
Atomization of Microfibrillated Cellulose and Its Incorporation into Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) by Reactive Extrusion

 and
and 
Abstract
:Featured Application
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Atomization of Cellulose Suspension
2.3. Preparation of Green Composite Films
2.3.1. Melt Mixing
2.3.2. Thermo-Compression
2.4. Material Characterization
2.4.1. Microscopy
2.4.2. Infrared Spectroscopy
2.4.3. Optical Evaluation
2.4.4. Thermal Analysis
2.4.5. Mechanical Characterization
2.4.6. Barrier Measurements
2.5. Statistical Analysis
3. Results and Discussion
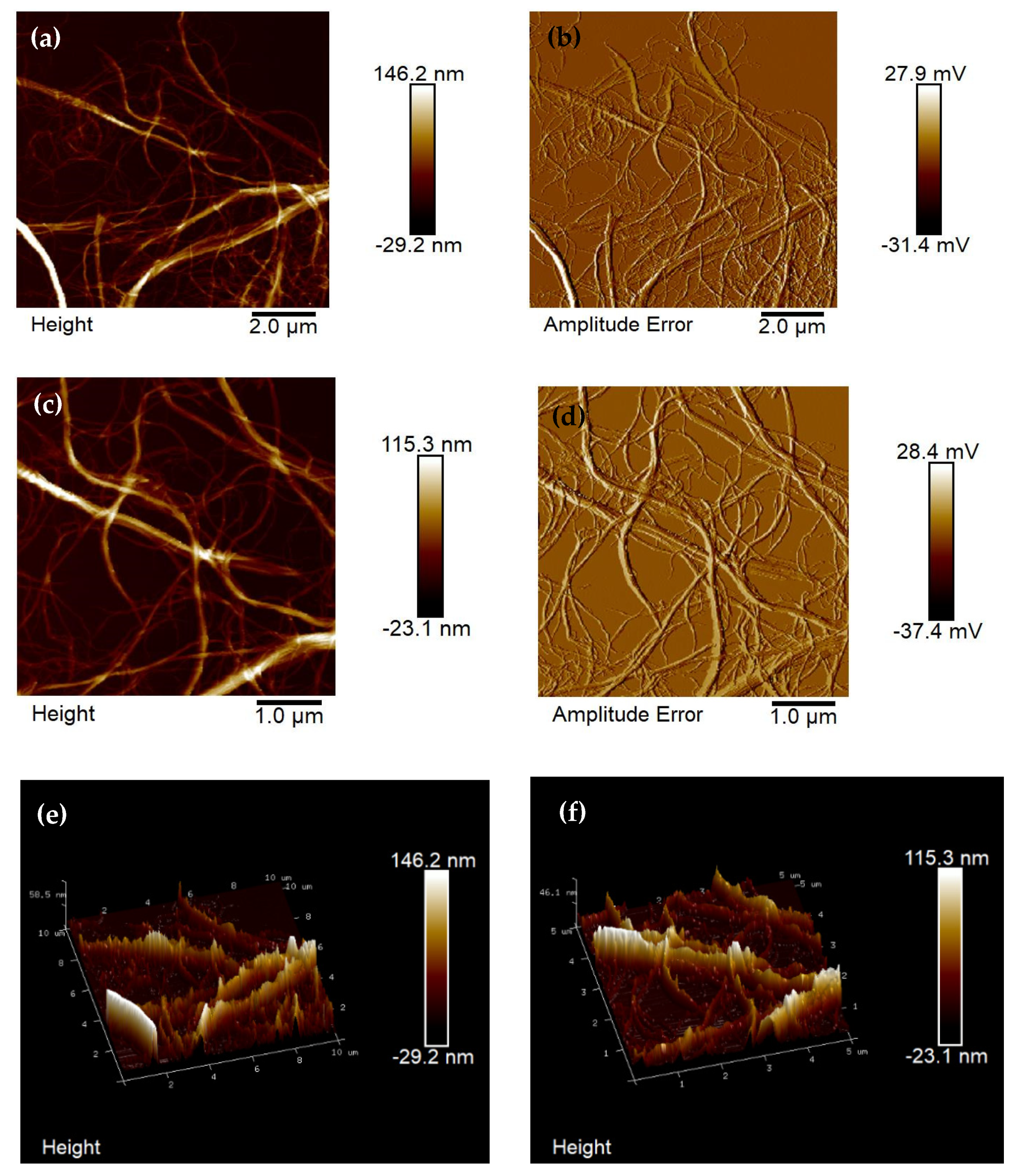
3.1. Atomization of Cellulose Suspension
3.2. Melt Processing of Green Composites
3.3. Chemical Analysis of Green Composites
3.4. Optical Properties of Green Composites
3.5. Morphological Properties of Green Composites
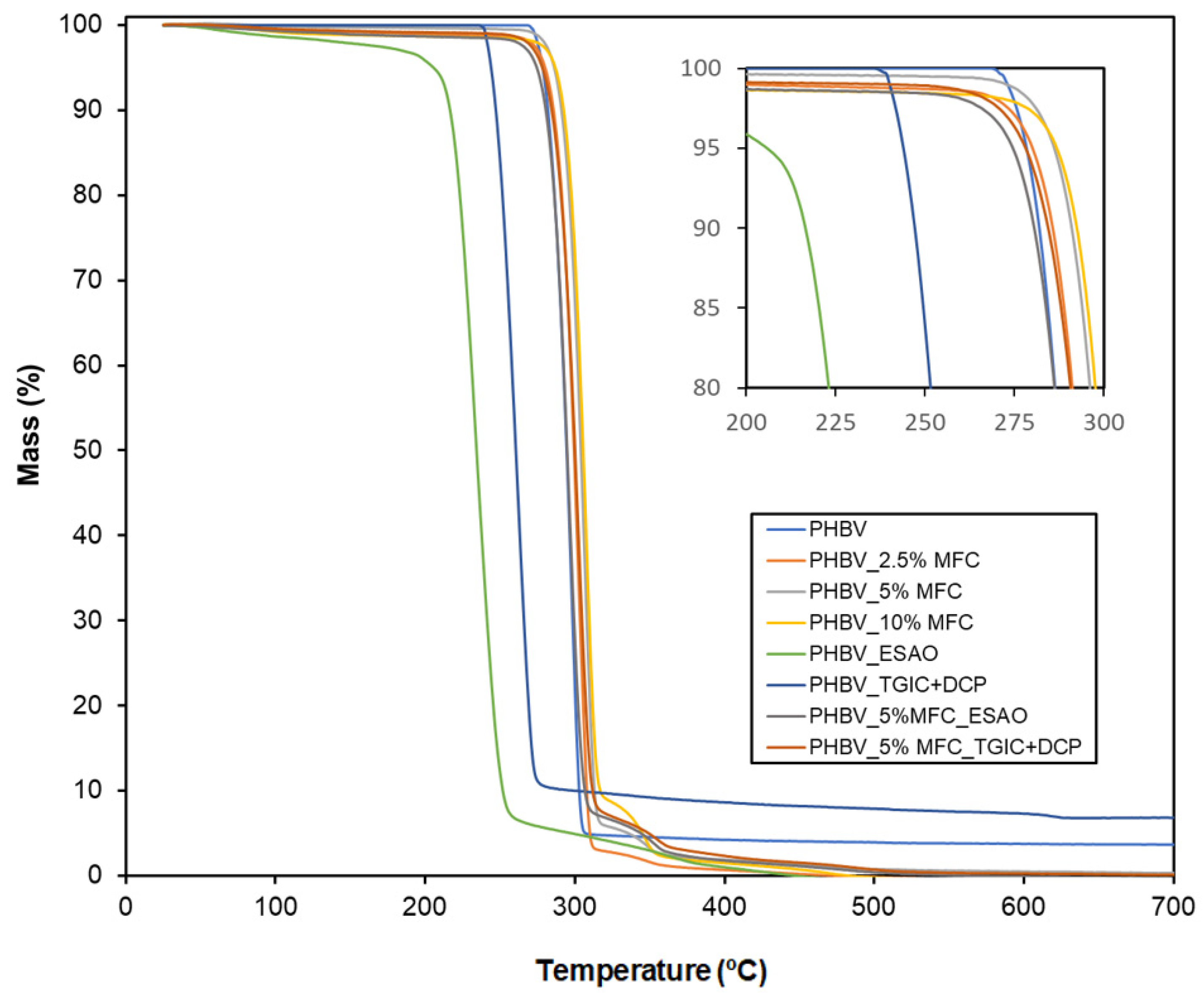
3.6. Thermal Properties of Green Composites
3.7. Mechanical Properties of Green Composites
3.8. Barrier Properties of Green Composites
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Geueke, B.; Groh, K.; Muncke, J. Food packaging in the circular economy: Overview of chemical safety aspects for commonly used materials. J. Clean. Prod. 2018, 193, 491–505. [Google Scholar] [CrossRef]
- Hottle, T.A.; Bilec, M.M.; Landis, A.E. Biopolymer production and end of life comparisons using life cycle assessment. Resour. Conserv. Recycl. 2017, 122, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Torres-Giner, S.; Figueroa-Lopez, K.J.; Melendez-Rodriguez, B.; Prieto, C.; Pardo-Figuerez, M.; Lagaron, J.M. Emerging Trends in Biopolymers for Food Packaging. In Sustainable Food Packaging Technology; Wiley: Hoboken, NJ, USA, 2021; pp. 1–33. [Google Scholar]
- Durkin, A.; Taptygin, I.; Kong, Q.; Gunam Resul, M.F.M.; Rehman, A.; Fernández, A.M.L.; Harvey, A.P.; Shah, N.; Guo, M. Scale-up and Sustainability Evaluation of Biopolymer Production from Citrus Waste Offering Carbon Capture and Utilisation Pathway. ChemistryOpen 2019, 8, 668–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini, S.N.; Pirsa, S.; Farzi, J. Biodegradable nano composite film based on modified starch-albumin/MgO; antibacterial, antioxidant and structural properties. Polym. Test. 2021, 97, 107182. [Google Scholar] [CrossRef]
- Sharifi, K.A.; Pirsa, S. Biodegradable film of black mulberry pulp pectin/chlorophyll of black mulberry leaf encapsulated with carboxymethylcellulose/silica nanoparticles: Investigation of physicochemical and antimicrobial properties. Mater. Chem. Phys. 2021, 267, 124580. [Google Scholar] [CrossRef]
- Mohammadi, B.; Pirsa, S.; Alizadeh, M. Preparing chitosan–polyaniline nanocomposite film and examining its mechanical, electrical, and antimicrobial properties. Polym. Polym. Compos. 2019, 27, 507–517. [Google Scholar] [CrossRef]
- Pirsa, S.; Mohammadi, B. Conducting/biodegradable chitosan-polyaniline film; Antioxidant, color, solubility and water vapor permeability properties. Main Group Chem. 2021, 20, 133–147. [Google Scholar] [CrossRef]
