
Microscopic Pair Potentials and the Physical Properties of the Condensed Phases of Parahydrogen


Abstract
:1. Introduction
2. Model and Methodology
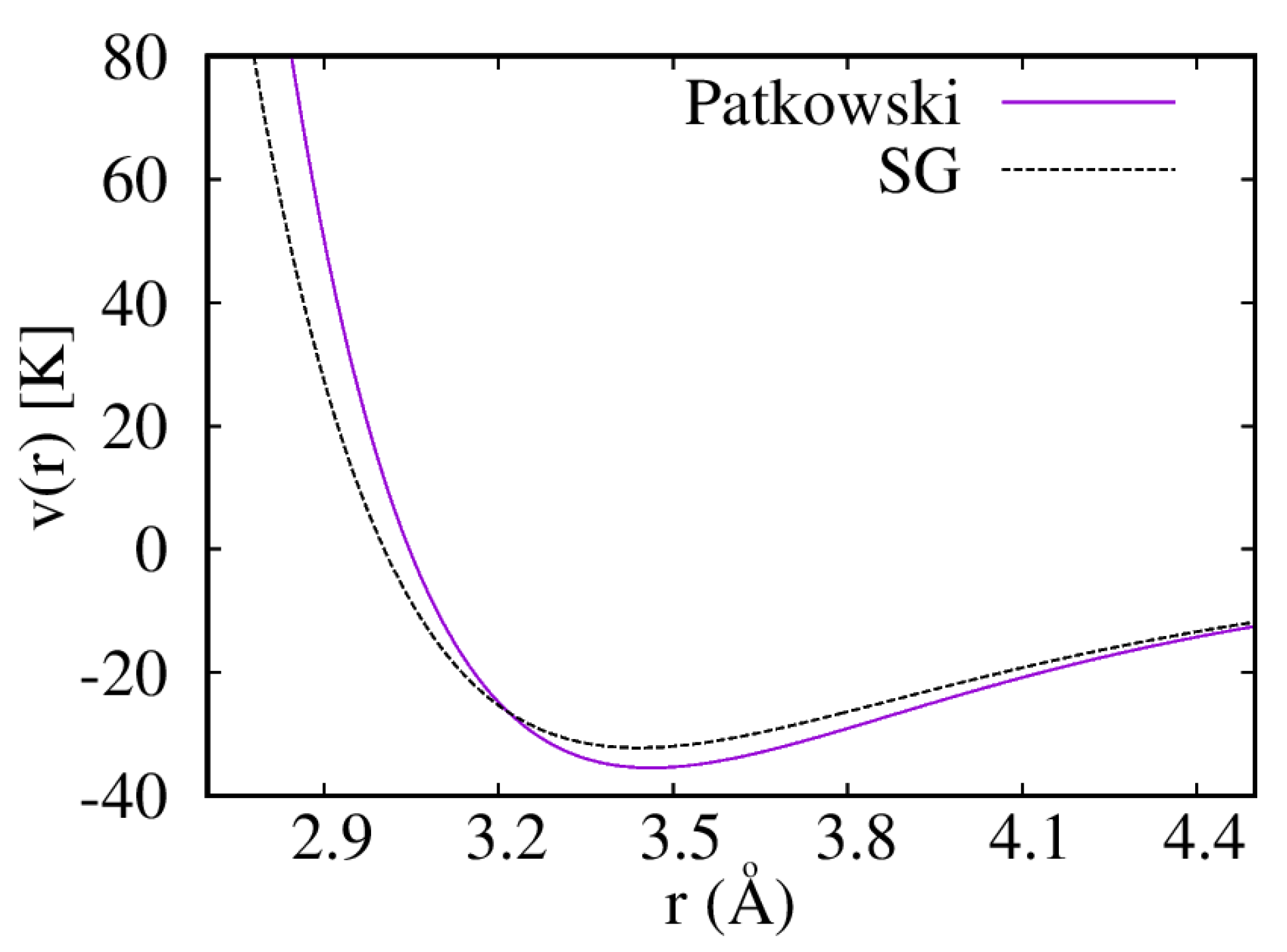
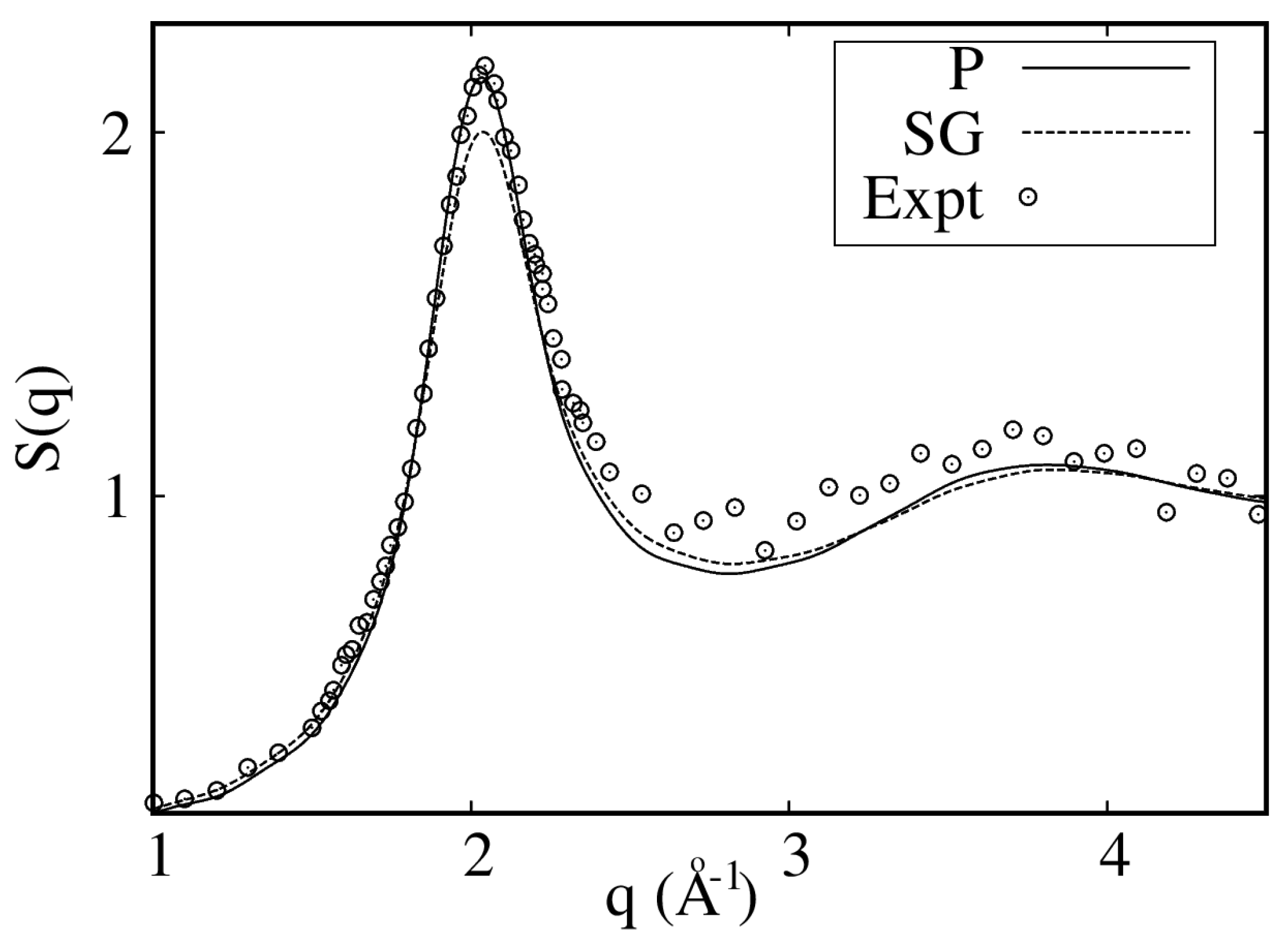
3. Results
3.1. Liquid
3.2. Solid
4. Discussion and Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Silvera, I.F. The solid molecular hydrogens in the condensed phase: Fundamentals and static properties. Rev. Mod. Phys. 1980, 52, 393–452. [Google Scholar] [CrossRef]
- Chabal, Y.J.; Patel, C.K.N. Molecular hydrogen in a-Si: H. Rev. Mod. Phys. 1987, 59, 835–844. [Google Scholar] [CrossRef]
- Myers, S.M.; Baskes, M.I.; Birnbaum, H.K.; Corbett, J.W.; DeLeo, G.G.; Estreicher, S.K.; Haller, E.E.; Jena, P.; Johnson, N.M.; Kirchheim, R.; et al. Hydrogen interactions with defects in crystalline solids. Rev. Mod. Phys. 1992, 64, 559–617. [Google Scholar] [CrossRef]
- Mao, H.K.; Hemley, R.J. Ultrahigh-pressure transitions in solid hydrogen. Rev. Mod. Phys. 1994, 66, 671–692. [Google Scholar] [CrossRef]
- Nellis, W.J.; Ross, M.; Holmes, N.C. Temperature Measurements of Shock-Compressed Liquid Hydrogen: Implications for the Interior of Jupiter. Science 1995, 269, 1249–1252. [Google Scholar] [CrossRef]
- Kohanoff, J. The status of the low-temperature phase diagram of hydrogen at the turn of the century. J. Low. Temp. Phys. 2001, 122, 297–311. [Google Scholar] [CrossRef]
- Guillot, T. The Interiors of Giant Planets: Models and Outstanding Questions. Ann. Rev. Earth Planet. Sci. 2005, 33, 493–530. [Google Scholar] [CrossRef]
- Silvera, I.F.; Cole, J.W. Metallic hydrogen: The most powerful rocket fuel yet to exist. J. Phys. Conf. Ser. 2010, 215, 012194. [Google Scholar] [CrossRef]
- McMahon, J.M.; Morales, M.A.; Pierleoni, C.; Ceperley, D.M. The properties of hydrogen and helium under extreme conditions. Rev. Mod. Phys. 2012, 84, 1607–1653. [Google Scholar] [CrossRef]
- Tozzini, V.; Pellegrini, V. Prospects for hydrogen storage in graphene. Phys. Chem. Chem. Phys. 2013, 15, 80–89. [Google Scholar] [CrossRef]
- Silvera, I.F.; Dias, R. Phases of the hydrogen isotopes under pressure: Metallic hydrogen. Adv. Phys. X 2021, 6, 1961607. [Google Scholar] [CrossRef]
- Dharma-wardana, M.W.C.; Perrot, F. Density-functional theory of hydrogen plasmas. Phys. Rev. A 1982, 26, 2096–2104. [Google Scholar] [CrossRef]
