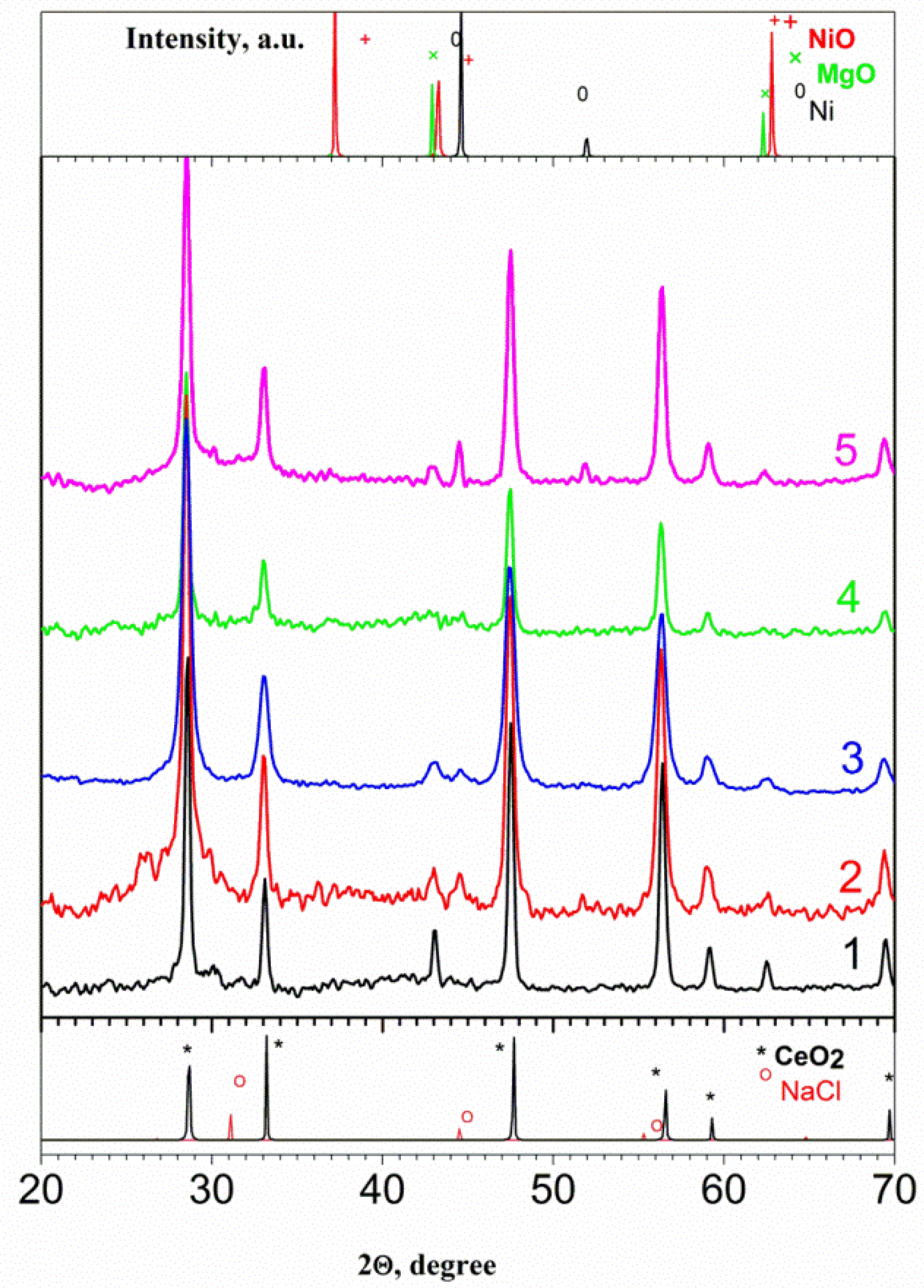
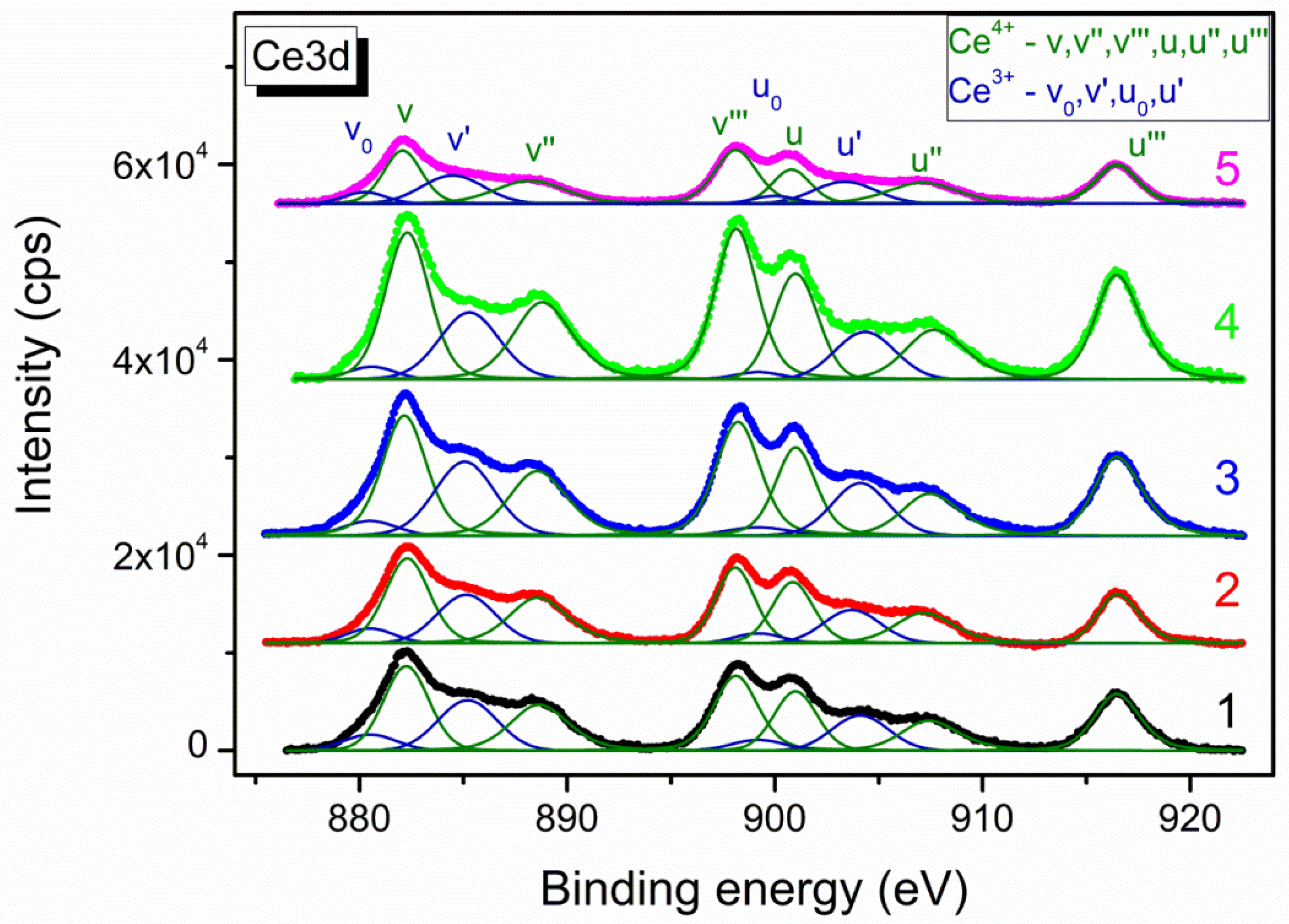
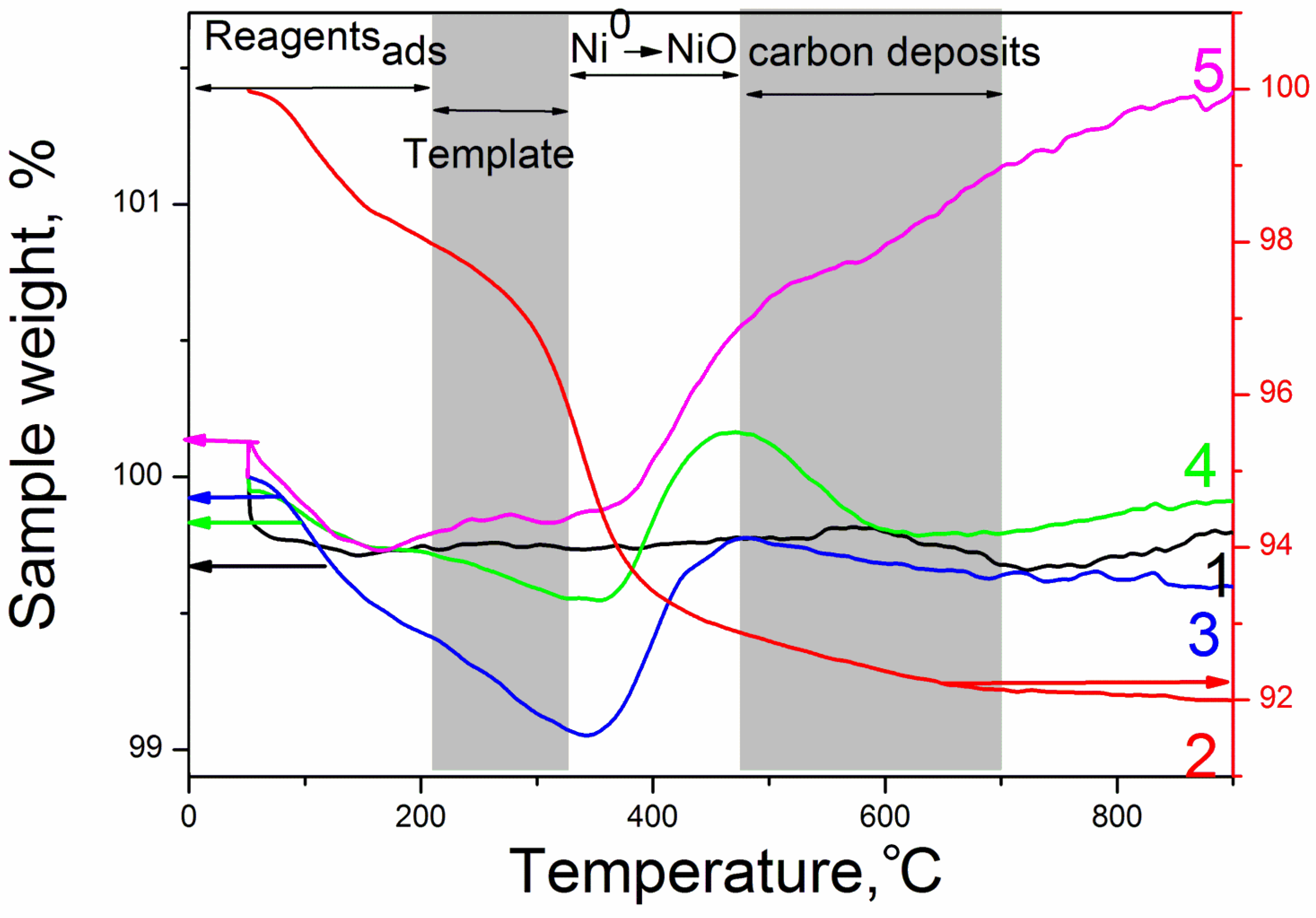
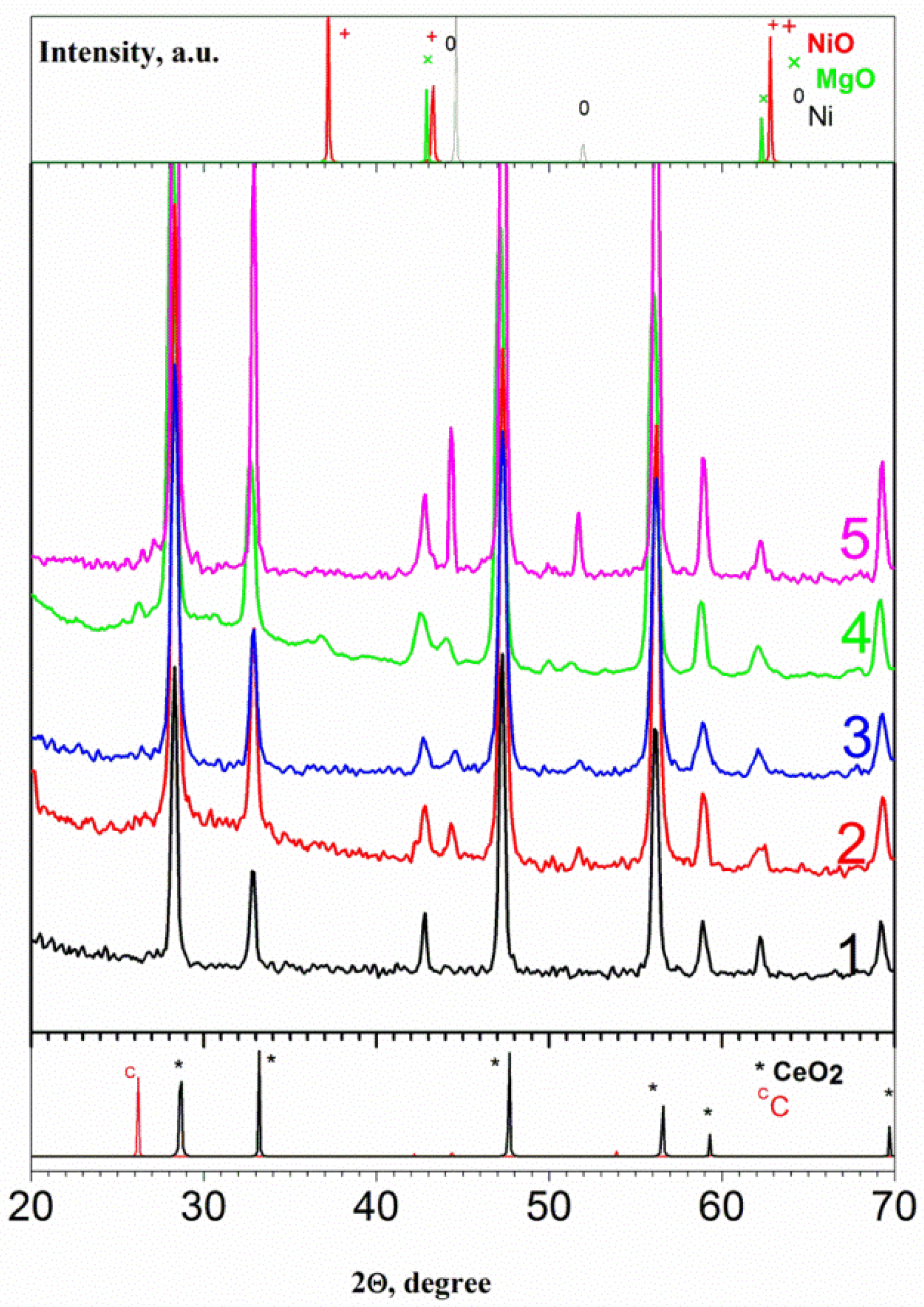
3.2. The Characterization of the Reduced Catalysts
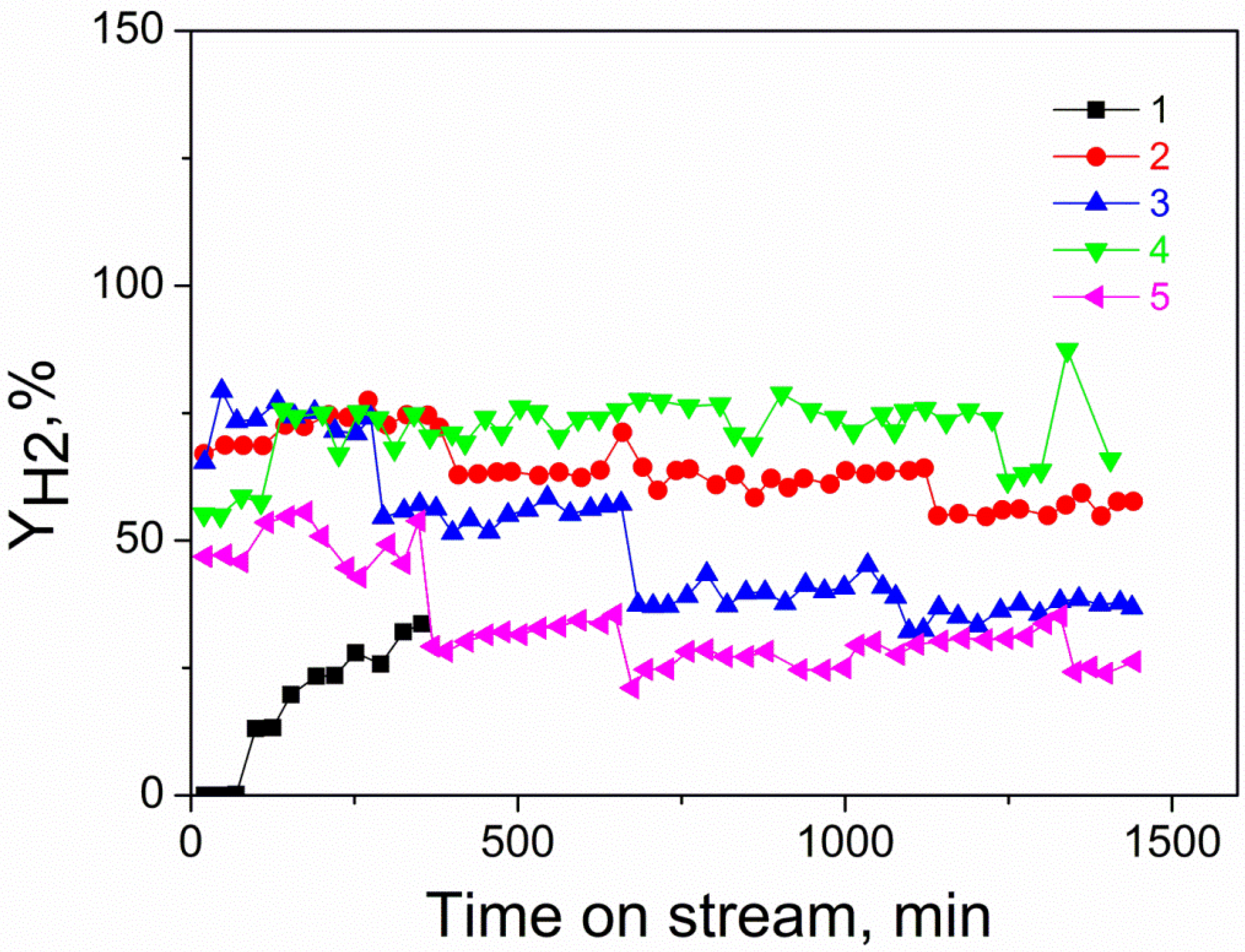
3.3. Catalytic Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hosseini, S.E.; Wahid, M.A. Hydrogen Production from Renewable and Sustainable Energy Resources: Promising Green Energy Carrier for Clean Development. Renew. Sustain. Energy Rev. 2016, 57, 850–866. [Google Scholar] [CrossRef]
- Nikolaidis, P.; Poullikkas, A. A Comparative Overview of Hydrogen Production Processes. Renew. Sustain. Energy Rev. 2017, 67, 597–611. [Google Scholar] [CrossRef]
- Usman, M.; Wan Daud, W.M.A.; Abbas, H.F. Dry Reforming of Methane: Influence of Process Parameters—A Review. Renew. Sustain. Energy Rev. 2015, 45, 710–744. [Google Scholar] [CrossRef] [Green Version]
- Da Fonseca, R.O.; Rabelo-Neto, R.C.; Simões, R.C.C.; Mattos, L.V.; Noronha, F.B. Pt Supported on Doped CeO2/Al2O3 as Catalyst for Dry Reforming of Methane. Int. J. Hydrog. Energy 2020, 45, 5182–5191. [Google Scholar] [CrossRef]
- Lu, Y.; Lee, T. Influence of the Feed Gas Composition on the Fischer-Tropsch Synthesis in Commercial Operations. J. Nat. Gas Chem. 2007, 16, 329–341. [Google Scholar] [CrossRef]
- Peng, X.; Cheng, K.; Kang, J.; Gu, B.; Yu, X.; Zhang, Q.; Wang, Y. Impact of Hydrogenolysis on the Selectivity of the Fischer-Tropsch Synthesis: Diesel Fuel Production over Mesoporous Zeolite-Y-Supported Cobalt Nanoparticles. Angew. Chemie —Int. Ed. 2015, 54, 4553–4556. [Google Scholar] [CrossRef]
- Xia, Y.; Lu, N.; Li, J.; Jiang, N.; Shang, K.; Wu, Y. Combined Steam and CO2 Reforming of CH4 for Syngas Production in a Gliding Arc Discharge Plasma. J. CO2 Util. 2020, 37, 248–259. [Google Scholar] [CrossRef]
- Batebi, D.; Abedini, R.; Mosayebi, A. Combined Steam and CO2 Reforming of Methane (CSCRM) over Ni–Pd/Al2O3 Catalyst for Syngas Formation. Int. J. Hydrog. Energy 2020, 45, 14293–14310. [Google Scholar] [CrossRef]
- De Araújo Moreira, T.G.; de Carvalho Filho, J.F.S.; Carvalho, Y.; de Almeida, J.M.A.R.; Nothaft Romano, P.; Falabella Sousa-Aguiar, E. Highly Stable Low Noble Metal Content Rhodium-Based Catalyst for the Dry Reforming of Methane. Fuel 2021, 287, 119536. [Google Scholar] [CrossRef]
- De Caprariis, B.; De Filippis, P.; Palma, V.; Petrullo, A.; Ricca, A.; Ruocco, C.; Scarsella, M. Rh, Ru and Pt Ternary Perovskites Type Oxides BaZr(1-x)MexO3 for Methane Dry Reforming. Appl. Catal. A Gen. 2016, 517, 47–55. [Google Scholar] [CrossRef]