- Pirsa, S.; Farshchi, E.; Roufegarinejad, L. Antioxidant/Antimicrobial Film Based on Carboxymethyl Cellulose/Gelatin/TiO2–Ag Nano-Composite. J. Polym. Environ. 2020, 28, 3154–3163. [Google Scholar] [CrossRef]
- Pirsa, S.; Chavoshizadeh, S. Design of an optical sensor for ethylene based on nanofiber bacterial cellulose film and its application for determination of banana storage time. Polym. Adv. Technol. 2018, 29, 1385–1393. [Google Scholar] [CrossRef]
- Pirsa, S. Nanocomposite base on carboxymethylcellulose hydrogel: Simultaneous absorbent of ethylene and humidity to increase the shelf life of banana fruit. Int. J. Biol. Macromol. 2021, 193, 300–310. [Google Scholar] [CrossRef]
- Yorghanlu, R.A.; Hemmati, H.; Pirsa, S.; Makhani, A. Production of biodegradable sodium caseinate film containing titanium oxide nanoparticles and grape seed essence and investigation of physicochemical properties. Polym. Bull. 2021. [Google Scholar] [CrossRef]
- Jabraili, A.; Pirsa, S.; Pirouzifard, M.K.; Amiri, S. Biodegradable Nanocomposite Film Based on Gluten/Silica/Calcium Chloride: Physicochemical Properties and Bioactive Compounds Extraction Capacity. J. Polym. Environ. 2021, 29, 2557–2571. [Google Scholar] [CrossRef]
- Rezaei, M.; Pirsa, S.; Chavoshizadeh, S. Photocatalytic/Antimicrobial Active Film Based on Wheat Gluten/ZnO Nanoparticles. J. Inorg. Organomet. Polym. Mater. 2019, 30, 2654–2665. [Google Scholar] [CrossRef]
- Arcos-Hernández, M.V.; Laycock, B.; Donose, B.C.; Pratt, S.; Halley, P.; Al-Luaibi, S.; Werker, A.; Lant, P.A. Physicochemical and mechanical properties of mixed culture polyhydroxyalkanoate (PHBV). Eur. Polym. J. 2013, 49, 904–913. [Google Scholar] [CrossRef]
- Thushari, G.G.N.; Senevirathna, J.D.M. Plastic pollution in the marine environment. Heliyon 2020, 6, e04709. [Google Scholar] [CrossRef]
- Bucci, D.Z.; Tavares, L.B.B.; Sell, I. Biodegradation and physical evaluation of PHB packaging. Polym. Test. 2007, 26, 908–915. [Google Scholar] [CrossRef]
- Torres-Giner, S.; Montanes, N.; Fombuena, V.; Boronat, T.; Sanchez-Nacher, L. Preparation and characterization of compression-molded green composite sheets made of poly(3-hydroxybutyrate) reinforced with long pita fibers. Adv. Polym. Technol. 2018, 37, 1305–1315. [Google Scholar] [CrossRef] [Green Version]
- Ray, S.S.; Bousmina, M. Biodegradable polymers and their layered silicate nanocomposites: In greening the 21st century materials world. Prog. Mater. Sci. 2005, 50, 962–1079. [Google Scholar]
- Keshavarz, T.; Roy, I. Polyhydroxyalkanoates: Bioplastics with a green agenda. Curr. Opin. Microbiol. 2010, 13, 321–326. [Google Scholar] [CrossRef]
- Laycock, B.; Arcos-Hernandez, M.V.; Langford, A.; Buchanan, J.; Halley, P.J.; Werker, A.; Lant, P.A.; Pratt, S. Thermal properties and crystallization behavior of fractionated blocky and random polyhydroxyalkanoate copolymers from mixed microbial cultures. J. Appl. Polym. Sci. 2014, 131, 40836. [Google Scholar] [CrossRef]
