
Review of Quantitative Methods for the Detection of Alzheimer’s Disease with Positron Emission Tomography

Abstract
:Featured Application
Abstract
1. Introduction
2. Alzheimer’s Disease—Epidemiology, Progression, ATN Biomarkers
3. Positron Emission Tomography & Tracers for the Detection of AD
3.1. Fluorodeoxyglucose (FDG)
3.2. Amyloid-Binding Tracers
3.3. Tau-Binding Tracers
4. Quantitative Methods for the Detection of AD
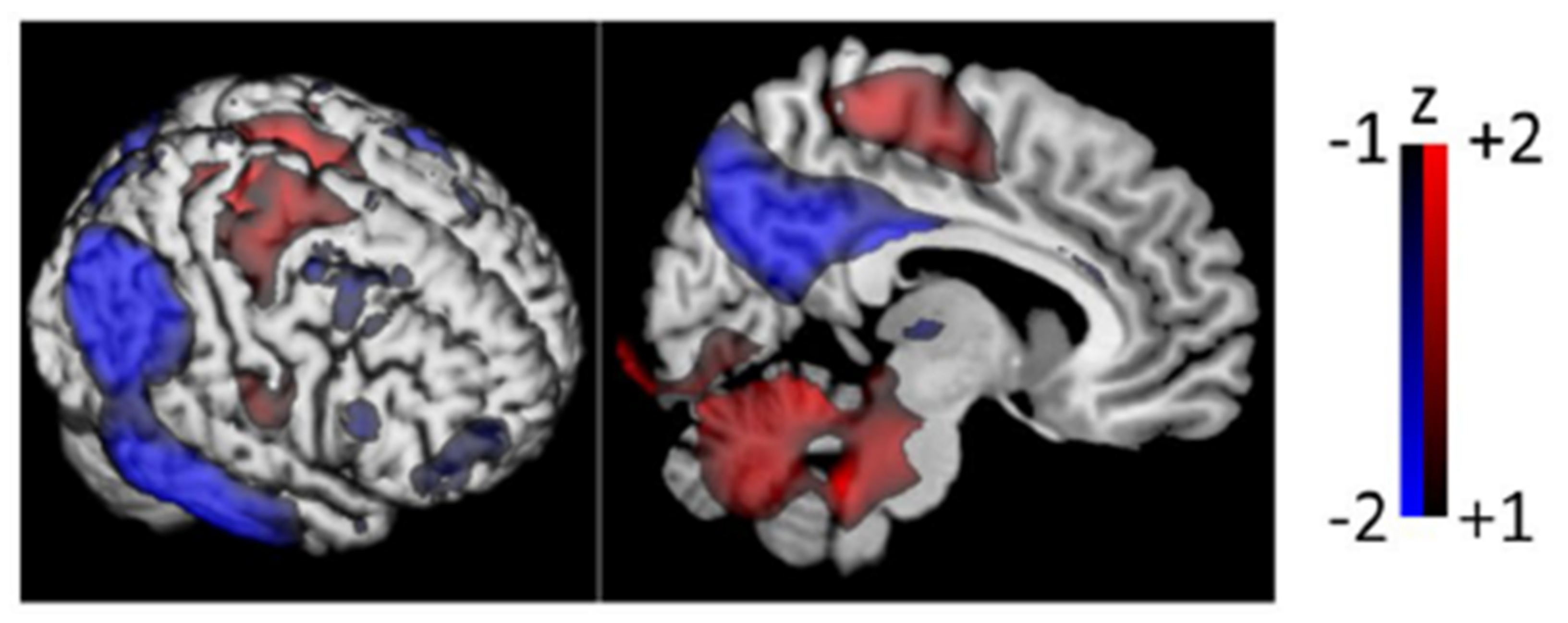
4.1. General Linear Models and Statistical Parametric Mapping
4.2. Stereotactic Surface Projection
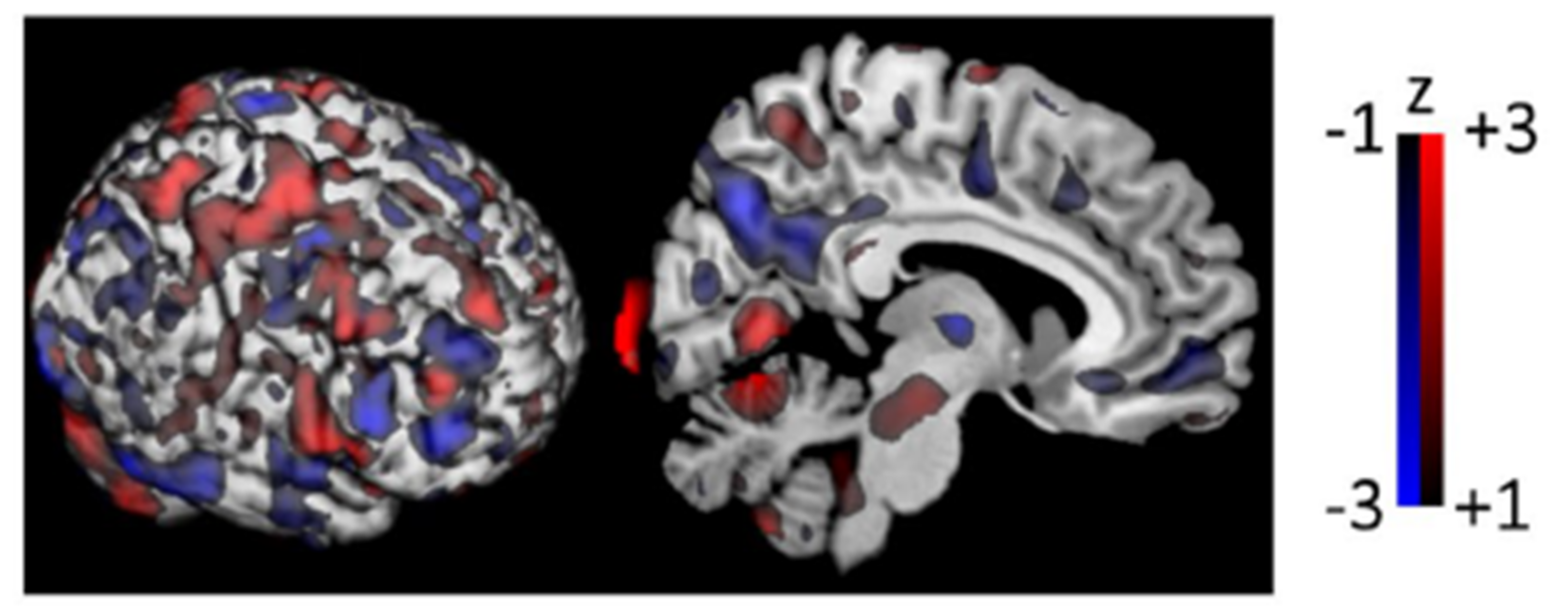
4.3. Principal Component Analysis & Scaled Subprofile Modeling
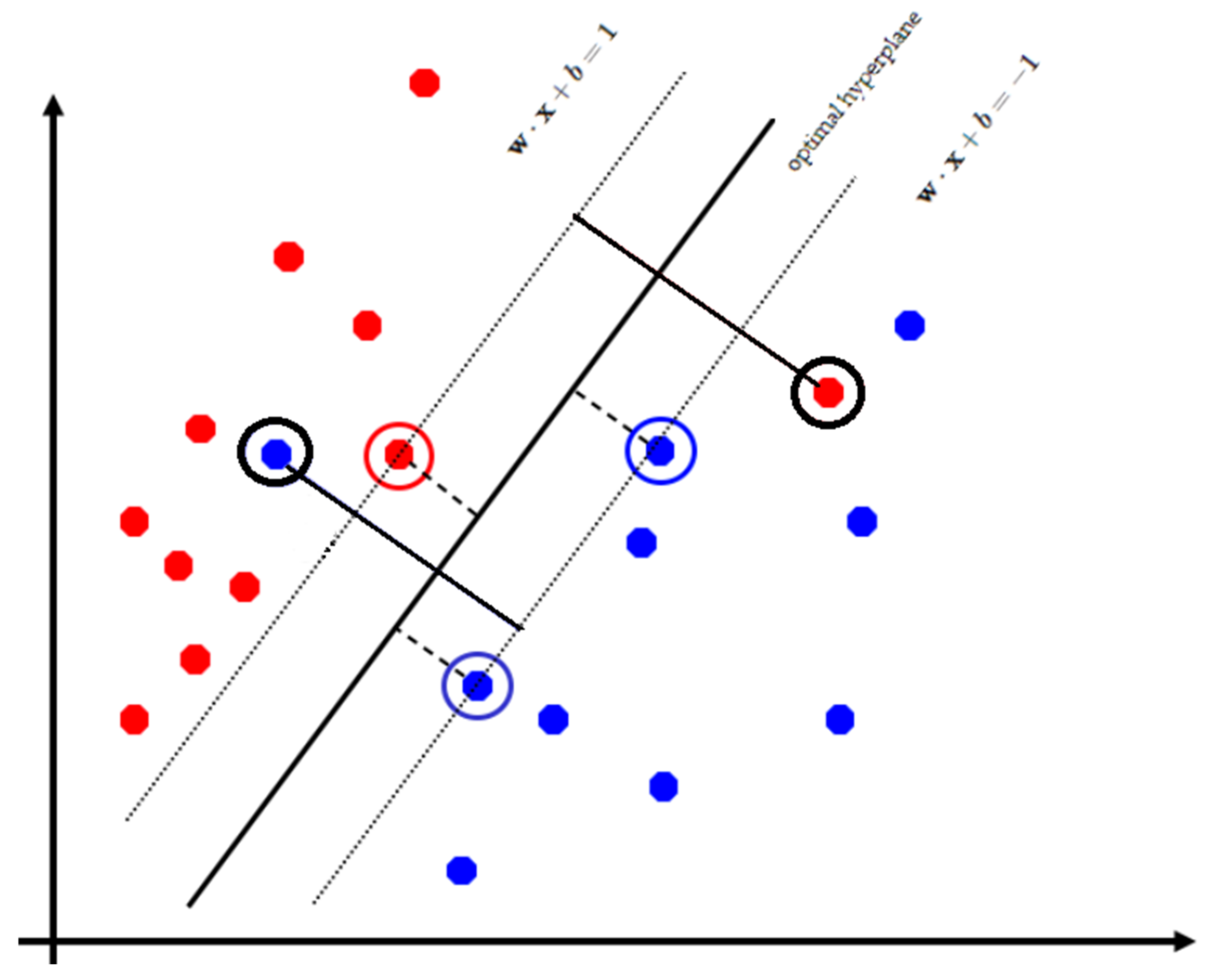
4.4. Support Vector Machines
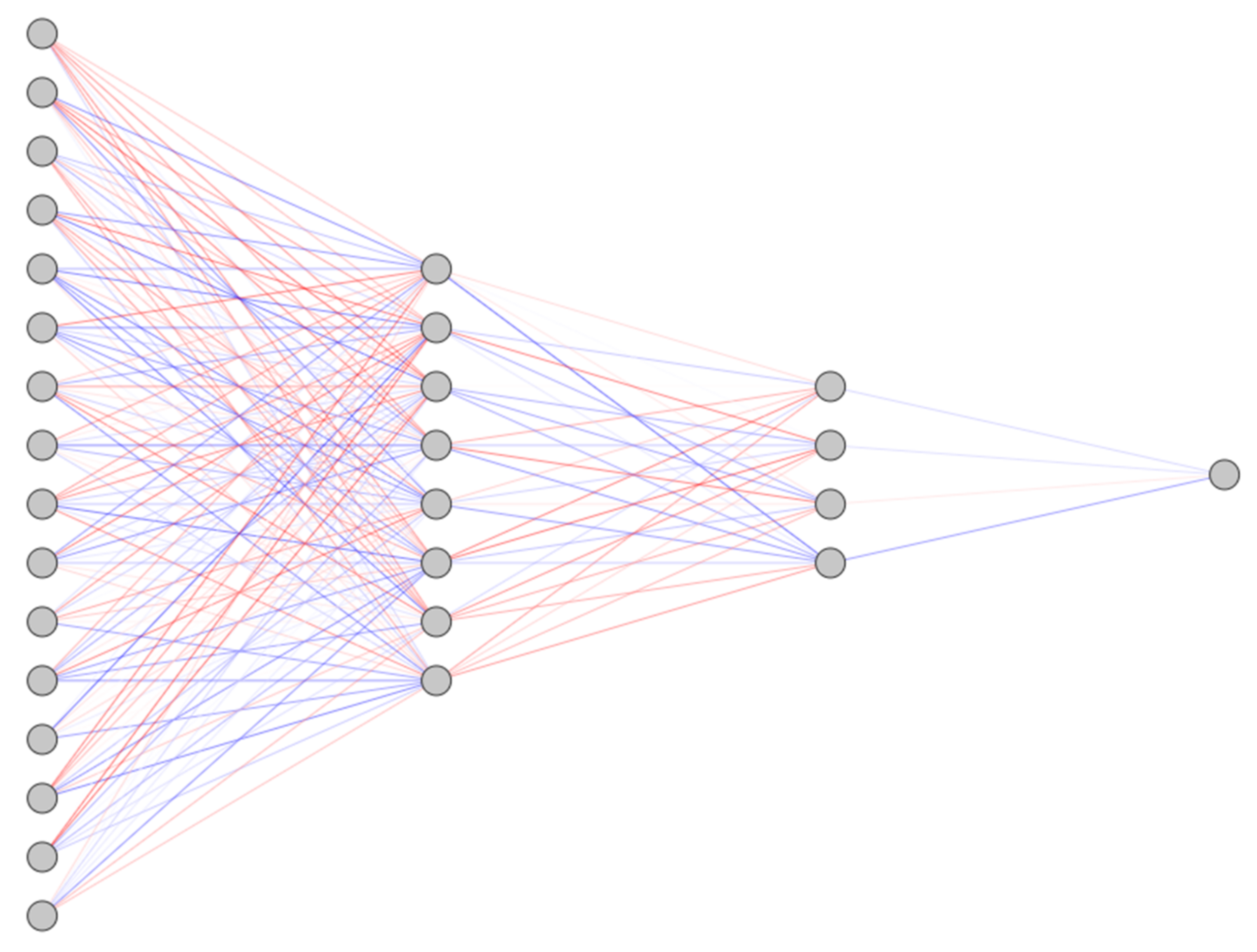
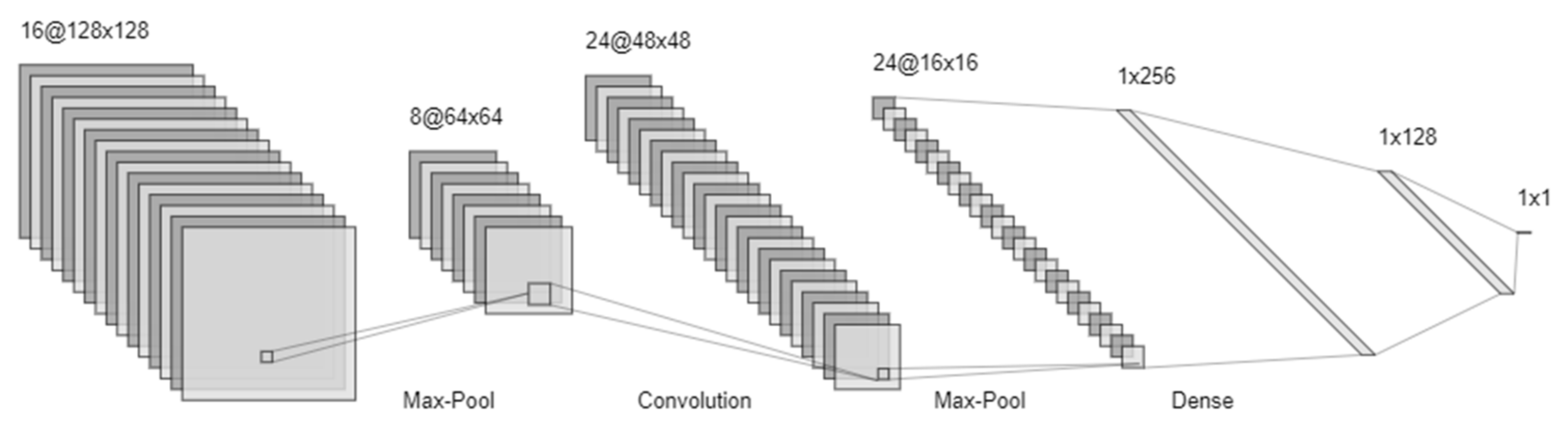
4.5. Neural Networks
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arvanitakis, Z.; Shah, R.C.; Bennett, D.A. Diagnosis and management of dementia. JAMA 2019, 322, 1589–1599. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; Monsell, S.E.; Phillips, L.E.; Kukull, W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005–2010. J. Neuropathol. Exp. Neurol. 2012, 71, 266–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunnström, H.; Englund, E. Clinicopathological concordance in dementia diagnostics. Am. J. Geriatr. Psychiatry 2009, 17, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Rockwood, K. How golden is the gold standard of neuropathology in dementia. Alzheimer’s Dement. 2011, 7, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.; Tardiff, S.; Dye, C.; Arrighi, H.M. Rate of conversion from prodromal Alzheimer’s disease to Alzheimer’s dementia: A systematic review of the literature. Dement. Geriatr. Cogn. Disord. Extra 2013, 3, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Petersen, R.C. Mild cognitive impairment. CONTINUUM Lifelong Learn. Neurol. 2016, 22, 404. [Google Scholar] [CrossRef] [Green Version]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Itwatsubo, T.; Jack, C.R.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [Green Version]
- Eschweiler, G.W.; Leyhe, T.; Klöppel, S.; Hüll, M. New developments in the diagnosis of dementia. Dtsch. Ärzteblatt Int. 2010, 107, 677. [Google Scholar] [CrossRef]
- Van Maurik, I.S.; Zwan, M.D.; Tijms, B.M.; Bouwman, F.H.; Teunissen, C.E.; Scheltens, P.; Wattjes, M.P.; Barkhof, F.; Berkhof, J.; van der Flier, W.M. Alzheimer’s Disease Neuroimaging Initiative. Interpreting biomarker results in individual patients with mild cognitive impairment in the Alzheimer’s biomarkers in daily practice (ABIDE) project. JAMA Neurol. 2017, 74, 1481–1491. [Google Scholar] [CrossRef]