- Mahfouz, R.; Estephane, J.; Gennequin, C.; Tidahy, L.; Aouad, S.; Abi-Aad, E. CO2 reforming of Methane over Ni and/or Ru Catalysts Supported on Mesoporous KIT-6: Effect of Promotion with Ce. J. Environ. Chem. Eng. 2021, 9, 104662. [Google Scholar] [CrossRef]
- Singha, R.K.; Yadav, A.; Shukla, A.; Kumar, M.; Bal, R. Low Temperature Dry Reforming of Methane over Pd-CeO2 Nanocatalyst. Catal. Commun. 2017, 92, 19–22. [Google Scholar] [CrossRef]
- Yue, L.; Li, J.; Chen, C.; Fu, X.; Gong, Y.; Xia, X.; Hou, J.; Xiao, C.; Chen, X.; Zhao, L.; et al. Thermal-Stable Pd@mesoporous Silica Core-Shell Nanocatalysts for Dry Reforming of Methane with Good Coke-Resistant Performance. Fuel 2018, 218, 335–341. [Google Scholar] [CrossRef]
- Anil, C.; Modak, J.M.; Madras, G. Syngas Production via CO2 Reforming of Methane over Noble Metal (Ru, Pt, and Pd) Doped LaAlO3 Perovskite Catalyst. Mol. Catal. 2020, 484, 110805. [Google Scholar] [CrossRef]
- Araiza, D.G.; Arcos, D.G.; Gómez-Cortés, A.; Díaz, G. Dry Reforming of Methane over Pt-Ni/CeO2 Catalysts: Effect of the Metal Composition on the Stability. Catal. Today 2021, 360, 46–54. [Google Scholar] [CrossRef]
- Bahari, M.B.; Setiabudi, H.D.; Ainirazali, N.; Vo, D.-V.N. A Short Review on Bimetallic Co-Based Catalysts for Carbon Dioxide Reforming of Methane. Mater. Today Proc. 2021, 42, 94–100. [Google Scholar] [CrossRef]
- Abasaeed, A.E.; Al-Fatesh, A.S.; Naeem, M.A.; Ibrahim, A.A.; Fakeeha, A.H. Catalytic Performance of CeO2 and ZrO2 Supported Co Catalysts for Hydrogen Production via Dry Reforming of Methane. Int. J. Hydrog. Energy 2015, 40, 6818–6826. [Google Scholar] [CrossRef]
- Sengupta, S.; Deo, G. Modifying Alumina with CaO or MgO in Supported Ni and Ni-Co Catalysts and Its Effect on Dry Reforming of CH4. J. CO2 Util. 2015, 10, 67–77. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, Z.; Zhu, Y.A.; Liu, Z.; Sui, Z.; Zhu, K.; Zhou, X. Dry Reforming of Methane on Ni-Fe-MgO Catalysts: Influence of Fe on Carbon-Resistant Property and Kinetics. Appl. Catal. B Environ. 2020, 264, 118497. [Google Scholar] [CrossRef]
- Kim, H.M.; Jang, W.J.; Yoo, S.Y.; Shim, J.O.; Jeon, K.W.; Na, H.S.; Lee, Y.L.; Jeon, B.H.; Bae, J.W.; Roh, H.S. Low Temperature Steam Reforming of Methane Using Metal Oxide Promoted Ni-Ce0.8Zr0.2O2 Catalysts in a Compact Reformer. Int. J. Hydrog. Energy 2018, 43, 262–270. [Google Scholar] [CrossRef]
- Solymosi, F.; Kutsán, G.; Erdöhelyi, A. Catalytic Reaction of CH4 with CO2 over Alumina-Supported Pt Metals. Catal. Letters 1991, 11, 149–156. [Google Scholar] [CrossRef]
