
Molecular Dynamics of Atomic Layer Deposition: Sticking Coefficient Investigation

 ,
,  ,
, 
Abstract
:1. Introduction
2. Computational Details
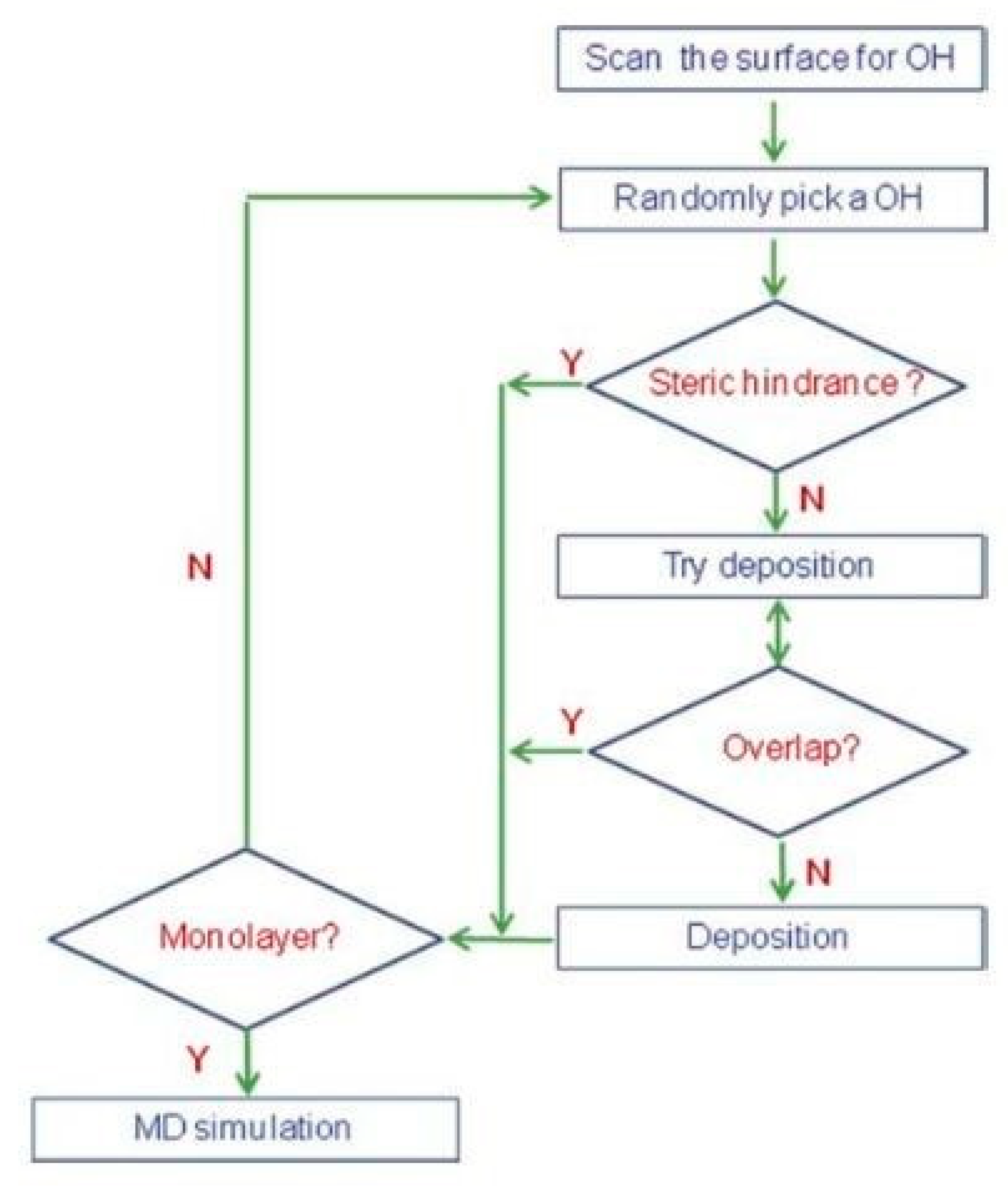
2.1. The Simulation Approach
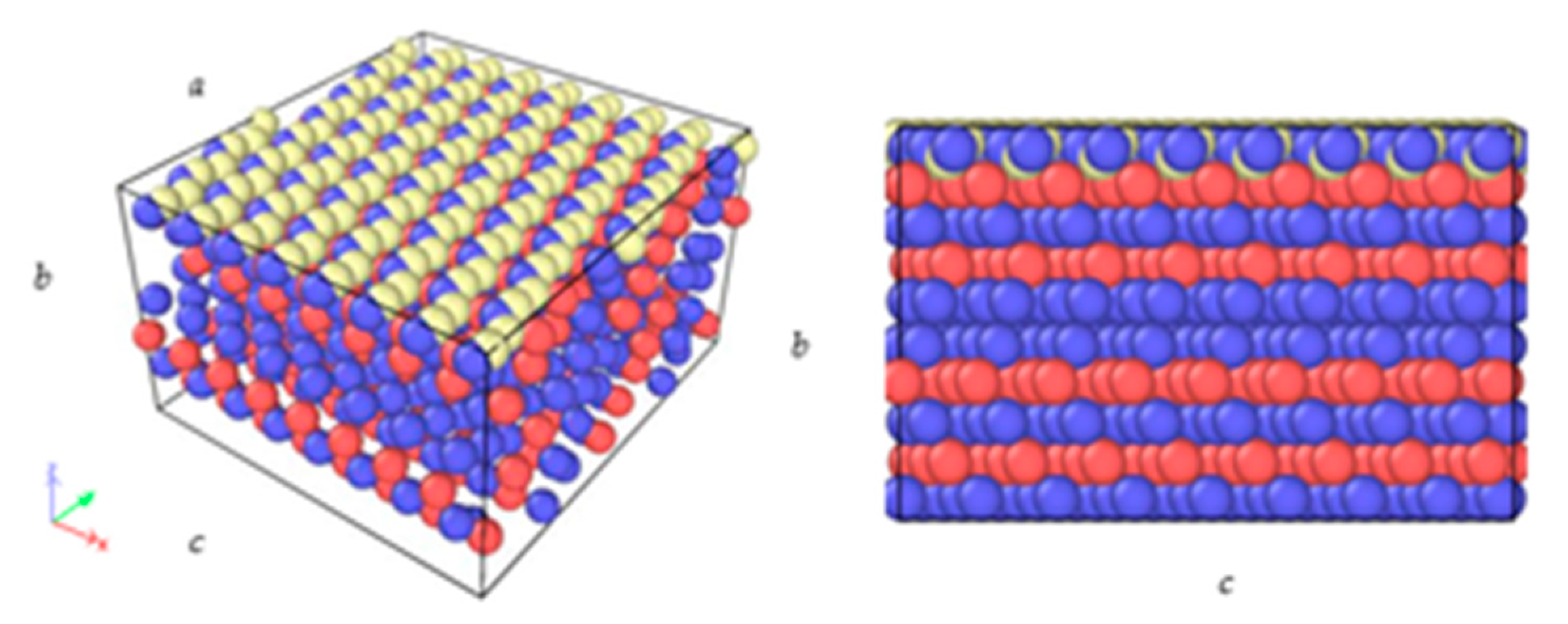
2.2. Substrate Preparation
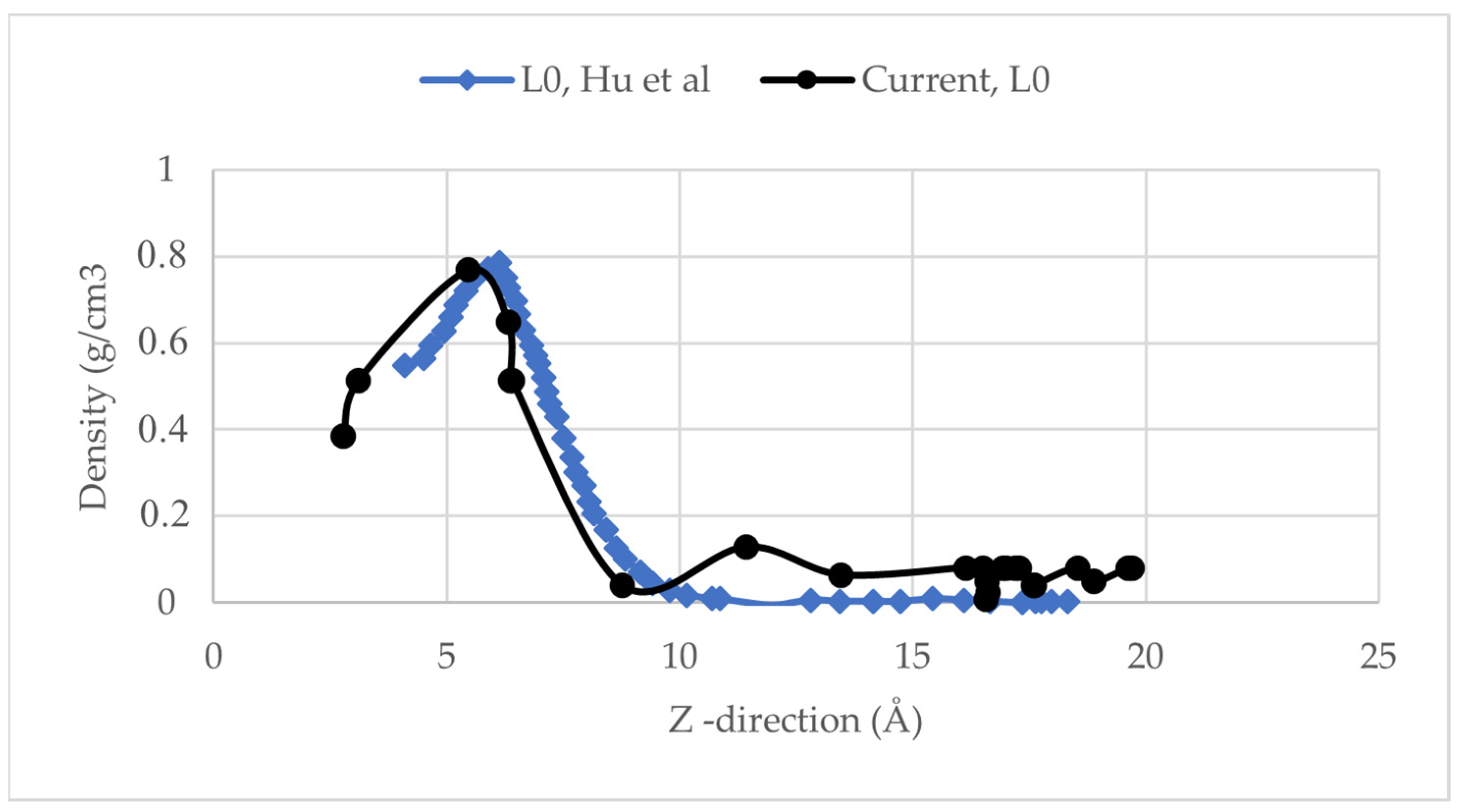
2.3. Model Validation
3. Results
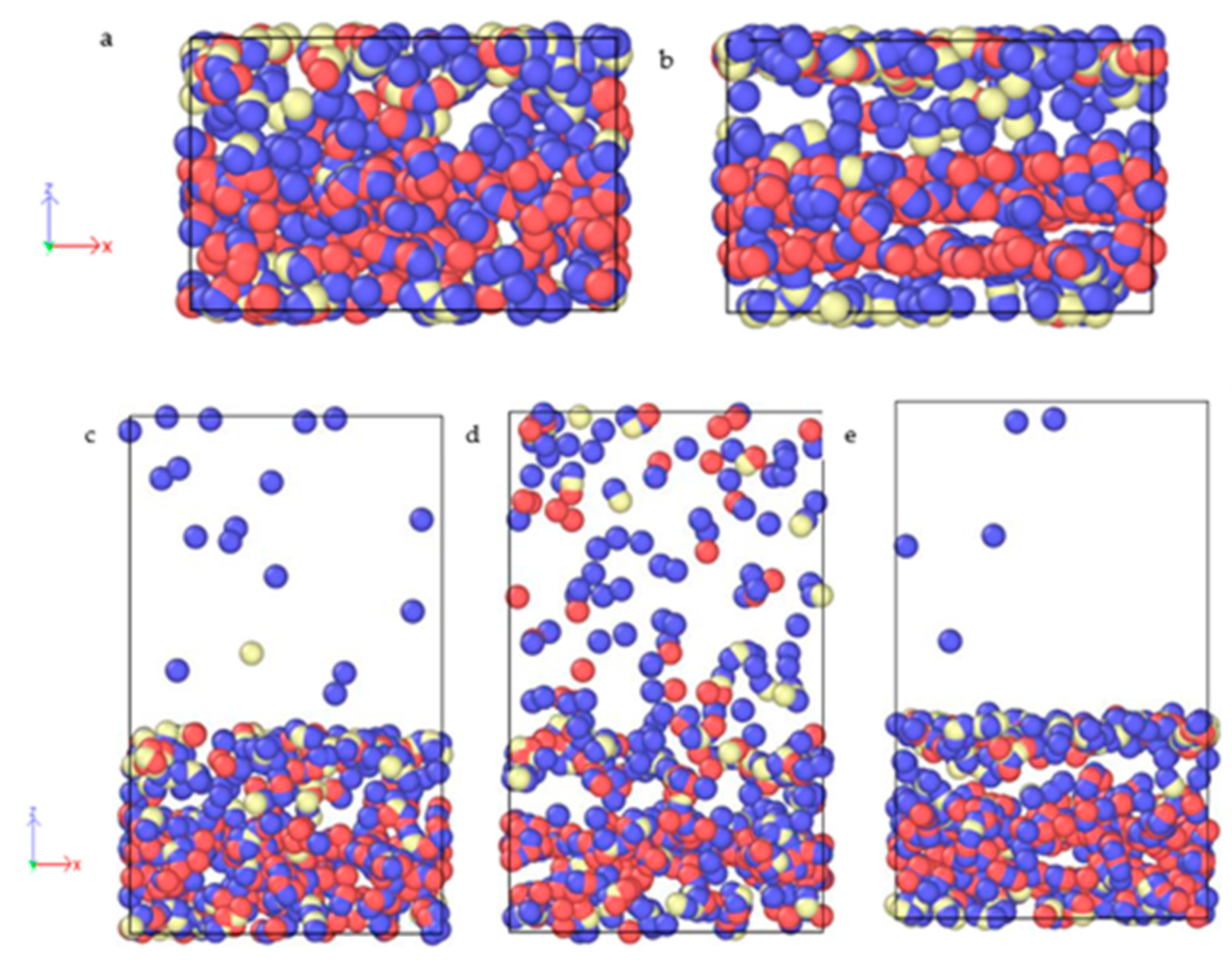
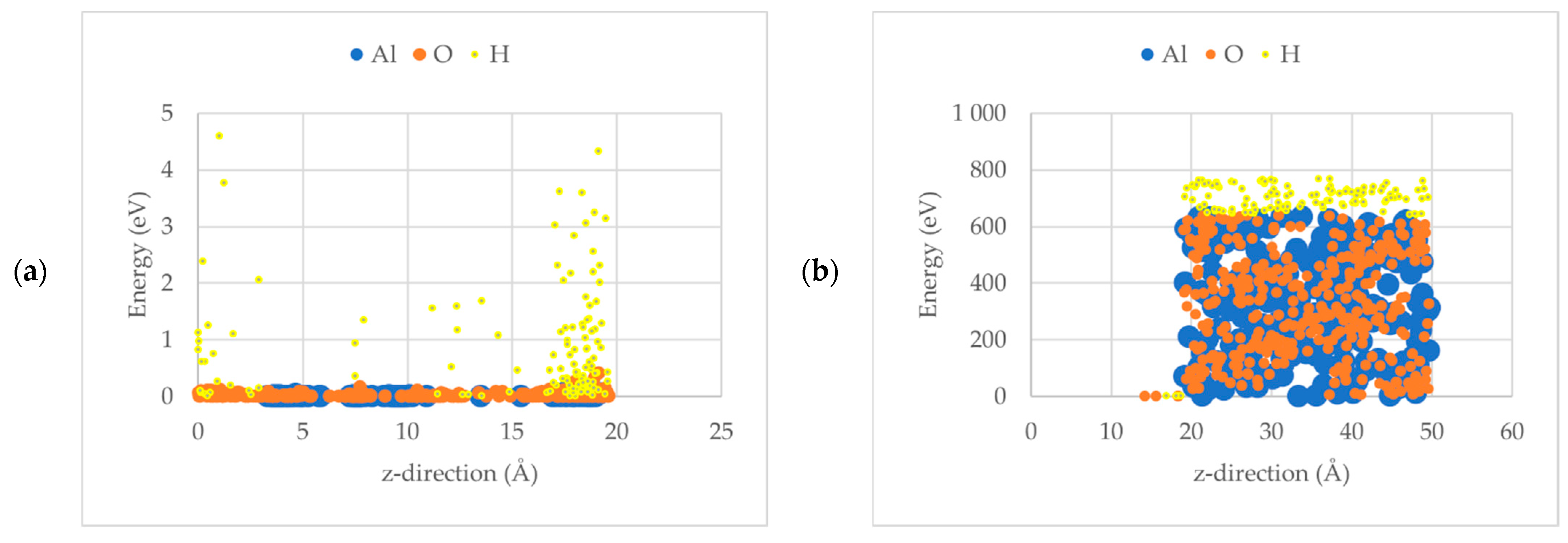
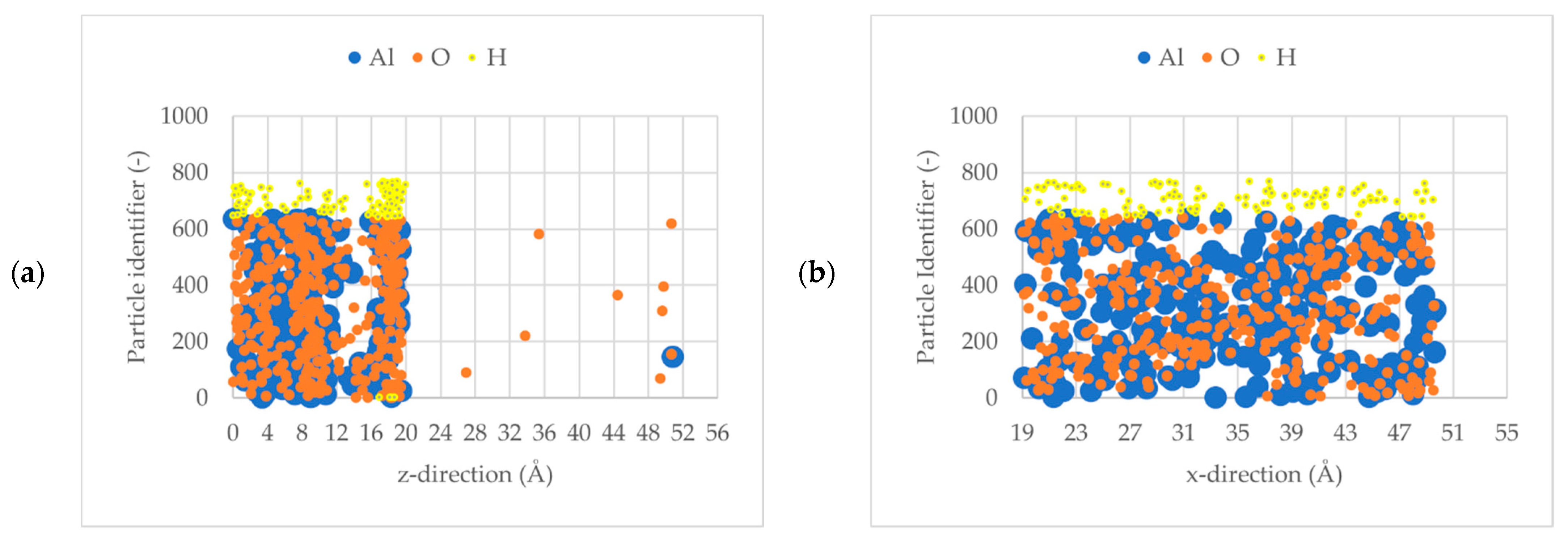
3.1. Al2O3 Film Thickness