- De Wilde, A.; Maurik, I.S.; Kunneman, M.; Bouwman, F.; Zwan, M.; Willemse, E.A.; Biessels, G.J.; Minkman, M.; Pel, R.; Schoonenboom, N.S.; et al. Alzheimer’s biomarkers in daily practice (ABIDE) project: Rationale and design. Alzheimer’s Dementia Diagn. Assess. Dis. Monit. 2017, 6, 143–151. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc. 2011, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimer’s Dement. 2007, 3, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.; Tan, C.-C.; Xu, W.; Hu, H.; Cao, X.-P.; Dong, Q.; Tan, L.; Yu, J.-T. The Prevalence of Dementia: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2020, 73, 1157–1166. [Google Scholar] [CrossRef]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Knight, R.; Khondoker, M.; Magill, N.; Stewart, R.; Landau, S. A systematic review and meta-analysis of the effectiveness of acetylcholinesterase inhibitors and memantine in treating the cognitive symptoms of dementia. Dement. Geriatr. Cogn. Disord. 2018, 45, 131–151. [Google Scholar] [CrossRef] [Green Version]
- Tolar, M.; Abushakra, S.; Hey, J.A.; Porsteinsson, A.; Sabbagh, M. Aducanumab, gantenerumab, BAN2401, and ALZ-801—The first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimer’s Res. Ther. 2020, 12, 95. [Google Scholar] [CrossRef]
- Gandy, S.; Knopman, D.S.; Sano, M. Talking points for physicians, patients and caregivers considering Aduhelm® infusion and the accelerated pathway for its approval by the FDA. Mol. Neurodegener. 2021, 16, 74. [Google Scholar] [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef]
- Acosta, C.; Anderson, H.D.; Anderson, C.M. Astrocyte dysfunction in Alzheimer disease. J. Neurosci. Res. 2017, 95, 2430–2447. [Google Scholar] [CrossRef]
- Pimlott, S.L.; Sutherland, A. Molecular tracers for the PET and SPECT imaging of disease. Chem. Soc. Rev. 2011, 40, 149–162. [Google Scholar] [CrossRef]
- Bao, W.; Xie, F.; Zuo, C.; Guan, Y.; Huang, Y.H. PET neuroimaging of Alzheimer’s disease: Radiotracers and their utility in clinical research. Front. Aging Neurosci. 2021, 13, 624330. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Nishii, R.; Kagawa, S.; Kishibe, Y.; Takahashi, M.; Okina, T.; Suzuki, N.; Hasegawa, H.; Nagahama, Y.; Ishizu, K.; et al. 18F-FPYBF-2, a new F-18-labelled amyloid imaging PET tracer: First experience in 61 volunteers and 55 patients with dementia. Ann. Nucl. Med. 2018, 32, 206–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minoshima, S.; Frey, K.A.; Cross, D.J.; Kuhl, D.E. Neurochemical imaging of dementias. Semin. Nucl. Med. 2004, 34, 70–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, A.; Klein, B.; Janssen, B.; Pedragosa, J.; Pepe, G.; Zinnhardt, B.; Vugts, D.J.; Gelosa, P.; Sironi, L.; Beaino, W.; et al. Identification of new molecular targets for PET imaging of the microglial anti-inflammatory activation state. Theranostics 2018, 8, 5400. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Ichise, M.; Ito, H.; Shimada, H.; Ikoma, Y.; Seki, C.; Takano, H.; Kitamura, S.; Shinotoh, H.; Kawamura, K.; et al. PET Quantification of Tau Pathology in Human Brain with 11C-PBB3. J. Nucl. Med. 2015, 56, 1359–1365. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.E.; Janssens, L.; Moechars, D.; Rombouts, F.J.; Timmers, M.; Barret, O.; Constantinescu, C.C.; Madonia, J.; Russell, D.S.; Sandiego, C.M.; et al. Clinical evaluation of [18F] JNJ-64326067, a novel candidate PET tracer for the detection of tau pathology in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 3176–3185. [Google Scholar] [CrossRef]
