
Brassinin Enhances Apoptosis in Hepatic Carcinoma by Inducing Reactive Oxygen Species Production and Suppressing the JAK2/STAT3 Pathway



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Cell Culture
2.2. MTT Assay
2.3. Western Blotting
2.4. ROS Detection
2.5. DNA Fragmentation
2.6. Immunofluorescence for Nuclear Translocation
2.7. Colony Assay
2.8. Cell Invasion
2.9. Cell Migration
2.10. TUNEL Assay
2.11. Transfection Assay
2.12. Statistics
3. Results
3.1. BSN Inhibited Cell Growth in HCC Cells
3.2. BSN Induced Apoptosis through Caspase-Mediated Activation
3.3. BSN Induced Caspase-Dependent Apoptosis in HCC Cells
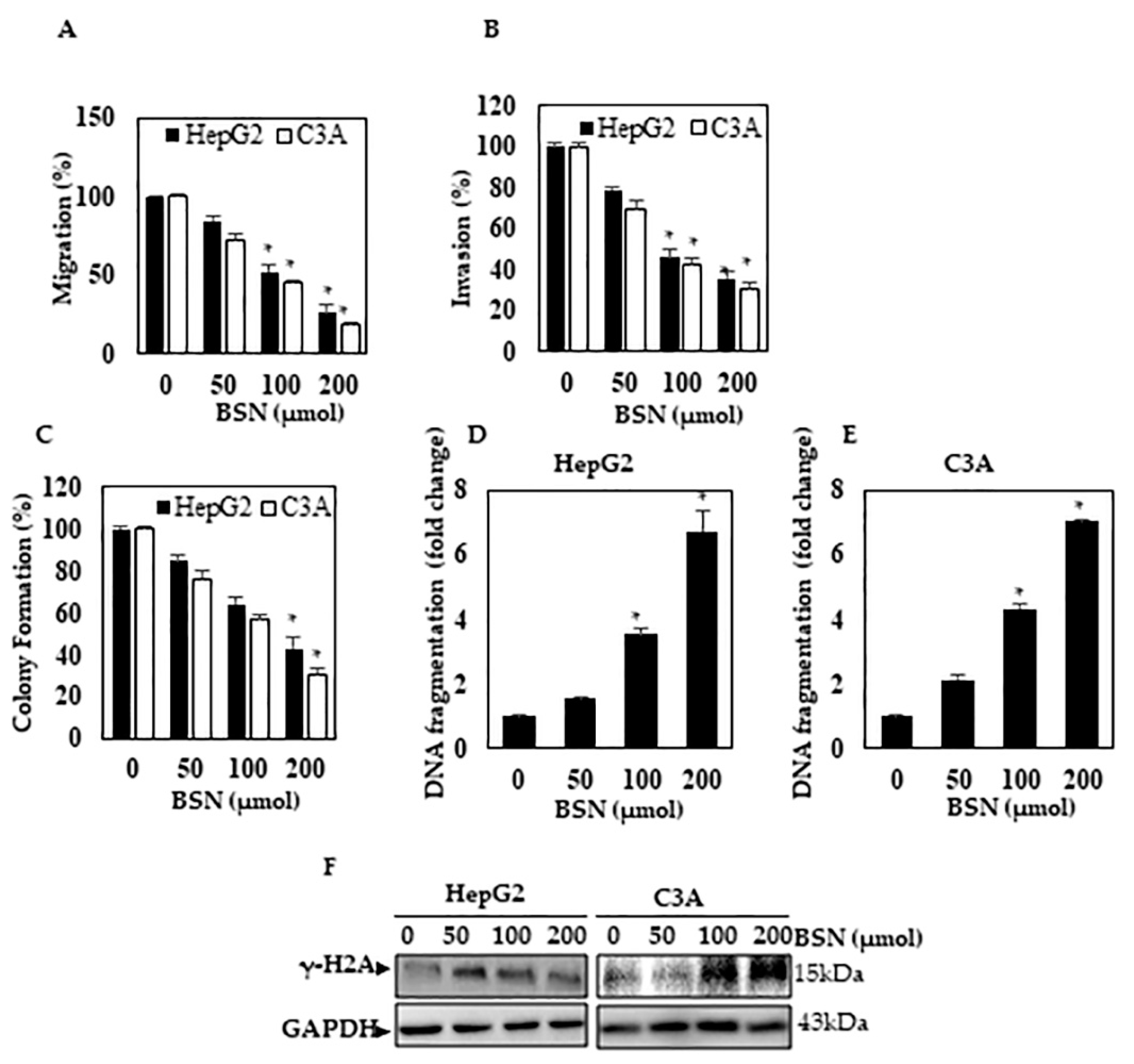
3.4. BSN Inhibited the HCC Migration and Invasion
3.5. BSN Inhibited HCC Colony Formation
3.6. BSN Induced DNA Damage in HCC Cells
3.7. BSN Promoted ROS Production and Inactivated STAT3 in HCC Cancer Cells
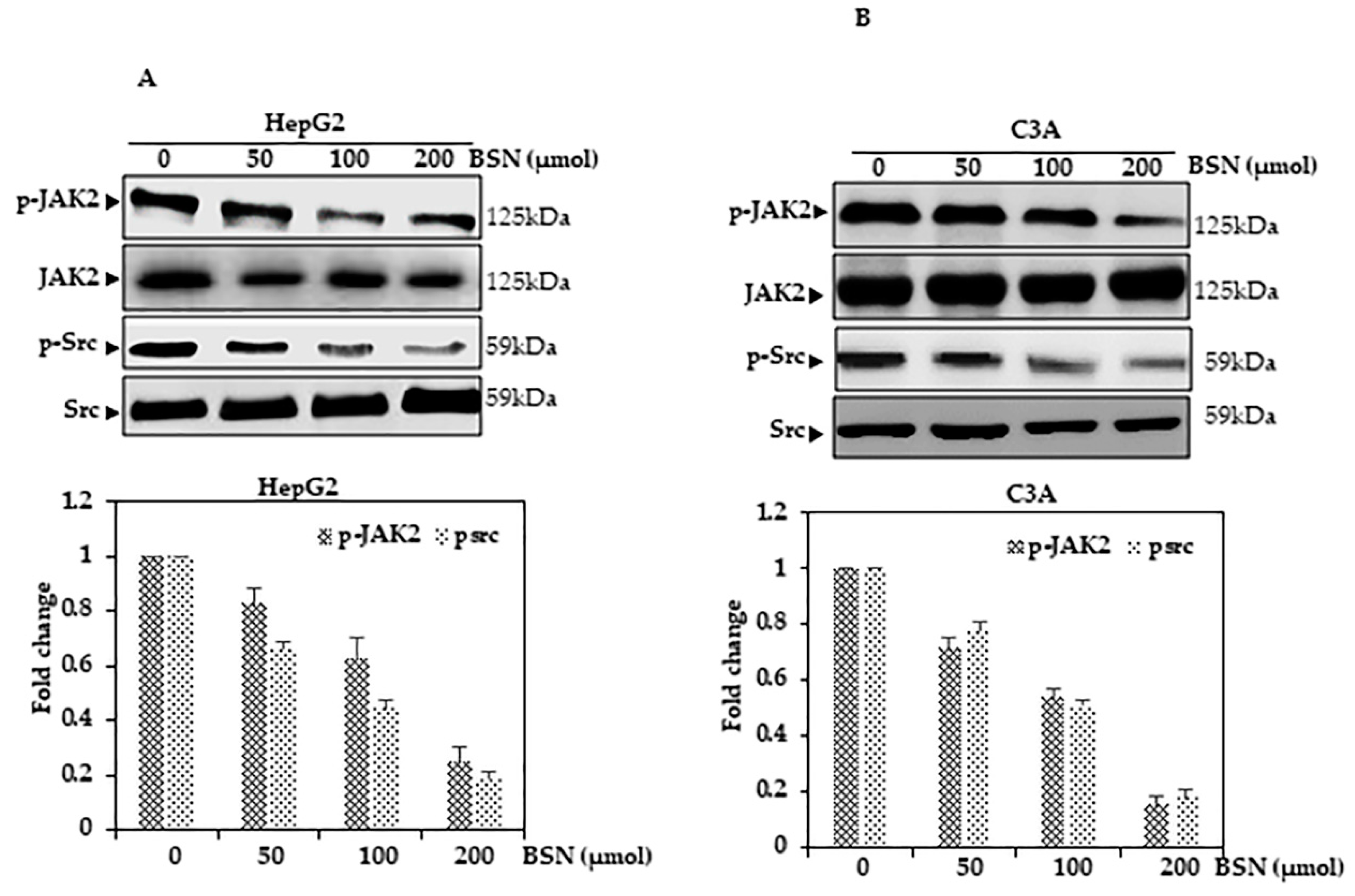
3.8. BSN Inhibited STAT3 Activation in HCC Cells
3.9. STAT3 Silencing Suppressed BSN-Mediated Apoptosis in HCC
3.10. BSN Inhibited Src Activation
3.11. BSN Promoted Apoptosis in HCC Cells
3.12. Gene Expression Profiling Interactive Analysis of STAT3 in Liver Cancer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mohan, C.D.; Yang, M.H.; Rangappa, S.; Chinnathambi, A.; Alharbi, S.A.; Alahmadi, T.A.; Deivasigamani, A.; Hui, K.M.; Sethi, G.; Rangappa, K.S. 3-Formylchromone Counteracts STAT3 Signaling Pathway by Elevating SHP-2 Expression in Hepatocellular Carcinoma. Biology 2021, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Mohan, C.D.; Bharathkumar, H.; Bulusu, K.C.; Pandey, V.; Rangappa, S.; Fuchs, J.E.; Shanmugam, M.K.; Dai, X.; Li, F.; Deivasigamani, A. Development of a novel azaspirane that targets the Janus kinase-signal transducer and activator of transcription (STAT) pathway in hepatocellular carcinoma in vitro and in vivo. J. Biol. Chem. 2014, 289, 34296–34307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-H.; Yeh, C.-W.; Lo, H.-C.; Su, S.-L.; Hseu, Y.-C.; Hsu, L.-S. Generation of reactive oxygen species mediates butein-induced apoptosis in neuroblastoma cells. Oncol. Rep. 2012, 27, 1233–1237. [Google Scholar] [CrossRef] [Green Version]