3.2. Initial Sticking Coefficient, So
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- George, S.M. Atomic Layer Deposition: An Overview. Chem. Rev. 2010, 110, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Kanarik, K.J.; Lill, T.; Hudson, E.A.; Sriraman, S.; Tan, S.; Marks, J.; Vahedi, V.; Gottscho, R.A. Overview of atomic layer etching in the semiconductor industry. J. Vac. Sci. Technol. A Vac. Surf. Film. 2015, 33, 020802. [Google Scholar] [CrossRef] [Green Version]
- Hagen, D.J.; Pemble, M.E.; Karppinen, M. Atomic layer deposition of metals: Precursors and film growth. Appl. Phys. Rev. 2019, 6, 041309. [Google Scholar] [CrossRef]
- Zhang, Y.; Ding, Y.; Christofides, P.D. Multiscale computational fluid dynamics modeling and reactor design of plasma-enhanced atomic layer deposition. Comput. Chem. Eng. 2020, 142, 107066. [Google Scholar] [CrossRef]
- Dominik Mark, J.H. Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods, 1st ed.; Cambridge University Press: New York, NY, USA, 2009. [Google Scholar]
- Masui, H. Simulation of texture formation of body-centered-cubic metals by three kinds of intersections of two {110} slip planes. IOP Conf. Ser. Mater. Sci. Eng. 2015, 82, 012010. [Google Scholar] [CrossRef] [Green Version]
- Kamberaj, H. Molecular Dynamics Simulations in Statistical Physics: Theory and Applications; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar] [CrossRef]
- Hu, Z.; Shi, J.; Turner, C.H. Molecular Simulation Molecular dynamics simulation of the Al2O3 film structure during atomic layer deposition Molecular dynamics simulation of the Al2O3 film structure during atomic layer deposition. Mol. Simul. 2009, 35, 270–279. [Google Scholar] [CrossRef]
- Brown, K.S.; Saggese, C.; Le Monnier, B.P.; Héroguel, F.; Luterbacher, J.S. Simulation of Gas- and Liquid-Phase Layer-By-Layer Deposition of Metal Oxides by Coarse-Grained Modeling. J. Phys. Chem. C 2018, 122, 6713–6720. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Musgrave, C.B. Atomic layer deposition of high- κ dielectrics on nitrided silicon surfaces. Appl. Phys. Lett. 2005, 86, 1–3. [Google Scholar] [CrossRef]
- Rose, M.; Bartha, J.W. Applied Surface Science Method to determine the sticking coefficient of precursor molecules in atomic layer deposition. Appl. Surf. Sci. 2009, 255, 6620–6623. [Google Scholar] [CrossRef]
- Langmuir, I. Nobel Lecture: Surface Chemistry. Nobelprize.org Nobel Media AB 2014. Available online: http://www.nobelprize.org/nobel_prizes/chemistry/laureates/1932/langmuir-lecture.html (accessed on 15 October 2020).