- Barbee, T.W.; Cohen, M.L.; Martins, J.L. Theory of high-pressure phases of hydrogen. Phys. Rev. Lett. 1989, 62, 1150–1153. [Google Scholar] [CrossRef] [PubMed]
- Pickard, C.J.; Needs, R.J. Structure of phase III of solid hydrogen. Nat. Phys. 2007, 3, 473–476. [Google Scholar] [CrossRef] [Green Version]
- Ceperley, D.M.; Alder, B.J. Ground state of solid hydrogen at high pressures. Phys. Rev. B 1987, 36, 2092–2106. [Google Scholar] [CrossRef]
- Natoli, V.; Martin, R.M.; Ceperley, D.M. Crystal structure of atomic hydrogen. Phys. Rev. Lett. 1993, 70, 1952–1955. [Google Scholar] [CrossRef]
- Pierleoni, C.; Ceperley, D.M.; Bernu, B.; Magro, W.R. Equation of State of the Hydrogen Plasma by Path Integral Monte Carlo Simulation. Phys. Rev. Lett. 1994, 73, 2145–2149. [Google Scholar] [CrossRef]
- Magro, W.R.; Ceperley, D.M.; Pierleoni, C.; Bernu, B. Molecular Dissociation in Hot, Dense Hydrogen. Phys. Rev. Lett. 1996, 76, 1240–1243. [Google Scholar] [CrossRef] [Green Version]
- Hohl, D.; Natoli, V.; Ceperley, D.M.; Martin, R.M. Molecular dynamics in dense hydrogen. Phys. Rev. Lett. 1993, 71, 541–544. [Google Scholar] [CrossRef]
- Kohanoff, J.; Scandolo, S.; Chiarotti, G.L.; Tosatti, E. Solid Molecular Hydrogen: The Broken Symmetry Phase. Phys. Rev. Lett. 1997, 78, 2783–2786. [Google Scholar] [CrossRef]
- Kitamura, H.; Tsuneyuki, S.; Ogitsu, T.; Miyake, T. Quantum distribution of protons in solid molecular hydrogen at megabar pressures. Nature 2000, 404, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Delaney, K.T.; Pierleoni, C.; Ceperley, D.M. Quantum Monte Carlo Simulation of the High-Pressure Molecular-Atomic Crossover in Fluid Hydrogen. Phys. Rev. Lett. 2006, 97, 235702. [Google Scholar] [CrossRef] [Green Version]
- Attaccalite, C.; Sorella, S. Stable Liquid Hydrogen at High Pressure by a Novel Ab Initio Molecular-Dynamics Calculation. Phys. Rev. Lett. 2008, 100, 114501. [Google Scholar] [CrossRef] [Green Version]
- Pierleoni, C.; Morales, M.A.; Rillo, G.; Holzmann, M.; Ceperley, D.M. Liquid–liquid phase transition in hydrogen by coupled electron–ion Monte Carlo simulations. Proc. Natl. Acad. Sci. USA 2016, 113, 4953–4957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzola, G.; Helled, R.; Sorella, S. Phase Diagram of Hydrogen and a Hydrogen-Helium Mixture at Planetary Conditions by Quantum Monte Carlo Simulations. Phys. Rev. Lett. 2018, 120, 025701. [Google Scholar] [CrossRef] [Green Version]