- Chripkova, M.; Drutovic, D.; Pilatova, M.; Mikes, J.; Budovska, M.; Vaskova, J.; Broggini, M.; Mirossay, L.; Mojzis, J. Brassinin and its derivatives as potential anticancer agents. Toxicol. Vitr. 2014, 28, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.; Ham, J.; Song, J.; Song, G.; Lim, W. Brassinin inhibits proliferation in human liver cancer cells via mitochondrial dysfunction. Cells 2021, 10, 332. [Google Scholar] [CrossRef]
- Takasugi, M.; Katsui, N.; Shirata, A. Isolation of three novel sulphur-containing phytoalexins from the chinese cabbage Brassica campestris L. ssp. pekinensis (cruciferae). J. Chem. Soc. Chem. Commun. 1986, 14, 1077–1078. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, C.; Sethi, G.; Ahn, K.S. Brassinin inhibits STAT3 signaling pathway through modulation of PIAS-3 and SOCS-3 expression and sensitizes human lung cancer xenograft in nude mice to paclitaxel. Oncotarget 2015, 6, 6386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.M.; Park, J.H.; Kim, K.D.; Nam, D.; Shim, B.S.; Kim, S.H.; Ahn, K.S.; Choi, S.H.; Ahn, K.S. Brassinin induces apoptosis in PC-3 human prostate cancer cells through the suppression of PI3K/Akt/mTOR/S6K1 signaling cascades. Phytother. Res. 2014, 28, 423–431. [Google Scholar] [CrossRef]
- Jung, Y.Y.; Ha, I.J.; Um, J.-Y.; Sethi, G.; Ahn, K.S. Fangchinoline diminishes STAT3 activation by stimulating oxidative stress and targeting SHP-1 protein in multiple myeloma model. J. Adv. Res. 2022, 35, 245–257. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Rajendran, P.; Li, F.; Kim, C.; Sikka, S.; Siveen, K.S.; Kumar, A.P.; Ahn, K.S.; Sethi, G. Abrogation of STAT3 signaling cascade by zerumbone inhibits proliferation and induces apoptosis in renal cell carcinoma xenograft mouse model. Mol. Carcinog. 2015, 54, 971–985. [Google Scholar] [CrossRef]
- Kannaiyan, R.; Hay, H.S.; Rajendran, P.; Li, F.; Shanmugam, M.K.; Vali, S.; Abbasi, T.; Kapoor, S.; Sharma, A.; Kumar, A.P. Celastrol inhibits proliferation and induces chemosensitization through down-regulation of NF-κB and STAT3 regulated gene products in multiple myeloma cells. Br. J. Pharmacol. 