- Fan, A.P.; An, H.; Moradi, F.; Rosenberg, J.; Ishii, Y.; Nariai, T.; Okazawa, H.; Zaharchuk, G. Quantification of brain oxygen extraction and metabolism with [15O]-gas PET: A technical review in the era of PET/MRI. NeuroImage 2020, 220, 117136. [Google Scholar] [CrossRef]
- Chételat, G.; Arbizu, J.; Barthel, H.; Garibotto, V.; Law, I.; Morbelli, S.; van de Giessen, E.; Agosta, F.; Barkhof, F.; Brooks, D.J.; et al. Amyloid-PET and 18F-FDG-PET in the diagnostic investigation of Alzheimer’s disease and other dementias. Lancet Neurol. 2020, 19, 951–962. [Google Scholar] [CrossRef]
- Herscovitch, P. Regulatory approval and insurance reimbursement: The final steps in clinical translation of amyloid brain imaging. Clin. Transl. Imaging 2015, 3, 75–77. [Google Scholar] [CrossRef]
- Portnow, L.H.; Vaillancourt, D.E.; Okun, M.S. The history of cerebral PET scanning: From physiology to cutting-edge technology. Neurology 2013, 80, 952–956. [Google Scholar] [CrossRef]
- Alavi, A.; Dann, R.; Chawluk, J.; Alavi, J.; Kushner, M.; Reivich, M. Positron emission tomography imaging of regional cerebral glucose metabolism. Semin. Nucl. Med. 1986, 16, 2–34. [Google Scholar] [CrossRef]
- Marcus, C.; Mena, E.; Subramaniam, R.M. Brain PET in the diagnosis of Alzheimer’s disease. Clin. Nucl. Med. 2014, 39, e413. [Google Scholar] [CrossRef] [Green Version]
- Nordberg, A.; Rinne, J.O.; Kadir, A.; Långström, B. The use of PET in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 78–87. [Google Scholar] [CrossRef]
- Minoshima, S.; Mosci, K.; Cross, D.; Thientunyakit, T. Brain [F-18] FDG PET for clinical dementia workup: Differential diagnosis of Alzheimer’s disease and other types of dementing disorders. Semin. Nucl. Med. 2021, 51, 230–240. [Google Scholar] [CrossRef]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, R.; Ropele, S.; Pendl, B.; Ofner, P.; Enzinger, C.; Schmidt, H.; Berghold, A.; Windisch, M.; Kolassa, H.; Fazekas, F. Longitudinal multimodal imaging in mild to moderate Alzheimer disease: A pilot study with memantine. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1312–1317. [Google Scholar] [CrossRef]
- Smith, G.S.; Laxton, A.W.; Tang-Wai, D.F.; McAndrews, M.P.; Diaconescu, A.O.; Workman, C.I.; Lozano, A.M. Increased cerebral metabolism after 1 year of deep brain stimulation in Alzheimer disease. Arch. Neurol. 2012, 69, 1141–1148. [Google Scholar] [CrossRef]
- Tzimopoulou, S.; Cunningham, V.J.; Nichols, T.E.; Searle, G.; Bird, N.P.; Mistry, P.; Ian, D.; William, H.; Brandon, W.; Andrew, B.; et al. A multi-center randomized proof-of-concept clinical trial applying [18F] FDG-PET for evaluation of metabolic therapy with rosiglitazone XR in mild to moderate Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 22, 1241–1256. [Google Scholar] [CrossRef]