- Diep, P.; Johnson, J.K. An accurate H2–H2 interaction potential from first principles. J. Chem. Phys. 2000, 112, 4465–4473. [Google Scholar] [CrossRef]
- Silvera, I.F.; Goldman, V.V. The isotropic intermolecular potential for H2 and D2 in the solid and gas phases. J. Chem. Phys. 1978, 69, 4209–4213. [Google Scholar] [CrossRef]
- Celli, M.; Colognesi, D.; Zoppi, M. Experimental determination of the translational kinetic energy of liquid and solid hydrogen. Eur. Phys. J. B 2000, 14, 239–244. [Google Scholar] [CrossRef]
- Operetto, F.; Pederiva, F. Diffusion Monte Carlo study of the equation of state of solid para- H2. Phys. Rev. B 2006, 73, 184124. [Google Scholar] [CrossRef] [Green Version]
- Gernoth, K.A.; Lindenau, T.; Ristig, M.L. Screening of particle exchange in quantum Boltzmann liquids. Phys. Rev. B 2007, 75, 174204. [Google Scholar] [CrossRef]
- Boninsegni, M. Quantum statistics and the momentum distribution of liquid parahydrogen. Phys. Rev. B 2009, 79, 174203. [Google Scholar] [CrossRef]
- Omiyinka, T.; Boninsegni, M. Pair potentials and equation of state of solid para-hydrogen to megabar pressure. Phys. Rev. B 2013, 88, 024112. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.W. Compression of solidified gases to 20,000 kg/cm2 at low temperature. J. Phys. Chem. Solids 1956, 1, 146–158. [Google Scholar] [CrossRef]
- Schnepp, O. One-Phonon Excited States of Solid H2 and D2 in the Ordered Phase. Phys. Rev. A 1970, 2, 2574. [Google Scholar] [CrossRef]
- Driessen, A.; Waal, J.A.; Silvera, I.F. Experimental determination of the equation of state of solid hydrogen and deuterium at high pressures. J. Low. Temp. Phys. 1979, 34, 255–305. [Google Scholar] [CrossRef]
- Dawidowski, J.; Bermejo, F.J.; Ristig, M.L.; Cabrillo, C.; Bennington, S.M. Density dependence of the momentum distributions in liquid para-hydrogen. Phys. Rev. B 2006, 73, 10. [Google Scholar] [CrossRef] [Green Version]
- Celli, M.; Bafile, U.; Cuello, G.J.; Formisano, F.; Guarini, E.; Magli, R.; Neumann, M.; Zoppi, M. Microscopic structure factor of liquid hydrogen by neutron-diffraction measurements. Phys. Rev. B 2005, 71, 014205. [Google Scholar] [CrossRef]
- Norman, M.J.; Watts, R.O.; Buck, U. A spherical potential for hydrogen from solid state and scattering data. J. Chem. Phys. 1984, 81, 3500–3504. [Google Scholar] [CrossRef]