2011, 164, 1506–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmugam, M.K.; Rajendran, P.; Li, F.; Nema, T.; Vali, S.; Abbasi, T.; Kapoor, S.; Sharma, A.; Kumar, A.P.; Ho, P.C. Ursolic acid inhibits multiple cell survival pathways leading to suppression of growth of prostate cancer xenograft in nude mice. J. Mol. Med. 2011, 89, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Li, F.; Manu, K.A.; Shanmugam, M.K.; Loo, S.Y.; Kumar, A.P.; Sethi, G. γ-Tocotrienol is a novel inhibitor of constitutive and inducible STAT3 signalling pathway in human hepatocellular carcinoma: Potential role as an antiproliferative, pro-apoptotic and chemosensitizing agent. Br. J. Pharmacol. 2011, 163, 283–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, S.H.; Ko, J.-H.; Lee, H.; Jung, J.; Kong, M.; Lee, J.-w.; Lee, J.; Chinnathambi, A.; Zayed, M.; Alharbi, S.A. Resveratrol inhibits STAT3 signaling pathway through the induction of SOCS-1: Role in apoptosis induction and radiosensitization in head and neck tumor cells. Phytomedicine 2016, 23, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Nandakumar, N.; Rengarajan, T.; Palaniswami, R.; Gnanadhas, E.N.; Lakshminarasaiah, U.; Gopas, J.; Nishigaki, I. Antioxidants and human diseases. Clin. Chim. Acta 2014, 436, 332–347. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative stress in cancer. Cancer cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Iyer, V.V.; Yang, H.; Ierapetritou, M.G.; Roth, C.M. Effects of glucose and insulin on HepG2-C3A cell metabolism. Biotechnol. Bioeng. 2010, 107, 347–356. [Google Scholar] [CrossRef]
- Takeno, A.; Kanazawa, I.; Notsu, M.; Tanaka, K.-i.; Sugimoto, T. Phloretin promotes adipogenesis via mitogen-activated protein kinase pathways in mouse marrow stromal ST2 cells. Int. J. Mol. Sci. 2018, 19, 1772. [Google Scholar] [CrossRef] [Green Version]
- Ismail, M.B.; Rajendran, P.; AbuZahra, H.M.; Veeraraghavan, V.P. Mangiferin Inhibits Apoptosis in Doxorubicin-Induced Vascular Endothelial Cells via the Nrf2 Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 4259. [Google Scholar] [CrossRef]
- Adaramoye, O.; Erguen, B.; Oyebode, O.; Nitzsche, B.; Höpfner, M.; Jung, K.; Rabien, A. Antioxidant, antiangiogenic and antiproliferative activities of root methanol extract of Calliandra portoricensis in human prostate cancer cells. J. Integr. Med. 2015, 13, 185–193. [Google Scholar] [CrossRef]