- Khajenoori, M.; Rezaei, M.; Meshkani, F. Characterization of CeO2 Promoter of a Nanocrystalline Ni/MgO Catalyst in Dry Reforming of Methane. Chem. Eng. Technol. 2014, 37, 957–963. [Google Scholar] [CrossRef]
- Cao, L.; Ni, C.; Yuan, Z.; Wang, S. Correlation between Catalytic Selectivity and Oxygen Storage Capacity in Autothermal Reforming of Methane over Rh/Ce0.45Zr0.45RE0.1 Catalysts (RE = La, Pr, Nd, Sm, Eu, Gd, Tb). Catal. Commun. 2009, 10, 1192–1195. [Google Scholar] [CrossRef]
- Hussain, I.; Tanimu, G.; Ahmed, S.; Aniz, C.U.; Alasiri, H.; Alhooshani, K. A Review of the Indispensable Role of Oxygen Vacancies for Enhanced CO2 Methanation Activity over CeO2-Based Catalysts: Uncovering, Influencing, and Tuning Strategies. Int. J. Hydrogen Energy 2022, in press. [Google Scholar] [CrossRef]
- Li, P.; Chen, X.; Li, Y.; Schwank, J.W. A Review on Oxygen Storage Capacity of CeO2-Based Materials: Influence Factors, Measurement Techniques, and Applications in Reactions Related to Catalytic Automotive Emissions Control. Catal. Today 2019, 327, 90–115. [Google Scholar] [CrossRef]
- Zhang, Q.H.; Li, Y.; Xu, B.Q. Reforming of Methane and Coalbed Methane over Nanocomposite Ni/ZrO2 Catalyst. Catal. Today 2004, 98, 601–605. [Google Scholar] [CrossRef]
- Roh, H.S.; Koo, K.Y.; Jeong, J.H.; Seo, Y.T.; Seo, D.J.; Seo, Y.S.; Yoon, W.L.; Park, S. Bin Combined Reforming of Methane over Supported Ni Catalysts. Catal. Letters 2007, 117, 85–90. [Google Scholar] [CrossRef]
- Matus, E.; Sukhova, O.; Kerzhentsev, M.; Ismagilov, I.; Yashnik, S.; Ushakov, V.; Stonkus, O.; Gerasimov, E.; Nikitin, A.; Bharali, P.; et al. Hydrogen Production through Bi-Reforming of Methane: Improving Ni Catalyst Performance via an Exsolution Approach. Catalysts 2022, 12, 1493. [Google Scholar] [CrossRef]
- Lucrédio, A.F.; Assaf, J.M.; Assaf, E.M. Methane Conversion Reactions on Ni Catalysts Promoted with Rh: Influence of Support. Appl. Catal. A Gen. 2011, 400, 156–165. [Google Scholar] [CrossRef]
- Özdemir, H.; Faruk Öksüzömer, M.A.; Ali Gürkaynak, M. Preparation and Characterization of Ni Based Catalysts for the Catalytic Partial Oxidation of Methane: Effect of Support Basicity on H2/CO Ratio and Carbon Deposition. Int. J. Hydrog. Energy 2010, 35, 12147–12160. [Google Scholar] [CrossRef]
- Jeon, K.W.; Kim, H.M.; Kim, B.J.; Lee, Y.L.; Na, H.S.; Shim, J.O.; Jang, W.J.; Roh, H.S. Synthesis Gas Production from Carbon Dioxide Reforming of Methane over Ni-MgO Catalyst: Combined Effects of Titration Rate during Co-Precipitation and CeO2 Addition. Fuel Process. Technol. 2021, 219, 106877. [Google Scholar] [CrossRef]