- Tajeddin, B. Cellulose-Based Polymers for Packaging Applications. In Lignocellulosic Polymer Composites; Wiley: Hoboken, NJ, USA, 2014; pp. 477–498. [Google Scholar]
- Henriksson, M.; Henriksson, G.; Berglund, L.A.; Lindström, T. An environmentally friendly method for enzyme-assisted preparation of microfibrillated cellulose (MFC) nanofibers. Eur. Polym. J. 2007, 43, 3434–3441. [Google Scholar] [CrossRef]
- Fortunati, E.; Armentano, I.; Zhou, Q.; Iannoni, A.; Saino, E.; Visai, L.; Berglund, L.A.; Kenny, J.M. Multifunctional bionanocomposite films of poly(lactic acid), cellulose nanocrystals and silver nanoparticles. Carbohydr. Polym. 2012, 87, 1596–1605. [Google Scholar] [CrossRef]
- Gatenholm, P.; Klemm, D. Bacterial Nanocellulose as a Renewable Material for Biomedical Applications. MRS Bull. 2010, 35, 208–213. [Google Scholar] [CrossRef]
- Suzuki, K.; Sato, A.; Okumura, H.; Hashimoto, T.; Nakagaito, A.N.; Yano, H. Novel high-strength, micro fibrillated cellulose-reinforced polypropylene composites using a cationic polymer as compatibilizer. Cellulose 2014, 21, 507–518. [Google Scholar] [CrossRef]
- Miao, C.; Hamad, W.Y. Cellulose reinforced polymer composites and nanocomposites: A critical review. Cellulose 2013, 20, 2221–2262. [Google Scholar] [CrossRef]
- Venkatraman, P.; Gohn, A.M.; Rhoades, A.M.; Foster, E.J. Developing high performance PA11/cellulose nanocomposites for industrial-scale melt processing. Compos. Part B Eng. 2019, 174, 106988. [Google Scholar] [CrossRef]
- Oksman, K.; Aitomäki, Y.; Mathew, A.P.; Siqueira, G.; Zhou, Q.; Butylina, S.; Tanpichai, S.; Zhou, X.; Hooshmand, S. Review of the recent developments in cellulose nanocomposite processing. Compos. Part A Appl. Sci. Manuf. 2016, 83, 2–18. [Google Scholar] [CrossRef] [Green Version]
- Fonseca-Valero, C.; Ochoa-Mendoza, A.; Arranz-Andrés, J.; González-Sánchez, C. Mechanical recycling and composition effects on the properties and structure of hardwood cellulose-reinforced high density polyethylene eco-composites. Compos. Part A Appl. Sci. Manuf. 2015, 69, 94–104. [Google Scholar] [CrossRef]
- Agüero, Á.; Garcia-Sanoguera, D.; Lascano, D.; Rojas-Lema, S.; Ivorra-Martinez, J.; Fenollar, O.; Torres-Giner, S. Evaluation of Different Compatibilization Strategies to Improve the Performance of Injection-Molded Green Composite Pieces Made of Polylactide Reinforced with Short Flaxseed Fibers. Polymers 2020, 12, 821. [Google Scholar] [CrossRef] [Green Version]
- Pothan, L.A.; Thomas, S. Polarity parameters and dynamic mechanical behaviour of chemically modified banana fiber reinforced polyester composites. Compos. Sci. Technol. 2003, 63, 1231–1240. [Google Scholar] [CrossRef]
- Bengtsson, M.; Gatenholm, P.; Oksman, K. The effect of crosslinking on the properties of polyethylene/wood flour composites. Compos. Sci. Technol. 2005, 65, 1468–1479. [Google Scholar] [CrossRef]