- Cao, K.; Tait, S.W. Apoptosis and cancer: Force awakens, phantom menace, or both? Int. Rev. Cell Mol. Biol. 2018, 337, 135–152. [Google Scholar] [PubMed]
- Prabhu, K.S.; Bhat, A.A.; Siveen, K.S.; Kuttikrishnan, S.; Raza, S.S.; Raheed, T.; Jochebeth, A.; Khan, A.Q.; Chawdhery, M.Z.; Haris, M. Sanguinarine mediated apoptosis in Non-Small Cell Lung Cancer via generation of reactive oxygen species and suppression of JAK/STAT pathway. Biomed. Pharmacother. 2021, 144, 112358. [Google Scholar] [CrossRef] [PubMed]
- Hseu, Y.-C.; Chiang, Y.-C.; Vudhya Gowrisankar, Y.; Lin, K.-Y.; Huang, S.-T.; Shrestha, S.; Chang, G.-R.; Yang, H.-L. The in vitro and in vivo anticancer properties of chalcone flavokawain b through induction of ros-mediated apoptotic and autophagic cell death in human melanoma cells. Cancers 2020, 12, 2936. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef]
- Rajendran, P.; Ben Ammar, R.; Al-Saeedi, F.J.; Elsayed Mohamed, M.; Islam, M.; Al-Ramadan, S.Y. Thidiazuron decreases epithelial-mesenchymal transition activity through the NF-kB and PI3K/AKT signalling pathways in breast cancer. J. Cell. Mol. Med. 2020, 24, 14525–14538. [Google Scholar] [CrossRef]
- Yang, H.-L.; Tsai, C.-H.; Shrestha, S.; Lee, C.-C.; Liao, J.-W.; Hseu, Y.-C. Coenzyme Q0, a novel quinone derivative of Antrodia camphorata, induces ROS-mediated cytotoxic autophagy and apoptosis against human glioblastoma cells in vitro and in vivo. Food Chem. Toxicol. 2021, 155, 112384. [Google Scholar] [CrossRef]
- Rajendran, P.; Ong, T.H.; Chen, L.; Li, F.; Shanmugam, M.K.; Vali, S.; Abbasi, T.; Kapoor, S.; Sharma, A.; Kumar, A.P. Suppression of signal transducer and activator of transcription 3 activation by butein inhibits growth of human hepatocellular carcinoma in vivo. Clin. Cancer Res. 2011, 17, 1425–1439. [Google Scholar] [CrossRef] [Green Version]
- Sikka, S.; Shanmugam, M.K.; Siveen, K.S.; Ong, T.H.; Yang, M.H.; Lee, J.H.; Rajendran, P.; Chinnathambi, A.; Alharbi, S.A.; Alahmadi, T.A. Diosgenin attenuates tumor growth and metastasis in transgenic prostate cancer mouse model by negatively regulating both NF-κB/STAT3 signaling cascades. Eur. J. Pharmacol. 2021, 906, 174274. [Google Scholar] [CrossRef]