- Wang, Y.H.; Liu, H.M.; Xu, B.Q. Durable Ni/MgO Catalysts for CO2 Reforming of Methane: Activity and Metal-Support Interaction. J. Mol. Catal. A Chem. 2009, 299, 44–52. [Google Scholar] [CrossRef]
- Bukhari, S.N.; Chin, C.Y.; Setiabudi, H.D.; Vo, D.V.N. Tailoring the Properties and Catalytic Activities of Ni/SBA-15 via Different TEOS/P123 Mass Ratios for CO2 Reforming of CH4. J. Environ. Chem. Eng. 2017, 5, 3122–3128. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhang, T.; Shi, Y.; Zhao, B.; Wang, M.; Liu, Q.; Wang, J.; Long, K.; Duan, Y.; Ning, P. A Sintering and Carbon-Resistant Ni-SBA-15 Catalyst Prepared by Solid-State Grinding Method for Dry Reforming of Methane. J. CO2 Util. 2017, 17, 10–19. [Google Scholar] [CrossRef]
- Muraleedharan Nair, M.; Kaliaguine, S. Structured Catalysts for Dry Reforming of Methane. New J. Chem. 2016, 40, 4049–4060. [Google Scholar] [CrossRef]
- Li, D.; Nakagawa, Y.; Tomishige, K. Methane Reforming to Synthesis Gas over Ni Catalysts Modified with Noble Metals. Appl. Catal. A Gen. 2011, 408, 1–24. [Google Scholar] [CrossRef]
- Pan, C.; Guo, Z.; Dai, H.; Ren, R.; Chu, W. Anti-Sintering Mesoporous Ni–Pd Bimetallic Catalysts for Hydrogen Production via Dry Reforming of Methane. Int. J. Hydrog. Energy 2020, 45, 16133–16143. [Google Scholar] [CrossRef]
- Nurunnabi, M.; Fujimoto, K.I.; Suzuki, K.; Li, B.; Kado, S.; Kunimori, K.; Tomishige, K. Promoting Effect of Noble Metals Addition on Activity and Resistance to Carbon Deposition in Oxidative Steam Reforming of Methane over NiO–MgO Solid Solution. Catal. Commun. 2006, 7, 73–78. [Google Scholar] [CrossRef]
- Jeong, J.H.; Lee, J.W.; Seo, D.J.; Seo, Y.; Yoon, W.L.; Lee, D.K.; Kim, D.H. Ru-Doped Ni Catalysts Effective for the Steam Reforming of Methane without the Pre-Reduction Treatment with H2. Appl. Catal. A Gen. 2006, 302, 151–156. [Google Scholar] [CrossRef]
- Trovarelli, A. Catalytic Properties of Ceria and CeO2-Containing Materials. Catal. Rev.—Sci. Eng. 1996, 38, 439–520. [Google Scholar] [CrossRef]
- Yoon, Y.; Kim, H.; Lee, J. Enhanced Catalytic Behavior of Ni Alloys in Steam Methane Reforming. J. Power Sources 2017, 359, 450–457. [Google Scholar] [CrossRef]