- Torres-Giner, S.; Montanes, N.; Boronat, T.; Quiles-Carrillo, L.; Balart, R. Melt grafting of sepiolite nanoclay onto poly(3-hydroxybutyrate-co-4-hydroxybutyrate) by reactive extrusion with multi-functional epoxy-based styrene-acrylic oligomer. Eur. Polym. J. 2016, 84, 693–707. [Google Scholar] [CrossRef]
- Melendez-Rodriguez, B.; Torres-Giner, S.; Aldureid, A.; Cabedo, L.; Lagaron, J.M. Reactive Melt Mixing of Poly(3-Hydroxybutyrate)/Rice Husk Flour Composites with Purified Biosustainably Produced Poly(3-Hydroxybutyrate-co-3-Hydroxyvalerate). Materials 2019, 12, 2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agüero, A.; Morcillo, M.d.C.; Quiles-Carrillo, L.; Balart, R.; Boronat, T.; Lascano, D.; Torres-Giner, S.; Fenollar, O. Study of the Influence of the Reprocessing Cycles on the Final Properties of Polylactide Pieces Obtained by Injection Molding. Polymers 2019, 11, 1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Giner, S.; Hilliou, L.; Melendez-Rodriguez, B.; Figueroa-Lopez, K.J.; Madalena, D.; Cabedo, L.; Covas, J.A.; Vicente, A.A.; Lagaron, J.M. Melt processability, characterization, and antibacterial activity of compression-molded green composite sheets made of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) reinforced with coconut fibers impregnated with oregano essential oil. Food Packag. Shelf Life 2018, 17, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Shariatnia, S.; Veldanda, A.; Obeidat, S.; Jarrahbashi, D.; Asadi, A. Atomization of cellulose nanocrystals aqueous suspensions in fused deposition modeling: A scalable technique to improve the strength of 3D printed polymers. Compos. Part B Eng. 2019, 177, 107291. [Google Scholar] [CrossRef]
- Larsson, M.; Markbo, O.; Jannasch, P. Melt processability and thermomechanical properties of blends based on polyhydroxyalkanoates and poly(butylene adipate-co-terephthalate). RSC Adv. 2016, 6, 44354–44363. [Google Scholar] [CrossRef] [Green Version]
- Torres-Giner, S. Preparation of conductive carbon black-filled polymer nanocomposites via melt compounding. In Conductive Materials and Composites; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2016; pp. 117–164. [Google Scholar]
- Quiles-Carrillo, L.; Montanes, N.; Lagaron, J.M.; Balart, R.; Torres-Giner, S. In Situ Compatibilization of Biopolymer Ternary Blends by Reactive Extrusion with Low-Functionality Epoxy-Based Styrene–Acrylic Oligomer. J. Polym. Environ. 2019, 27, 84–96. [Google Scholar] [CrossRef]
- Quiles-Carrillo, L.; Duart, S.; Montanes, N.; Torres-Giner, S.; Balart, R. Enhancement of the mechanical and thermal properties of injection-molded polylactide parts by the addition of acrylated epoxidized soybean oil. Mater. Des. 2018, 140, 54–63. [Google Scholar] [CrossRef]
- Melendez-Rodriguez, B.; Castro-Mayorga, J.L.; Reis, M.A.M.; Sammon, C.; Cabedo, L.; Torres-Giner, S.; Lagaron, J.M. Preparation and Characterization of Electrospun Food Biopackaging Films of Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) Derived From Fruit Pulp Biowaste. Front. Sustain. Food Syst. 2018, 2, 38. [Google Scholar] [CrossRef]