- Xia, M.; Wang, J.; He, Y. BrainNet Viewer: A Network Visualization Tool for Human Brain Connectomics. PLoS ONE 2013, 8, e68910. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, U.; Kayed, R. Amyloid β, Tau, and α-Synuclein aggregates in the pathogenesis, prognosis, and therapeutics for neurodegenerative diseases. Prog. Neurobiol. 2022, 23, 102270. [Google Scholar] [CrossRef]
- Grochowska, K.M.; Yuanxiang, P.; Bär, J.; Raman, R.; Brugal, G.; Sahu, G.; Schweizer, M.; Bikbaev, A.; Schilling, S.; Demuth, H.; et al. Posttranslational modification impact on the mechanism by which amyloid-β induces synaptic dysfunction. EMBO Rep. 2017, 18, 962–981. [Google Scholar] [CrossRef] [PubMed]
- Penke, B.; Szűcs, M.; Bogár, F. Oligomerization and conformational change turn monomeric β-amyloid and tau proteins toxic: Their role in Alzheimer’s pathogenesis. Molecules 2020, 25, 1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jack, R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Villemagne, V.L.; Ong, K.; Mulligan, R.S.; Holl, G.; Pejoska, S.; Jones, G.; O’Keefe, G.; Ackerman, U.; Tochon-Danguy, H.; Chan, J.G.; et al. Amyloid Imaging with 18F-Florbetaben in Alzheimer Disease and Other Dementias. J. Nucl. Med. 2011, 52, 1210–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engler, H.; Forsberg, A.; Almkvist, O.; Blomquist, G.; Larsson, E.; Savitcheva, I.; Wall, A.; Ringheim, A.; Långström, B.; Nordberg, A. Two-year follow-up of amyloid deposition in patients with Alzheimer’s disease. Brain 2006, 129, 2856–2866. [Google Scholar] [CrossRef] [Green Version]
- Klunk, W.E. Amyloid imaging as a biomarker for cerebral β-amyloidosis and risk prediction for Alzheimer dementia. Neurobiol. Aging 2011, 32, S20–S36. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.T.; Veenith, T.; Dewar, D.; Outtrim, J.G.; Mani, V.; Williams, C.; Pimlott, S.; Hutchinson, P.; Tavares, A.; Canales, R.; et al. Amyloid imaging with carbon 11–labeled Pittsburgh compound B for traumatic brain injury. JAMA Neurol. 2014, 71, 23–31. [Google Scholar] [CrossRef]
- Svedberg, M.M.; Hall, H.; Hellström-Lindahl, E.; Estrada, S.; Guan, Z.; Nordberg, A.; Långström, B. [11C] PIB-amyloid binding and levels of Aβ40 and Aβ42 in postmortem brain tissue from Alzheimer patients. Neurochem. Int. 2009, 54, 347–357. [Google Scholar] [CrossRef]
- Kemppainen, N.M.; Aalto, S.; Wilson, I.A.; Någren, K.; Helin, S.; Brück, A.; Oikonen, V.; Kailajärvi, M.; Scheinin, M.; Viitanen, M.; et al. PET amyloid ligand [11C] PIB uptake is increased in mild cognitive impairment. Neurology 2007, 68, 1603–1606. [Google Scholar] [CrossRef]
- Chamberlain, R.; Reyes, D.; Curran, G.L.; Marjanska, M.; Wengenack, T.M.; Poduslo, J.F.; Jack, C.R., Jr. Comparison of amyloid plaque contrast generated by T2-weighted, T-weighted, and susceptibility-weighted imaging methods in transgenic mouse models of Alzheimer’s disease. Magn. Reson. Med. 2009, 61, 1158–1164. [Google Scholar] [CrossRef]
- Okello, A.; Koivunen, J.; Edison, P.; Archer, H.A.; Turkheimer, F.E.; Någren, K.U.; Bullock, R.; Walker, Z.; Kennedy, A.; Fox, N.C. Conversion of amyloid positive and negative MCI to AD over 3 years: An 11C-PIB PET study. Neurology 2009, 73, 754–760. [Google Scholar] [CrossRef] [Green Version]