- Rose, M.; Bartha, J.W.; Endler, I. Temperature dependence of the sticking coefficient in atomic layer deposition. Appl. Surf. Sci. 2010, 256, 3778–3782. [Google Scholar] [CrossRef]
- Schwille, M.C.; Schössler, T.; Schön, F.; Oettel, M.; Bartha, J.W. Temperature dependence of the sticking coefficients of bis-diethyl aminosilane and trimethylaluminum in atomic layer deposition. J. Vac. Sci. Technol. A Vac. Surf. Film. 2017, 35, 01B119. [Google Scholar] [CrossRef]
- Harra, D.J. Review of Sticking Coefficients and Sorption Capacities of Gases on Titanium Films. J. Vac. Sci. Technol. 1975, 13, 471–475. [Google Scholar] [CrossRef]
- Schwille, M.C.; Schössler, T.; Barth, J.; Knaut, M.; Schön, F.; Höchst, A.; Oettel, M.; Bartha, J.W. Experimental and simulation approach for process optimization of atomic layer deposited thin films in high aspect ratio 3D structures. J. Vac. Sci. Technol. A Vac. Surf. Film. 2017, 35, 01B118. [Google Scholar] [CrossRef]
- Arts, K.; Vandalon, V.; Puurunen, R.L.; Utriainen, M.; Gao, F.; Kessels, W.M.; Knoops, H.C. Sticking probabilities of H2O and Al(CH3)3 during atomic layer deposition of Al2O3 extracted from their impact on film conformality. J. Vac. Sci. Technol. A 2019, 37, 030908. [Google Scholar] [CrossRef]
- Träskelin, P.; Salonen, E.; Nordlund, K.; Krasheninnikov, A.V.; Keinonen, J.; Wu, C.H. Molecular dynamics simulations of CH3 sticking on carbon surfaces. J. Appl. Phys. 2003, 93, 1826–1831. [Google Scholar] [CrossRef] [Green Version]
- Argyris, D.; Tummala, N.R.; Striolo, A.; Cole, D.R. Molecular Structure and Dynamics in Thin Water Films at the Silica and Graphite Surfaces. J. Phys. Chem. C 2008, 112, 13587–13599. [Google Scholar] [CrossRef]
- Argyris, D.; Cole, D.R.; Striolo, A. Dynamic Behavior of Interfacial Water at the Silica Surface. J. Phys. Chem. C 2009, 113, 19591–19600. [Google Scholar] [CrossRef]
- Cremers, V.; Puurunen, R.L.; Dendooven, J. Conformality in atomic layer deposition: Current status overview of analysis and modelling. Appl. Phys. Rev. 2019, 6, 021302. [Google Scholar] [CrossRef] [Green Version]
- Matsui, M. A Transferable Interatomic Potential Model for Crystals and Melts in the System CaO-MgO-Al2O3-SiO2. Mineral. Mag. 1994, 58, 571–572. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short- Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–42. [Google Scholar] [CrossRef] [Green Version]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO-the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tuckerman, M.E.; Tobias, D.J.; Klein, M.L. Explicit reversible integrators for extended systems dynamics. Mol. Phys. 1996, 87, 1117–1157. [Google Scholar] [CrossRef]
- Arnold, C.; Nilsson, E.; Bengtsson, A. Establishing simulations of shockwaves in InSb using molecular dynamics. Lund Rep. At. Phys. (LRAP) 2021, 571–585. [Google Scholar]
- Adiga, S.P.; Zapol, P.; Curtiss, L.A. Atomistic simulations of amorphous alumina surfaces. Phys. Rev. B 2006, 74, 064204. [Google Scholar] [CrossRef]
- Tichmann, K.; Von Toussaint, U.; Jacob, W. Determination of the sticking coefficient of energetic hydrocarbon molecules by molecular dynamics. J. Nucl. Mater. 2012, 420, 291–296. [Google Scholar] [CrossRef] [Green Version]
- Ukoba, O.K.; Jen, T.C. Review of Atomic Layer Deposition of Nanostructured Solar Cells 4. J. Phys. Conf. Ser. 2019, 1378, 042060. [Google Scholar] [CrossRef]
- Stenstr, S. A model-based methodology for the analysis and design of atomic layer deposition processes—Part II: Experimental validation and mechanistic analysis. Chem. Eng. Sci. 2013, 94, 316–329. [Google Scholar] [CrossRef]
- Liang, T.; Shan, T.R.; Cheng, Y.T.; Devine, B.D.; Noordhoek, M.; Li, Y.; Lu, Z.; Phillpot, S.R.; Sinnott, S.B. Classical atomistic simulations of surfaces and heterogeneous interfaces with the charge-optimized many body (COMB) potentials. Mater. Sci. Eng. R Rep. 2013, 74, 255–279. [Google Scholar] [CrossRef]
- Pal, S.; Snehanshu Ray, B.C. Molecular Dynamics Simulation of Nanostructured Materials: An Understanding of Mechanical Behavior. 2016, Volume 1107. Available online: https://www.routledge.com/Molecular-Dynamics-Simulation-of-Nanostructured-Materials-An-Understanding/Pal-Ray/p/book/9780367029821 (accessed on 15 October 2020).