- Rajendran, P.; Li, F.; Shanmugam, M.K.; Kannaiyan, R.; Goh, J.N.; Wong, K.F.; Wang, W.; Khin, E.; Tergaonkar, V.; Kumar, A.P. Celastrol suppresses growth and induces apoptosis of human hepatocellular carcinoma through the modulation of STAT3/JAK2 signaling cascade in vitro and in vivo. Cancer Prev. Res. 2012, 5, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Lu, N.-N.; Feng, L. Apigetrin inhibits gastric cancer progression through inducing apoptosis and regulating ROS-modulated STAT3/JAK2 pathway. Biochem. Biophys. Res. Commun. 2018, 498, 164–170. [Google Scholar] [CrossRef]
- Yang, M.H.; Lee, J.H.; Ko, J.-H.; Jung, S.H.; Sethi, G.; Ahn, K.S. Brassinin represses invasive potential of lung carcinoma cells through deactivation of PI3K/Akt/mTOR signaling cascade. Molecules 2019, 24, 1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, C.F.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019, 10, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opferman, J.T.; Kothari, A. Anti-apoptotic BCL-2 family members in development. Cell Death Differ. 2018, 25, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.H.; Baek, S.H.; Ha, I.J.; Um, J.Y.; Ahn, K.S. Brassinin enhances the anticancer actions of paclitaxel by targeting multiple signaling pathways in colorectal cancer cells. Phytother. Res. 2021, 35, 3875–3885. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, C.; Ding, Y.; Zhao, Q.; Yang, L.; Ling, J.; Liu, L.; Ji, H.; Zhang, Y. The activation of p38 and JNK by ROS, contribute to OLO-2-mediated intrinsic apoptosis in human hepatocellular carcinoma cells. Food Chem. Toxicol. 2014, 63, 38–47. [Google Scholar] [CrossRef]
- Tischlerova, V.; Kello, M.; Budovska, M.; Mojzis, J. Indole phytoalexin derivatives induce mitochondrial-mediated apoptosis in human colorectal carcinoma cells. World J. Gastroenterol. 2017, 23, 4341. [Google Scholar] [CrossRef]
- Mezencev, R.; Updegrove, T.; Kutschy, P.; Repovská, M.; McDonald, J.F. Camalexin induces apoptosis in T-leukemia Jurkat cells by increased concentration of reactive oxygen species and activation of caspase-8 and caspase-9. J. Nat. Med. 2011, 65, 488–499. [Google Scholar] [CrossRef]