- Drzezga, A.; Grimmer, T.; Henriksen, G.; Mühlau, M.; Perneczky, R.; Miederer, I.; Praus, C.; Sorg, C.; Wohlschläger, A.; Riemenschneider, M.; et al. Effect of APOE genotype on amyloid plaque load and gray matter volume in Alzheimer disease. Neurology 2009, 72, 1487–1494. [Google Scholar] [CrossRef]
- Clark, C.M.; Schneider, J.A.; Bedell, B.J.; Beach, T.G.; Bilker, W.B.; Mintun, M.A.; Pontecorvo, M.; Hefti, F.; Carpenter, A.; Flitter, M.; et al. AV45-A07 Study Group. Use of florbetapir-PET for imaging β-amyloid pathology. JAMA 2011, 305, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Klunk, W.E.; Koeppe, R.A.; Price, J.C.; Benzinger, T.L.; Devous Sr, M.D.; Jagust, W.J.; Johnson, K.A.; Mathis, C.A.; Minhas, D.; Pontecorvo, M.J.; et al. The Centiloid Project: Standardizing quantitative amyloid plaque estimation by PET. Alzheimer’s Dement. 2015, 11, 1–15.e4. [Google Scholar] [CrossRef] [Green Version]
- Ossenkoppele, R.; van Berckel, B.N.; Prins, N.D. Amyloid imaging in prodromal Alzheimer’s disease. Alzheimer’s Res. Ther. 2011, 3, 26. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.R., Jr.; Knopman, D.S.; Chételat, G.; Dickson, D.; Fagan, A.M.; Frisoni, G.B.; Jagust, W.; Mormino, E.C.; Petersen, R.C.; Sperling, R.A.; et al. Suspected non-Alzheimer disease pathophysiology—Concept and controversy. Nat. Rev. Neurol. 2016, 12, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Braak, E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol. Aging 1997, 18, 351–357. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Ricci, M.; Cimini, A.; Camedda, R.; Chiaravalloti, A.; Schillaci, O. Tau Biomarkers in Dementia: Positron Emission Tomography Radiopharmaceuticals in Tauopathy Assessment and Future Perspective. Int. J. Mol. Sci. 2021, 22, 13002. [Google Scholar] [CrossRef]
- Whitwell, J.L.; Graff-Radford, J.; Tosakulwong, N.; Weigand, S.D.; Machulda, M.M.; Senjem, M.L.; Spychalla, A.J.; Vemuri, P.; Jones, D.T.; Drubach, D.A.; et al. Imaging correlations of tau, amyloid, metabolism, and atrophy in typical and atypical Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 1005–1014. [Google Scholar] [CrossRef]
- Ishiki, A.; Okamura, N.; Furukawa, K.; Furumoto, S.; Harada, R.; Tomita, N.; Hiraoka, K.; Watanuki, S.; Ishikawa, Y.; Tago, T.; et al. Longitudinal assessment of tau pathology in patients with Alzheimer’s disease using [18F] THK-5117 positron emission tomography. PLoS ONE 2015, 10, e0140311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schöll, M.; Ossenkoppele, R.; Strandberg, O.; Palmqvist, S.; Jögi, J.; Ohlsson, T.; Smith, R.; Hansson, O.; The Swedish BioFINDER study. Distinct 18F-AV-1451 tau PET retention patterns in early- and late-onset Alzheimer’s disease. Brain 2017, 140, 2286–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ossenkoppele, R.; Schonhaut, D.R.; Schöll, M.; Lockhart, S.N.; Ayakta, N.; Baker, S.L.; O’Neil, J.P.; Janabi, M.; Lazaris, A.; Cantwell, A.; et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 