- Xi, J.; Wang, Q.; Duan, X.; Zhang, N.; Yu, J.; Sun, H.; Wang, S. Continuous Flow Reduction of Organic Dyes over Pd-Fe Alloy Based Fibrous Catalyst in a Fixed-Bed System. Chem. Eng. Sci. 2021, 231, 116303. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, J.; Dong, Z.; Zhan, Y.; Xi, J.; Xiao, J.; Huang, S.; Tian, F. Pd–Fe Bimetallic Nanoparticles Anchored on N-Doped Carbon-Modified Graphene for Efficient Catalytic Organic Reactions. Carbon Lett. 2022, 33, 77–87. [Google Scholar] [CrossRef]
- Wang, D.; Li, Y.; Wen, L.; Xi, J.; Liu, P.; Hansen, T.W.; Li, P. Ni-Pd-Incorporated Fe3O4 Yolk-Shelled Nanospheres as Efficient Magnetically Recyclable Catalysts for Reduction of N-Containing Unsaturated Compounds. Catalysts 2023, 13, 190. [Google Scholar] [CrossRef]
- Zhang, N.; Qiu, Y.; Sun, H.; Hao, J.; Chen, J.; Xi, J.; Liu, J.; He, B.; Bai, Z.-W. Substrate-Assisted Encapsulation of Pd-Fe Bimetal Nanoparticles on Functionalized Silica Nanotubes for Catalytic Hydrogenation of Nitroarenes and Azo Dyes. ACS Appl. Nano Mater. 2021, 4, 5854–5863. [Google Scholar] [CrossRef]
- Wen, L.; Wang, D.; Xi, J.; Tian, F.; Liu, P.; Bai, Z.W. Heterometal Modified Fe3O4 Hollow Nanospheres as Efficient Catalysts for Organic Transformations. J. Catal. 2022, 413, 779–785. [Google Scholar] [CrossRef]
- Matus, V.; Sukhova, O.B.; Ismagilov, I.Z.; Ushakov, V.A.; Yashnik, S.A.; Kerzhentsev, M.A.; Ismagilov, Z.R. Steam/CO2 Reforming of Methane Over Impregnated Ni/CeO2 Catalysts: Effect of Sample Composition on Their Activity and Stability. Eurasian Chem. J. 2022, 24, 191–202. [Google Scholar] [CrossRef]
- Okhlopkova, L.B.; Kerzhentsev, M.A.; Tuzikov, F.V.; Larichev, Y.V.; Prosvirin, I.P.; Ismagilov, Z.R. Palladium-Zinc Catalysts on Mesoporous Titania Prepared by Colloid Synthesis. I. Size Control Synthesis of PdZn Nanoclusters by a Polyol Method. J. Nanoparticle Res. 2012, 14, 1089. [Google Scholar] [CrossRef]
- Matus, E.V.; Sukhova, O.B.; Ismagilov, I.Z.; Kerzhentsev, M.A.; Li, L.; Ismagilov, Z.R. Bi-Reforming of Methane: Thermodynamic Equilibrium Analysis and Selection of Preferable Reaction Conditions. J. Phys. Conf. Ser. 2021, 1749, 012023. [Google Scholar] [CrossRef]
- Nguyen-Phan, T.D.; Song, M.B.; Kim, E.J.; Shin, E.W. The Role of Rare Earth Metals in Lanthanide-Incorporated Mesoporous Titania. Microporous Mesoporous Mater. 2009, 119, 290–298. [Google Scholar] [CrossRef]
- Yao, S.Y.; Xu, W.Q.; Johnston-Peck, A.C.; Zhao, F.Z.; Liu, Z.Y.; Luo, S.; Senanayake, S.D.; Martínez-Arias, A.; Liu, W.J.; Rodriguez, J.A. Morphological Effects of the Nanostructured Ceria Support on the Activity and Stability of CuO/CeO2 Catalysts for the Water-Gas Shift Reaction. Phys. Chem. Chem. Phys. 2014, 16, 17183–17195. [Google Scholar] [CrossRef] [PubMed]