- Moraldi, M. Effective Pair Potential for Solid Molecular Hydrogen at High Pressures. J. Low Temp. Phys. 2012, 168, 275–284. [Google Scholar] [CrossRef]
- Patkowski, K.; Cencek, W.; Jankowski, P.; Szalewicz, K.; Mehl, J.B.; Garberoglio, G.; Harvey, A.H. Potential energy surface for interactions between two hydrogen molecules. J. Chem. Phys. 2008, 129, 094304. [Google Scholar] [CrossRef]
- Mehl, J.B.; Huber, M.L.; Harvey, A.H. Ab Initio Transport Coefficients of Gaseous Hydrogen. Int. J. Thermophys. 2010, 31, 740–755. [Google Scholar] [CrossRef]
- Sindzingre, P.; Ceperley, D.M.; Klein, M.L. Superfluidity in clusters of p-H2 molecules. Phys. Rev. Lett. 1991, 67, 1871–1874. [Google Scholar] [CrossRef]
- Mezzacapo, F.; Boninsegni, M. Classical and quantum physics of hydrogen clusters. J. Phys. Condens. Matter 2009, 21, 164205. [Google Scholar] [CrossRef] [PubMed]
- Boninsegni, M. Computer Simulation Study of Nanoscale Size Parahydrogen Clusters. J. Low Temp. Phys. 2019, 195, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Omiyinka, T.; Boninsegni, M. Enhanced superfluid response of parahydrogen in nanoscale confinement. Phys. Rev. B 2014, 90, 064511. [Google Scholar] [CrossRef] [Green Version]
- Omiyinka, T.; Boninsegni, M. Quasi-one-dimensional parahydrogen in nanopores. Phys. Rev. B 2016, 93, 104501. [Google Scholar] [CrossRef] [Green Version]
- Boninsegni, M.; Pierleoni, C.; Ceperley, D.M. Isotopic shift of helium melting pressure: Path integral Monte Carlo study. Phys. Rev. Lett. 1994, 72, 1854–1857. [Google Scholar] [CrossRef] [Green Version]
- Mezzacapo, F.; Boninsegni, M. Superfluidity and Quantum Melting of p-H2 Clusters. Phys. Rev. Lett. 2006, 97, 045301. [Google Scholar] [CrossRef] [Green Version]
- Mezzacapo, F.; Boninsegni, M. Structure, superfluidity, and quantum melting of hydrogen clusters. Phys. Rev. A 2007, 75, 033201. [Google Scholar] [CrossRef] [Green Version]
- Boninsegni, M.; Prokof’ev, N.; Svistunov, B. Worm Algorithm for Continuous-Space Path Integral Monte Carlo Simulations. Phys. Rev. Lett. 2006, 96, 070601. [Google Scholar] [CrossRef]
- Boninsegni, M.; Prokof’ev, N.V.; Svistunov, B.V. Worm algorithm and diagrammatic Monte Carlo: A new approach to continuous-space path integral Monte Carlo simulations. Phys. Rev. E 2006, 74, 036701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feynman, R.; Hibbs, A. Quantum Mechanics and Path Integrals; McGraw-Hill: New York, NY, USA, 1965; Chapter 10. [Google Scholar]