2016, 139, 1551–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schöll, M.; Lockhart, S.N.; Schonhaut, D.R.; O’Neil, J.P.; Janabi, M.; Ossenkoppele, R.; Baker, S.L.; Vogel, J.W.; Faria, J.; Schwimmer, H.D.; et al. PET Imaging of Tau Deposition in the Aging Human Brain. Neuron 2016, 89, 971–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agdeppa, E.D.; Kepe, V.; Liu, J.; Flores-Torres, S.; Satyamurthy, N.; Petric, A.; Cole, G.M.; Small, G.W.; Huang, S.C.; Barrio, J.R. Binding characteristics of radiofluorinated 6-dialkylamino-2-naphthylethylidene derivatives as positron emission tomography imaging probes for β-amyloid plaques in Alzheimer’s disease. J. Neurosci. 2001, 21, RC189. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Goedert, M. Tau pathology and neurodegeneration. Lancet Neurol. 2013, 12, 609–622. [Google Scholar] [CrossRef]
- Ushizima, D.; Chen, Y.; Alegro, M.; Ovando, D.; Eser, R.; Lee, W.; Poon, K.; Shankar, A.; Kantamneni, N.; Satrawada, S.; et al. Deep learning for Alzheimer’s disease: Mapping large-scale histological tau protein for neuroimaging biomarker validation. NeuroImage 2021, 248, 118790. [Google Scholar] [CrossRef]
- Declercq, L.; Rombouts, F.; Koole, M.; Fierens, K.; Mariën, J.; Langlois, X.; Andrés, J.I.; Schmidt, M.; Macdonald, G.; Moechars, D.; et al. Preclinical evaluation of 18F-JNJ64349311, a novel PET tracer for tau imaging. J. Nucl. Med. 2017, 58, 975–981. [Google Scholar] [CrossRef] [Green Version]
- Rombouts, F.J.; Declercq, L.; Andrés, J.I.; Bottelbergs, A.; Chen, L.; Iturrino, L.; Leenaerts, J.E.; Marien, J.; Song, F.; Wintmolders, C.; et al. Discovery of N-(4-[18F] fluoro-5-methylpyridin-2-yl) isoquinolin-6-amine (JNJ-64326067), a new promising tau positron emission tomography imaging tracer. J. Med. Chem. 2019, 62, 2974–2987. [Google Scholar] [CrossRef]
- Teng, E.; Ward, M.; Manser, P.T.; Sanabria-Bohorquez, S.; Ray, R.D.; Wildsmith, K.R.; Baker, S.; Kerchner, G.A.; Weimer, R.M. Cross-sectional associations between [18F] GTP1 tau PET and cognition in Alzheimer’s disease. Neurobiol. Aging 2019, 81, 138–145. [Google Scholar] [CrossRef]
- Ossenkoppele, R.; Smith, R.; Mattsson-Carlgren, N.; Groot, C.; Leuzy, A.; Strandberg, O.; Palmqvist, S.; Olsson, T.; Jögi, J.; Stormrud, E.; et al. Accuracy of tau positron emission tomography as a prognostic marker in preclinical and prodromal Alzheimer disease: A head-to-head comparison against amyloid positron emission tomography and magnetic resonance imaging. JAMA Neurol. 2021, 78, 961–971. [Google Scholar] [CrossRef]
- Harrison, T.M.; La Joie, R.; Maass, A.; Baker, S.L.; Bs, K.S.; Fenton, L.; Bs, T.J.M.; Edwards, L.; Pham, J.; Miller, B.L.; et al. Longitudinal tau accumulation and atrophy in aging and alzheimer disease. Ann. Neurol. 2018, 85, 229–240. [Google Scholar] [CrossRef]
- Kuntner, C.; Stout, D. Quantitative preclinical PET imaging: Opportunities and challenges. Front. Phys. 2014, 2, 12. [Google Scholar] [CrossRef] [Green Version]