- Rojas-Lema, S.; Arevalo, J.; Gomez-Caturla, J.; Garcia-Garcia, D.; Torres-Giner, S. Peroxide-Induced Synthesis of Maleic Anhydride-Grafted Poly(butylene succinate) and Its Compatibilizing Effect on Poly(butylene succinate)/Pistachio Shell Flour Composites. Molecules 2021, 26, 5927. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Mekonnen, T.; Misra, M.; Mohanty, A.K. Novel Biodegradable Cast Film from Carbon Dioxide Based Copolymer and Poly(Lactic Acid). J. Polym. Environ. 2016, 24, 23–36. [Google Scholar] [CrossRef] [Green Version]
- Invernizzi, C.; Rovetta, T.; Licchelli, M.; Malagodi, M. Mid and Near-Infrared Reflection Spectral Database of Natural Organic Materials in the Cultural Heritage Field. Int. J. Anal. Chem. 2018, 2018, 7823248. [Google Scholar] [CrossRef]
- Colthup, N. Introduction to Infrared and Raman Spectroscopy; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Villalobos, M.; Awojulu, A.; Greeley, T.; Turco, G.; Deeter, G. Oligomeric chain extenders for economic reprocessing and recycling of condensation plastics. Energy 2006, 31, 3227–3234. [Google Scholar] [CrossRef]
- Samaniego, K.; Matos, A.; Sánchez-Safont, E.; Candal, M.V.; Lagaron, J.M.; Cabedo, L.; Famez-Perez, J. Role of Plasticizers on PHB/bio-TPE Blends Compatibilized by Reactive Extrusion. Materials 2022, 15, 1226. [Google Scholar] [CrossRef]
- Quiles-Carrillo, L.; Montanes, N.; Lagaron, J.M.; Balart, R.; Torres-Giner, S. On the use of acrylated epoxidized soybean oil as a reactive compatibilizer in injection-molded compostable pieces consisting of polylactide filled with orange peel flour. Polym. Int. 2018, 67, 1341–1351. [Google Scholar] [CrossRef]
- Wei, L.; McDonald, A.G.; Stark, N.M. Grafting of Bacterial Polyhydroxybutyrate (PHB) onto Cellulose via In Situ Reactive Extrusion with Dicumyl Peroxide. Biomacromolecules 2015, 16, 1040–1049. [Google Scholar] [CrossRef]
- Ortega-Toro, R.; Jiménez, A.; Talens, P.; Chiralt, A. Properties of starch–hydroxypropyl methylcellulose based films obtained by compression molding. Carbohydr. Polym. 2014, 109, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Villalobos, R.; Chanona, J.; Hernández, P.; Gutiérrez, G.; Chiralt, A. Gloss and transparency of hydroxypropyl methylcellulose films containing surfactants as affected by their microstructure. Food Hydrocoll. 2005, 19, 53–61. [Google Scholar] [CrossRef]
- Ivorra-Martinez, J.; Quiles-Carrillo, L.; Boronat, T.; Torres-Giner, S.; Covas, J.A. Assessment of the Mechanical and Thermal Properties of Injection-Molded Poly(3-hydroxybutyrate-co-3-hydroxyhexanoate)/Hydroxyapatite Nanoparticles Parts for Use in Bone Tissue Engineering. Polymers 2020, 12, 1389. [Google Scholar] [CrossRef]