- Khalesi, A.; Arandiyan, H.R.; Parvari, M. Production of Syngas by CO2 Reforming on MxLa1−xNi0.3Al0.7O3−d (M = Li, Na, K) Catalysts. Ind. Eng. Chem. Res. 2008, 47, 5892–5898. [Google Scholar] [CrossRef]
- Matus, E.V.; Okhlopkova, L.B.; Sukhova, O.B.; Ismagilov, I.Z.; Kerzhentsev, M.A.; Ismagilov, Z.R. Effects of Preparation Mode and Doping on the Genesis and Properties of Ni/Ce1−xMxOy Nanocrystallites (M = Gd, La, Mg) for Catalytic Applications. J. Nanoparticle Res. 2019, 21, 11. [Google Scholar] [CrossRef]
- Zhiming, G.A.O.; Shan, Z.; Hongwei, M.A.; Zhanping, L.I. Surface Composition Change of Chlorine-Doped Catalyst Ni (Clx)/CeO2 in Methanation Reaction. J. Rare Earths 2017, 35, 977–983. [Google Scholar] [CrossRef]
- Huang, Y.; Long, B.; Tang, M.; Rui, Z.; Balogun, M.S.; Tong, Y.; Ji, H. Bifunctional Catalytic Material: An Ultrastable and High-Performance Surface Defect CeO2 Nanosheets for Formaldehyde Thermal Oxidation and Photocatalytic Oxidation. Appl. Catal. B Environ. 2016, 181, 779–787. [Google Scholar] [CrossRef]
- Huang, T.J.; Lin, H.J.; Yu, T.C. A Comparison of Oxygen-Vacancy Effect on Activity Behaviors of Carbon Dioxide and Steam Reforming of Methane over Supported Nickel Catalysts. Catal. Letters 2005, 105, 239–247. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, L.; Zhang, R.; Li, L.; Cheng, Z.; Zhou, Z. Selective Hydrogenation of Phenylacetylene over Bimetallic Pd-Cu/Al2O3 and Pd-Zn/Al2O3 Catalysts. Catal. Today 2016, 264, 37–43. [Google Scholar] [CrossRef]
- Löfberg, A.; Guerrero-Caballero, J.; Kane, T.; Rubbens, A.; Jalowiecki-Duhamel, L. Ni/CeO2 Based Catalysts as Oxygen Vectors for the Chemical Looping Dry Reforming of Methane for Syngas Production. Appl. Catal. B Environ. 2017, 212, 159–174. [Google Scholar] [CrossRef]
- Özkara-Aydnolu, Ş. Thermodynamic Equilibrium Analysis of Combined Carbon Dioxide Reforming with Steam Reforming of Methane to Synthesis Gas. Int. J. Hydrog. Energy 2010, 35, 12821–12828. [Google Scholar] [CrossRef]
- Maestri, M.; Vlachos, D.G.; Beretta, A.; Groppi, G.; Tronconi, E. Steam and Dry Reforming of Methane on Rh: Microkinetic Analysis and Hierarchy of Kinetic Models. J. Catal. 2008, 259, 211–222. [Google Scholar] [CrossRef]
- Shinde, V.M.; Madras, G. Catalytic Performance of Highly Dispersed Ni/TiO2 for Dry and Steam Reforming of Methane. RSC Adv. 2014, 4, 4817–4826. [Google Scholar] [CrossRef]
- Wei, J.; Iglesia, E. Structural Requirements and Reaction Pathways in Methane Activation and Chemical Conversion Catalyzed by Rhodium. J. Catal. 2004, 225, 116–127. [Google Scholar] [CrossRef]