- Gunn, R.N.; Slifstein, M.; Searle, G.E.; Price, J.C. Quantitative imaging of protein targets in the human brain with PET. Phys. Med. Biol. 2015, 60, R363. [Google Scholar] [CrossRef] [Green Version]
- Heurling, K.; Leuzy, A.; Jonasson, M.; Frick, A.; Zimmer, E.R.; Nordberg, A.; Lubberink, M. Quantitative positron emission tomography in brain research. Brain Res. 2017, 1670, 220–234. [Google Scholar] [CrossRef]
- Friston, K.J.; Holmes, A.P.; Worsley, K.J.; Poline, J.P.; Frith, C.D.; Frackowiak, R.S. Statistical parametric maps in functional imaging: A general linear approach. Hum. Brain Mapp. 1994, 2, 189–210. [Google Scholar] [CrossRef]
- Friston, K.J.; Frith, C.D.; Liddle, P.F.; Dolan, R.J.; Lammertsma, A.A.; Frackowiak, R.S.J. The relationship between global and local changes in PET scans. J. Cereb. Blood Flow Metab. 1990, 10, 458–466. [Google Scholar] [CrossRef] [Green Version]
- Worsley, K.J.; Evans, A.C.; Marrett, S.; Neelin, P. A three-dimensional statistical analysis for CBF activation studies in human brain. J. Cereb. Blood Flow Metab. 1992, 12, 900–918. [Google Scholar] [CrossRef] [Green Version]
- Pagani, M.; Nobili, F.; Morbelli, S.; Arnaldi, D.; Giuliani, A.; Öberg, J.; Girtler, N.; Brugnolo, A.; Picco, A.; Bauckneht, M.; et al. Early identification of MCI converting to AD: A FDG PET study. Eur. J. Pediatr. 2017, 44, 2042–2052. [Google Scholar] [CrossRef]
- Della Rosa, P.A.; The EADC-PET Consortium; Cerami, C.; Gallivanone, F.; Prestia, A.; Caroli, A.; Castiglioni, I.; Gilardi, M.C.; Frisoni, G.; Friston, K.; et al. A Standardized [18F]-FDG-PET Template for Spatial Normalization in Statistical Parametric Mapping of Dementia. Neuroinformatics 2014, 12, 575–593. [Google Scholar] [CrossRef]
- Perani, D.; Della Rosa, P.A.; Cerami, C.; Gallivanone, F.; Fallanca, F.; Vanoli, E.G.; Panzacchi, A.; Nobili, F.; Pappatà, S.; Marcone, A.; et al. Validation of an optimized SPM procedure for FDG-PET in dementia diagnosis in a clinical setting. NeuroImage Clin. 2014, 6, 445–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, C.; Suppa, P.; Frings, L.; Brenner, W.; Spies, L.; Buchert, R.; Alzheimer’s Disease Neuroimaging Initiative. Optimization of statistical single subject analysis of brain FDG PET for the prognosis of mild cognitive impairment-to-Alzheimer’s disease conversion. J. Alzheimer’s Dis. 2016, 49, 945–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presotto, L.; Ballarini, T.; Caminiti, S.P.; Bettinardi, V.; Gianolli, L.; Perani, D. Validation of 18F–FDG-PET Single-subject optimized SPM procedure with different PET scanners. Neuroinformatics 2017, 15, 151–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sörensen, A.; Blazhenets, G.; Rücker, G.; Schiller, F.; Meyer, P.T.; Frings, L.; Alzheimer’s Disease Neuroimaging Initiative. Prognosis of conversion of mild cognitive impairment to Alzheimer’s dementia by voxel-wise Cox regression based on FDG PET data. NeuroImage Clin. 2019, 21, 101637. [Google Scholar] [CrossRef] [PubMed]