- Jun, D.; Guomin, Z.; Mingzhu, P.; Leilei, Z.; Dagang, L.; Rui, Z. Crystallization and mechanical properties of reinforced PHBV composites using melt compounding: Effect of CNCs and CNFs. Carbohydr. Polym. 2017, 168, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Melendez-Rodriguez, B.; Figueroa-Lopez, K.J.; Bernardos, A.; Martínez-Máñez, R.; Cabedo, L.; Torres-Giner, S.; Lagaron, J.M. Electrospun Antimicrobial Films of Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) Containing Eugenol Essential Oil Encapsulated in Mesoporous Silica Nanoparticles. Nanomaterials 2019, 9, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugnicourt, E.; Cinelli, P.; Lazzeri, A.; Alvarez, V.A. Polyhydroxyalkanoate (PHA): Review of synthesis, characteristics, processing and potential applications in packaging; Budapest University of Technology and Economics. Express Polym. Lett. 2014, 8, 791–808. [Google Scholar] [CrossRef] [Green Version]
- De Koning, G.J.M.; Lemstra, P.J. Crystallization phenomena in bacterial poly[(R)-3-hydroxybutyrate]: 2. Embrittlement and rejuvenation. Polymer 1993, 34, 4089–4094. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Salazar, J.; Sanchez-Cuesta, M.; Barham, P.J.; Keller, A. Thermal expansion and spherulite cracking in 3-hydroxybutyrate/3-hydroxyvalerate copolymers. J. Mater. Sci. Lett. 1989, 8, 490–492. [Google Scholar] [CrossRef]
- Barham, P.J.; Keller, A. The relationship between microstructure and mode of fracture in polyhydroxybutyrate. J. Polym. Sci. B Polym. Phys. 1986, 24, 69–77. [Google Scholar] [CrossRef]
- Biddlestone, F.; Harris, A.; Hay, J.N.; Hammond, T. The physical ageing of amorphous poly(hydroxybutyrate). Polym. Int. 1996, 39, 221–229. [Google Scholar] [CrossRef]
- Di Lorenzo, M.L.; Righetti, M.C. Effect of thermal history on the evolution of crystal and amorphous fractions of poly[(R)-3-hydroxybutyrate] upon storage at ambient temperature. Eur. Polym. J. 2013, 49, 510–517. [Google Scholar] [CrossRef]
- Torres-Giner, S.; Gil, L.; Pascual-Ramírez, L.; Garde-Belza, J.A. Packaging: Food waste reduction. Encycl. Polym. Appl. 2018, 3, 1990–2009. [Google Scholar]
- Gabirondo, E.; Melendez-Rodriguez, B.; Arnal, C.; Lagaron, J.M.; de Ilarduya, A.M.; Sardon, H.; Torres-Giner, S. Organocatalyzed closed-loop chemical recycling of thermo-compressed films of poly(ethylene furanoate). Polym. Chem. 2021, 12, 1571–1580. [Google Scholar] [CrossRef]
- Lagarón, J.M. 1-Multifunctional and nanoreinforced polymers for food packaging. In Multifunctional and Nanoreinforced Polymers for Food Packaging; Lagarón, J.-M., Ed.; Woodhead Publishing: Sawston, UK, 2011; pp. 1–28. [Google Scholar]
- Arrieta, M.P.; Fortunati, E.; Dominici, F.; Rayón, E.; López, J.; Kenny, J.M. PLA-PHB/cellulose based films: Mechanical, barrier and disintegration properties. Polym. Degrad. Stab. 2014, 107, 139–149. [Google Scholar] [CrossRef]
- Sanchez-Garcia, M.D.; Gimenez, E.; Lagaron, J.M. Novel PET Nanocomposites of Interest in Food Packaging Applications and Comparative Barrier Performance with Biopolyester Nanocomposites. J. Plast. Film Sheeting 2007, 23, 133–148. [Google Scholar] [CrossRef]
- Cherpinski, A.; Torres-Giner, S.; Vartiainen, J.; Peresin, M.S.; Lahtinen, P.; Lagaron, J.M. Improving the water resistance of nanocellulose-based films with polyhydroxyalkanoates processed by the electrospinning coating technique. Cellulose 2018, 25, 1291–1307. [Google Scholar] [CrossRef]