- Halasi, M.; Wang, M.; Chavan, T.S.; Gaponenko, V.; Hay, N.; Gartel, A.L. ROS inhibitor N-acetyl-L-cysteine antagonizes the activity of proteasome inhibitors. Biochem. J. 2013, 454, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Siveen, K.S.; Sikka, S.; Surana, R.; Dai, X.; Zhang, J.; Kumar, A.P.; Tan, B.K.; Sethi, G.; Bishayee, A. Targeting the STAT3 signaling pathway in cancer: Role of synthetic and natural inhibitors. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2014, 1845, 136–154. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Li, J.; Liu, T.; Li, S.; Feng, J.; Yu, Q.; Zhang, J.; Chen, J.; Zhou, Y.; Ji, J. Quercetin shows anti-tumor effect in hepatocellular carcinoma LM3 cells by abrogating JAK2/STAT3 signaling pathway. Cancer Med. 2019, 8, 4806–4820. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, M.; Pathak, A.K.; Nair, A.S.; Kunnumakkara, A.B.; Guha, S.; Sethi, G.; Aggarwal, B.B. Capsaicin is a novel blocker of constitutive and interleukin-6–inducible STAT3 activation. Clin. Cancer Res. 2007, 13, 3024–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seitz, S.J.; Schleithoff, E.S.; Koch, A.; Schuster, A.; Teufel, A.; Staib, F.; Stremmel, W.; Melino, G.; Krammer, P.H.; Schilling, T. Chemotherapy-induced apoptosis in hepatocellular carcinoma involves the p53 family and is mediated via the extrinsic and the intrinsic pathway. Int. J. Cancer 2010, 126, 2049–2066. [Google Scholar] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajendran, P.; Elsawy, H.; Alfwuaires, M.; Sedky, A. Brassinin Enhances Apoptosis in Hepatic Carcinoma by Inducing Reactive Oxygen Species Production and Suppressing the JAK2/STAT3 Pathway. Appl. Sci. 2022, 12, 4733. https://doi.org/10.3390/app12094733
Rajendran P, Elsawy H, Alfwuaires M, Sedky A. Brassinin Enhances Apoptosis in Hepatic Carcinoma by Inducing Reactive Oxygen Species Production and Suppressing the JAK2/STAT3 Pathway. Applied Sciences. 2022; 12(9):4733. https://doi.org/10.3390/app12094733
Chicago/Turabian StyleRajendran, Peramaiyan, Hany Elsawy, Manal Alfwuaires, and Azza Sedky. 2022. "Brassinin Enhances Apoptosis in Hepatic Carcinoma by Inducing Reactive Oxygen Species Production and Suppressing the JAK2/STAT3 Pathway" Applied Sciences 12, no. 9: 4733. https://doi.org/10.3390/app12094733