- Boninsegni, M. Search for superfluidity in supercooled liquid parahydrogen. Phys. Rev. B 2018, 97, 054517. [Google Scholar] [CrossRef] [Green Version]
- Boninsegni, M. Low-temperature phase diagram of condensed para-hydrogen in two dimensions. Phys. Rev. B 2004, 70, 193411. [Google Scholar] [CrossRef] [Green Version]
- Boninsegni, M. Ground State Phase Diagram of Parahydrogen in One Dimension. Phys. Rev. Lett. 2013, 111, 235303. [Google Scholar] [CrossRef] [Green Version]
- Boninsegni, M. Permutation Sampling in Path Integral Monte Carlo. J. Low Temp. Phys. 2005, 141, 27–46. [Google Scholar] [CrossRef] [Green Version]
- Ceperley, D.M. Path integrals in the theory of condensed helium. Rev. Mod. Phys. 1995, 67, 279–355. [Google Scholar] [CrossRef]
- Azuah, R.T.; Stirling, W.R.; Glyde, H.R.; Boninsegni, M.; Sokol, P.E.; Bennington, S.M. Condensate and final-state effects in superfluid 4He. Phys. Rev. B 1997, 56, 14620–14630. [Google Scholar] [CrossRef]
- Zoppi, M.; Colognesi, D.; Celli, M. Density dependence of mean kinetic energy in liquid and solid hydrogen at 19.3 K. Eur. Phys. J. B 2001, 23, 171–178. [Google Scholar] [CrossRef]
- Diallo, S.O.; Pearce, J.V.; Azuah, R.T.; Albergamo, F.; Glyde, H.R. Condensate fraction and atomic kinetic energy of liquid He3-He4 mixtures. Phys. Rev. B 2006, 74, 144503. [Google Scholar] [CrossRef]
- Boninsegni, M. Kinetic energy and momentum distribution of isotopic liquid helium mixtures. J. Chem. Phys. 2018, 148, 102308. [Google Scholar] [CrossRef]
- Fernandez-Alonso, F.; Cabrillo, C.; Fernández-Perea, R.; Bermejo, F.J.; González, M.A.; Mondelli, C.; Farhi, E. Solid para-hydrogen as the paradigmatic quantum crystal: Three observables probed by ultrahigh-resolution neutron spectroscopy. Phys. Rev. B 2012, 86, 144524. [Google Scholar] [CrossRef] [Green Version]
- Axilrod, B.M.; Teller, E. Interaction of the van der Waals Type between Three Atoms. J. Chem. Phys. 1943, 11, 299. [Google Scholar] [CrossRef]
- Moroni, S.; Pederiva, F.; Fantoni, S.; Boninsegni, M. Equation of State of Solid 3He. Phys. Rev. Lett. 2000, 84, 2650–2653. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.Y.; Boninsegni, M. Ab initio potentials and the equation of state of condensed helium at high pressure. J. Chem. Phys. 2001, 115, 2629–2633. [Google Scholar] [CrossRef]