- Cao, X.; Wang, Y.; Chen, H.; Hu, J.; Cui, L. Preparation of different morphologies cellulose nanocrystals from waste cotton fibers and its effect on PLLA/PDLA composites films. Compos. Part B Eng. 2021, 217, 108934. [Google Scholar] [CrossRef]
- Jiang, G.; Yu, L.; Zhang, M.; Wang, F.; Zhang, S. Poly(propylene carbonate)/poly(3-hydroxybutyrate)-based bionanocomposites reinforced with cellulose nanocrystal for potential application as a packaging material. Polym. Adv. Technol. 2020, 31, 853–863. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | PHBV (wt %) | MFC (wt %) | MAH (phr) | ESAO (phr) | TGIC (phr) | DCP (phr) |
|---|---|---|---|---|---|---|
| PHBV | 100 | 0 | 0 | 0 | 0 | 0 |
| PHBV_2.5%MFC | 97.5 | 2.5 | 0 | 0 | 0 | 0 |
| PHBV_5%MFC | 95 | 5 | 0 | 0 | 0 | 0 |
| PHBV_10%MFC | 90 | 10 | 0 | 0 | 0 | 0 |
| PHBV_ESAO | 100 | 0 | 0 | 1 | 0 | 0 |
| PHBV_TGIC+DCP | 100 | 0 | 0 | 0 | 1 | 0.25 |
| PHBV_5%MFC_ESAO | 95 | 5 | 0 | 1 | 0 | 0 |
| PHBV_5%MFC_TGIC+DCP | 95 | 5 | 0 | 0 | 1 | 0.25 |
| Film | L* | a* | b* | Cab* | hab* | ∆E* |
|---|---|---|---|---|---|---|
| PHBV | 88.41 ± 0.11 a | −0.94 ± 0.01 a | 10.20 ± 0.15 a | 10.24 ± 0.15 a | 95.25 ± 0.11 a | - |
| PHBV_2.5%MFC | 87.75 ± 0.38 a | −1.12 ± 0.03 b | 11.52 ± 0.87 b | 11.57 ± 0.86 b | 95.59± 0.47 a | 1.53 ± 0.87 a |
| PHBV_5%MFC | 88.40 ± 0.27 a | −0.91 ± 0.04 a | 10.22 ± 0.98 a | 10.25 ± 0.99 a | 95.10 ± 0.32 a | 1.27 ± 0.41 a |
| PHBV_10%MFC | 87.89 ± 0.50 a | −0.86 ± 0.05 a | 11.42 ± 0.64 b | 11.45 ± 0.64 b | 94.34 ± 0.41 b | 1.35 ± 0.75 a |
| PHBV_ESAO | 89.11 ± 0.24 a | −1.10 ± 0.05 b | 8.75 ± 0.23 c | 8.82 ± 0.23 c | 97.15 ± 0.17 c | 1.66 ± 0.34 a |
| PHBV_TGIC+DCP | 89.39 ± 0.15 a | −1.22 ± 0.04 c | 8.80 ± 0.44 c | 8.88 ± 0.44 c | 97.91 ± 0.38 c | 1.75 ± 0.52 a |
| PHBV_5%MFC_ESAO | 88.73 ± 0.07 a | −1.14 ± 0.02 b | 11.24± 0.24 b | 11.30 ± 0.24 a | 95.76 ± 0.12 a | 1.12 ± 0.17 a |
| PHBV_5%MFC_TGIC+DCP | 88.86 ± 0.09 a | −1.38 ± 0.05 d | 9.30 ± 0.22 d | 9.40 ± 0.22 c | 98.47 ± 0.23 c | 1.12 ± 0.20 a |
| Film | Tg (°C) | Tc (°C) | ∆Hc (J/g) | Tm (°C) | ∆Hm (J/g) | χc (%) |
|---|---|---|---|---|---|---|
| PHBV | −1.73 ± 0.11 a | 78.65 ± 0.36 a | 79.07 ± 0.70 a | 170.53 ± 0.16 a | 95.81 ± 8.64 a | 65.35 ± 1.89 a |
| PHBV_2.5%MFC | −1.62 ± 0.07 a | 82.26 ± 0.57 b | 78.63 ± 0.64 a | 167.80 ± 0.11 b | 91.61 ± 8.42 b | 65.78 ± 6.04 a |
| PHBV_5%MFC | −1.73 ± 0.04 a | 76.40 ± 0.66 c | 71.17 ± 0.23 b | 168.81 ± 0.04 b | 95.09 ± 1.22 a | 68.28 ± 0.87 b |
| PHBV_10%MFC | −1.71 ± 0.21 a | 75.59 ± 0.56 c | 63.59 ± 0.71 c | 169.06 ± 0.79 b | 84.45 ± 1.60 c | 64.01 ± 1.21 c |
| PHBV_ESAO | −1.72 ± 0.06 a | 73.07 ± 0.54 d | 73.18 ± 0.73 b | 168.16 ± 0.49 b | 91.92 ± 0.24 b | 63.34 ± 0.16 d |
| PHBV_TGIC+DCP | −1.73 ± 0.14 a | 77.07 ± 4.24 c | 75.79 ± 0.19 d | 166.31 ± 0.25 c | 90.95 ± 2.18 b | 62.85 ± 1.51 d |
| PHBV_5%MFC_ESAO | −1.77 ± 0.07 a | 76.90 ± 1.66 c | 78.87 ± 1.40 a | 167.40 ± 0.56 c | 93.66 ± 0.13 d | 67.25 ± 0.10 b |