- Jones, G.; Jakobsen, J.G.; Shim, S.S.; Kleis, J.; Andersson, M.P.; Rossmeisl, J.; Abild-Pedersen, F.; Bligaard, T.; Helveg, S.; Hinnemann, B.; et al. First Principles Calculations and Experimental Insight into Methane Steam Reforming over Transition Metal Catalysts. J. Catal. 2008, 259, 147–160. [Google Scholar] [CrossRef]
- Jang, W.J.; Jeong, D.W.; Shim, J.O.; Kim, H.M.; Roh, H.S.; Son, I.H.; Lee, S.J. Combined Steam and Carbon Dioxide Reforming of Methane and Side Reactions: Thermodynamic Equilibrium Analysis and Experimental Application. Appl. Energy 2016, 173, 80–91. [Google Scholar] [CrossRef]
- Jang, W.J.; Kim, H.M.; Shim, J.O.; Yoo, S.Y.; Jeon, K.W.; Na, H.S.; Lee, Y.L.; Jeong, D.W.; Bae, J.W.; Nah, I.W.; et al. Key Properties of Ni-MgO-CeO2, Ni-MgO-ZrO2, and Ni-MgO-Ce(1−x)Zr(x)O2 Catalysts for the Reforming of Methane with Carbon Dioxide. Green Chem. 2018, 20, 1621–1633. [Google Scholar] [CrossRef]
- Xiang, X.; Zhao, H.; Yang, J.; Zhao, J.; Yan, L.; Song, H.; Chou, L. Nickel Based Mesoporous Silica-Ceria-Zirconia Composite for Carbon Dioxide Reforming of Methane. Appl. Catal. A Gen. 2016, 520, 140–150. [Google Scholar] [CrossRef]
- Da Fonseca, R.O.; Ponseggi, A.R.; Rabelo-Neto, R.C.; Simões, R.C.C.; Mattos, L.V.; Noronha, F.B. Controlling Carbon Formation over Ni/CeO2 Catalyst for Dry Reforming of CH4 by Tuning Ni Crystallite Size and Oxygen Vacancies of the Support. J. CO2 Util. 2022, 57, 101880. [Google Scholar] [CrossRef]
- Ruckenstein, E. Binary MgO-Based Solid Solution Catalysts for Methane Conversion to Syngas. Catal. Rev.—Sci. Eng. 2002, 44, 423–453. [Google Scholar] [CrossRef]
- Singha, R.K.; Shukla, A.; Sandupatla, A.; Deo, G.; Bal, R. Synthesis and Catalytic Activity of a Pd Doped Ni-MgO Catalyst for Dry Reforming of Methane. J. Mater. Chem. A 2017, 5, 15688–15699. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Textural Properties | ||
|---|---|---|---|
| SBET, m2/g | Vpore, cm3/g | Dpore, nm | |
| CeMg-A-S-V | 77.9 | 0.12 | 5.1 |
| Ni/CeMg-C-S-O | 23.0 | 0.12 | 18.4 |
| Ni/CeMg-A-S-V | 13.9 | 0.05 | 9.4 |
| Ni/CeMg-A-S-O | 48.2 | 0.278 | 17.4 |
| Ni-Pd/CeMg-A-S-V | 21.1 | 0.06 | 8.3 |
| Ni/CeMg0.5-A-P-O | 34.3 | 0.104 | 7.6 |
| Sample | 500 °C | 700 °C | ||||||
|---|---|---|---|---|---|---|---|---|
| aCeO2, Å | dCeO2, nm | aNiO, Å | dNiO, nm | aCeO2, Å | dCeO2, nm | aNiO, Å | dNiO, nm | |
| Ni/CeMg-C-S-O | 5.393 | 21 | 4.20 | 17 | 5.394 | 29 | 4.196 | 40 |
| Ni/CeMg-A-S-V | 5.403 | 6 | ND 1 | ND 1 | 5.401 | 15 | 4.212 | 9 |