- Wind, P.; Røeggen, I. Ab initio calculation of three-body interaction in the (H2)3 trimer. Chem. Phys. 1996, 211, 179–189. [Google Scholar] [CrossRef]
- Hinde, R.J. Three-body interactions in solid parahydrogen. Chem. Phys. Lett. 2008, 460, 141–145. [Google Scholar] [CrossRef]
- Anatole von Lilienfeld, O.; Tkatchenko, A. Two- and three-body interatomic dispersion energy contributions to binding in molecules and solids. J. Chem. Phys. 2010, 132, 234109. [Google Scholar] [CrossRef]
- Huang, Y.; Beran, G.J.O. Reliable prediction of three-body intermolecular interactions using dispersion-corrected second-order Møller-Plesset perturbation theory. J. Chem. Phys. 2015, 143, 044113. [Google Scholar] [CrossRef]
- Ibrahim, A.; Roy, P.N. Three-body potential energy surface for para-hydrogen. J. Chem. Phys. 2022, 156, 044301. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Potential | (K) | Pressure (Bars) |
|---|---|---|
| Å, K | ||
| Silvera–Goldman | 61.7(2) | |
| Patkowski et al. | 66.7(2) | |
| Experiment [28] | ||
| Å, K | ||
| Silvera–Goldman | 61.8(2) | |
| Buck | 62.6(1) | |
| Patkowski et al. | 67.1(2) | 6.9(3) |
| Diep–Johnson | 67.0(1) | 20(1) |
| Experiment [36] | ~ | ~1 |
| Experiment [28] | ||
| Å, K | ||
| Silvera–Goldman | 64.1(1) | 9.6(4) |
| Patkowski et al. | 69.7(2) | 39.1(5) |
| Experiment [59] | − | |
| Å, K | ||
| Silvera–Goldman | 63.0(1) | |
| Patkowski et al. | 22.1(5) | |
| Experiment [59] | ||
| Potential | (K) | E (K) | Pressure (Bars) |
|---|---|---|---|
| Silvera–Goldman | |||
| Buck | |||
| Patkowski et al. | |||
| Diep–Johnson | |||
| Experiment [34] | |||
| Experiment [35] | ~6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, J.; Boninsegni, M. Microscopic Pair Potentials and the Physical Properties of the Condensed Phases of Parahydrogen. Appl. Sci. 2023, 13, 270. https://doi.org/10.3390/app13010270
Hu J, Boninsegni M. Microscopic Pair Potentials and the Physical Properties of the Condensed Phases of Parahydrogen. Applied Sciences. 2023; 13(1):270. https://doi.org/10.3390/app13010270
Chicago/Turabian StyleHu, Jieru, and Massimo Boninsegni. 2023. "Microscopic Pair Potentials and the Physical Properties of the Condensed Phases of Parahydrogen" Applied Sciences 13, no. 1: 270. https://doi.org/10.3390/app13010270