| PHBV_5%MFC_TGIC+DCP | −1.76 ± 0.30 a | 79.93 ± 0.85 e | 68.49 ± 0.34 e | 165.76 ± 0.45 d | 89.59 ± 1.23 b | 65.15 ± 0.89 c |
| Film | Tonset (°C) | Tdeg (°C) | Residual Mass (%) |
|---|---|---|---|
| PHBV | 277.50 ± 1.54 a | 296.25 ± 0.59 a | 2.26 ± 0.04 a |
| PHBV_2.5%MFC | 280.42 ± 1.77 b | 301.67 ± 1.18 b | 2.76 ± 0.12 a |
| PHBV_5%MFC | 282.92 ± 1.12 b | 301.67 ± 1.71 b | 2.07 ± 0.57 a |
| PHBV_10%MFC | 288.75 ± 2.95 c | 305.83 ± 2.47 c | 1.91 ± 0.84 a |
| PHBV_ESAO | 227.08 ± 3.23 d | 255.00 ± 2.46 d | 2.02 ± 0.08 a |
| PHBV_TGIC+DCP | 256.25 ± 1.27 e | 276.67 ± 2.39 e | 2.11 ± 0.15 a |
| PHBV_5%MFC_ESAO | 274.17 ± 2.35 a | 296.67 ± 1.18 d | 2.16 ± 0.69 a |
| PHBV_5%MFC_TGIC+DCP | 277.08 ± 0.59 a | 300.83 ± 1.17 d | 2.01 ± 0.11 a |
| Film | E (MPa) | σy (MPa) | ɛb (%) |
|---|---|---|---|
| PHBV | 2613.5 ± 92.7 a | 27.4 ± 2.1 a | 1.37 ± 0.21 a |
| PHBV_2.5%MFC | 2684.9 ± 77.1 b | 22.2 ± 1.6 b | 1.09 ± 0.13 b |
| PHBV_5%MFC | 2746.5 ± 69.7 c | 21.6 ± 0.9 c | 0.92 ± 0.20 c |
| PHBV_10%MFC | 2873.1 ± 73.5 d | 20.1 ± 0.7 d | 0.78 ± 0.14 d |
| PHBV_ESAO | 2670.2 ± 21.7 b | 27.3 ± 1.2 a | 1.27 ± 0.11 a |
| PHBV_TGIC+DCP | 2710.9 ± 76.4 ab | 27.6 ± 1.7 a | 1.29 ± 0.15 a |
| PHBV_5%MFC_ESAO | 2863.1 ± 55.6 d | 27.8 ± 1.1 a | 0.91 ± 0.09 c |
| PHBV_5%MFC_TGIC+DCP | 3070.7 ± 41.7 e | 29.3 ± 2.3 e | 1.43 ± 0.08 a |
| Film | Thickness (µm) | WVP × 1015 (kg·m·m−2·Pa−1·s−1) | LP × 1014 (kg·m·m−2·Pa−1·s−1) | OP × 1019 (m3·m·m−2·Pa−1·s−1) |
|---|---|---|---|---|
| PHBV | 126 ± 5 | 1.84 ± 0.39 a | 1.41 ± 0.12 a | 2.09 ± 0.48 a |
| PHBV_2.5%MFC | 133 ± 4 | 4.19 ± 0.40 b | 1.34 ± 0.06 a | 1.60 ± 0.19 b |
| PHBV_5%MFC | 128 ± 7 | 5.27 ± 0.53 c | 1.07 ± 0.09 b | 1.24 ± 0.36 c |
| PHBV_10%MFC | 129 ± 6 | 5.89 ± 0.47 c | 1.66 ± 0.15 c | 1.08 ± 0.12 d |
| PHBV_ESAO | 137 ± 2 | 4.46 ± 0.19 b | 2.04 ± 0.17 d | 2.27 ± 0.15 a |
| PHBV_TGIC+DCP | 135 ± 8 | 5.69 ± 0.29 c | 3.80 ± 0.31 e | 2.70 ± 0.26 a |
| PHBV_5%MFC_ESAO | 136 ± 7 | 5.48 ± 0.58 c | 0.99 ± 0.39 b | 1.36 ± 0.12 c |
| PHBV_5%MFC_TGIC+DCP | 128 ± 3 | 3.29 ± 0.30 d | 0.16 ± 0.01 f | 0.98 ± 0.17 f |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freitas, P.A.V.; Barrrasa, H.; Vargas, F.; Rivera, D.; Vargas, M.; Torres-Giner, S. Atomization of Microfibrillated Cellulose and Its Incorporation into Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) by Reactive Extrusion. Appl. Sci. 2022, 12, 2111. https://doi.org/10.3390/app12042111
Freitas PAV, Barrrasa H, Vargas F, Rivera D, Vargas M, Torres-Giner S. Atomization of Microfibrillated Cellulose and Its Incorporation into Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) by Reactive Extrusion. Applied Sciences. 2022; 12(4):2111. https://doi.org/10.3390/app12042111
Chicago/Turabian StyleFreitas, Pedro A. V., Hector Barrrasa, Fátima Vargas, Daniel Rivera, Maria Vargas, and Sergio Torres-Giner. 2022. "Atomization of Microfibrillated Cellulose and Its Incorporation into Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) by Reactive Extrusion" Applied Sciences 12, no. 4: 2111. https://doi.org/10.3390/app12042111