- Dwivedi, V.; Adomaitis, R.A. Multiscale modeling of atomic layer deposition processes. Proc. Am. Control Conf. 2009, 2495–2500. [Google Scholar] [CrossRef]
- Richey, N.E.; De Paula, C.; Bent, S.F. Understanding chemical and physical mechanisms in atomic layer deposition. J. Chem. Phys. 2020, 152, 40902. [Google Scholar] [CrossRef]
- Ylilammi, M.; Ylivaara, O.M.E.; Puurunen, R.L. Modeling growth kinetics of thin films made by atomic layer deposition in lateral high-aspect-ratio structures. J. Appl. Phys. 2018, 123, 205301. [Google Scholar] [CrossRef] [Green Version]
- Yim, J.; Ylivaara, O.M.; Ylilammi, M.; Korpelainen, V.; Haimi, E.; Verkama, E.; Utriainen, M.; Puurunen, R.L. Saturation Profile Based Conformality Analysis for Atomic Layer Deposition: Aluminum Oxide in Lateral High-Aspect-Ratio Channels. Phys. Chem. Chem. Phys. 2020, 22, 23107–23120. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | ||||
|---|---|---|---|---|
| Al | 1.4175 | 0.78520 | 0.03400 | 36.82 |
| O | −0.9450 | 1.82150 | 0.13800 | 90.61 |
| H | 0.4725 | −0.17607 | −0.02462 | 1.86285 |
| Method | Researchers | S0 |
|---|---|---|
| Diffusion model, thickness profiles, lateral high-aspect-ratio structure (LHAR) | Ylilammi et al., Ref. [35] | 5.27 × 10−3 |
| 1D structures, thickness profiles, lateral high-aspect-ratio structure (LHAR) | Arts et al., Ref. [17] | (2−7) × 10−3 |
| LHAR, thickness profiles, 3D structures, experiments and Monte Carlo simulations | Schwille et al., Ref. [14] | 2 × 10−2 |
| LHAR, thickness profiles, 3D structures | Yim et al., Ref. [36] | 4 × 10−3 |
| Molecular Dynamics (All-atom approach), thickness profiles, as Arts et al. Ref (Arts et al., 2019) | Current study | 4.257 × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunene, T.J.; Tartibu, L.K.; Karimzadeh, S.; Oviroh, P.O.; Ukoba, K.; Jen, T.-C. Molecular Dynamics of Atomic Layer Deposition: Sticking Coefficient Investigation. Appl. Sci. 2022, 12, 2188. https://doi.org/10.3390/app12042188
Kunene TJ, Tartibu LK, Karimzadeh S, Oviroh PO, Ukoba K, Jen T-C. Molecular Dynamics of Atomic Layer Deposition: Sticking Coefficient Investigation. Applied Sciences. 2022; 12(4):2188. https://doi.org/10.3390/app12042188
Chicago/Turabian StyleKunene, Thokozane Justin, Lagouge Kwanda Tartibu, Sina Karimzadeh, Peter Ozaveshe Oviroh, Kingsley Ukoba, and Tien-Chien Jen. 2022. "Molecular Dynamics of Atomic Layer Deposition: Sticking Coefficient Investigation" Applied Sciences 12, no. 4: 2188. https://doi.org/10.3390/app12042188