| Ni/CeMg-A-S-O | 5.400 | 17 | 4.17 | 14 | 5.406 | 21 | 4.185 | 19 |
| Ni-Pd/CeMg-A-S-V | 5.401 | 10 | ND 1 | ND 1 | 5.400 | 35 | 4.194 | 30 |
| Ni/CeMg-A-P-O | 5.397 | 19 | 4.17 | 18 | 5.404 | 36 | 4.185 | 40 |
| Sample | 600 °C | 800 °C | ||||||
|---|---|---|---|---|---|---|---|---|
| aCeO2, Å | dCeO2, nm | aNiO, Å | dNiO, nm | aCeO2, Å | dCeO2, nm | aNiO, Å | dNiO, nm | |
| Ni/CeMg-C-S-O | 5.399 | 44 | 4.20 | 49 | - | 5.410 | 50 | 4.20 |
| Ni/CeMg-A-S-V | 5.400 | 12 | ND 1 | ND 1 | ND 1 | 5.408 | 23 | 4.20 |
| Ni/CeMg-A-S-O | 5.401 | 12 | ND 1 | ND 1 | 12 | 5.406 | 32 | 4.18 |
| Ni-Pd/CeMg-A-S-V | 5.401 | 31 | ND 1 | ND 1 | 17 | 5.404 | 36 | ND |
| Ni/CeMg-A-P-O | 5.400 | 25 | ND 1 | ND 1 | 25 | 5.404 | 34 | 4.21 |
| Sample | Ce3+/Ce4+ 1 | Concentration of Defective Oxygen (%) 2 | Ni/Ce | Ni0/Ni2+ 3 |
|---|---|---|---|---|
| Ni/CeMg-C-S-V | 0.19 | 17.3 | 0.06 | 0 |
| Ni/CeMg-A-S-V | 0.25 | 28.4 | 0.52 | 0.72 |
| Ni/CeMg-A-S-O | 0.23 | 26.6 | 0.32 | 0.67 |
| Ni-Pd/CeMg-A-S-V | 0.28 | 32.7 | 0.41 | 0.63 |
| Ni/CeMg-A-P-O | 0.22 | 28.1 | 1.3 | 0.77 |
| Sample | Indicators of the Reaction at 750 °C | XRD Data | TA Data after Reaction | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| XCH4 | XCO2 | YH2 | YCO | H2/CO | aCeO2,Å | dCeO2, nm | dNiO, nm | dNi, nm | Δm/m3 % | Δm/m4 % | |
| Ni/CeMg-C-S-O | 12 | 22 | 0 | 8 | 0 | 5.402 | 39 | 51 | No phase | 0.04 | −0.09 |
| Ni/CeMg-A-S-V | 72 | 87 | 67 | 77 | 1.2 | 5.399 | 31 | ND 1 | 31 | −3.0 | −0.76 |
| Ni/CeMg-A-S-O | 74 | 75 | 65 | 72 | 1.3 | 5.402 | 35 | ND 1 | 46 | 0.71 | −0.15 |
| Ni-Pd/CeMg-A-S-V | 57 | 64 | 55 | 62 | 1.4 | 5.399 | 26 | 21 | 16 | 0.6 | −0.36 |
| Ni/CeMg-A-P-O | 51 | 71 | 49 | 59 | 1.1 | 5.400 | 47 | ND 1 | 83 | 0.69 | 0.59 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okhlopkova, L.; Prosvirin, I.; Kerzhentsev, M.; Ismagilov, Z. Combined Steam and CO2 Reforming of Methane over Ni-Based CeO2-MgO Catalysts: Impacts of Preparation Mode and Pd Addition. Appl. Sci. 2023, 13, 4689. https://doi.org/10.3390/app13084689
Okhlopkova L, Prosvirin I, Kerzhentsev M, Ismagilov Z. Combined Steam and CO2 Reforming of Methane over Ni-Based CeO2-MgO Catalysts: Impacts of Preparation Mode and Pd Addition. Applied Sciences. 2023; 13(8):4689. https://doi.org/10.3390/app13084689
Chicago/Turabian StyleOkhlopkova, Lyudmila, Igor Prosvirin, Mikhail Kerzhentsev, and Zinfer Ismagilov. 2023. "Combined Steam and CO2 Reforming of Methane over Ni-Based CeO2-MgO Catalysts: Impacts of Preparation Mode and Pd Addition" Applied Sciences 13, no. 8: 4689. https://doi.org/10.3390/app13084689