- Katako, A.; Shelton, P.; Goertzen, A.L.; Levin, D.; Bybel, B.; Aljuaid, M.; Yoon, H.J.; Kang, D.Y.; Kim, S.M.; Lee, C.S.; et al. Machine learning identified an Alzheimer’s disease-related FDG-PET pattern which is also expressed in Lewy body dementia and Parkinson’s disease dementia. Sci. Rep. 2018, 8, 13236. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Paranjpe, M.D.; Zhou, X.; Duy, P.Q.; Goyal, M.S.; Benzinger, T.L.; Lu, J.; Wang, R.; Zhou, Y. Sex modulates the ApoE ε4 effect on brain tau deposition measured by 18F-AV-1451 PET in individuals with mild cognitive impairment. Theranostics 2019, 9, 4959–4970. [Google Scholar] [CrossRef]
- Ottoy, J.; Niemantsverdriet, E.; Verhaeghe, J.; De Roeck, E.; Struyfs, H.; Somers, C.; Wyffels, L.; Ceyssens, S.; Van Mossevelde, S.; Van den Bossche, T.; et al. Association of short-term cognitive decline and MCI-to-AD dementia conversion with CSF, MRI, amyloid- and 18F-FDG-PET imaging. NeuroImage Clin. 2019, 22, 101771. [Google Scholar] [CrossRef]
- Nordberg, A.; Carter, S.F.; Rinne, J.; Drzezga, A.; Brooks, D.J.; Vandenberghe, R.; Perani, D.; Forsberg, A.; Långström, B.; Scheinin, N.; et al. A European multicentre PET study of fibrillar amyloid in Alzheimer’s disease. Eur. J. Pediatr. 2012, 40, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Saint-Aubert, L.; Almkvist, O.; Chiotis, K.; Almeida, R.; Wall, A.; Nordberg, A. Regional tau deposition measured by [18F] THK5317 positron emission tomography is associated to cognition via glucose metabolism in Alzheimer’s disease. Alzheimer’s Res. Ther. 2016, 8, 38. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.; Kang, J.M.; Seo, S.; Jeong, H.J.; Funck, T.; Lee, S.Y.; Yeon, B.K.; Ido, T.; Okamura, N. Topographical heterogeneity of Alzheimer’s disease based on MR imaging, tau PET, and amyloid PET. Front. Aging Neurosci. 2019, 11, 211. [Google Scholar] [CrossRef]
- Halawa, O.A.; Gatchel, J.R.; Amariglio, R.E.; Rentz, D.M.; Sperling, R.A.; Johnson, K.A.; Marshall, G.A. Inferior and medial temporal tau and cortical amyloid are associated with daily functional impairment in Alzheimer’s disease. Alzheimer’s Res. Ther. 2019, 11, 14. [Google Scholar] [CrossRef]
- Ossenkoppele, R.; Tolboom, N.; Foster-Dingley, J.C.; Adriaanse, S.F.; Boellaard, R.; Yaqub, M.; Windhorst, A.D.; Barkhof, F.; Lammertsma, A.A.; Scheltens, P.; et al. Longitudinal imaging of Alzheimer pathology using [11C] PIB, [18F] FDDNP and [18F] FDG PET. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 990–1000. [Google Scholar] [CrossRef]
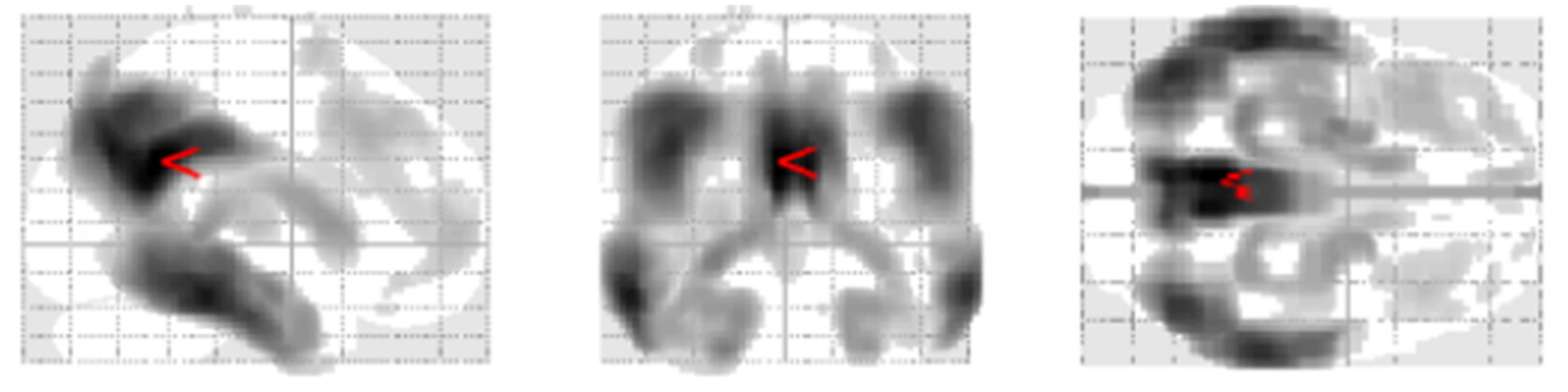
- Minoshima, S.; Frey, K.A.; Koeppe, R.A.; Foster, N.L.; Kuhl, D.E. A diagnostic approach in Alzheimer’s disease using three-dimensional stereotactic surface projections of fluorine-18-FDG PET. J. Nucl. Med. 1995, 36, 1238–1248. [Google Scholar]
- Ishii, K.; Willoch, F.; Minoshima, S.; Drzezga, A.; Ficaro, E.P.; Cross, D.J.; E Kuhl, D.; Schwaiger, M. Statistical brain mapping of 18F-FDG PET in Alzheimer’s disease: Validation of anatomic standardization for atrophied brains. J. Nucl. Med. 2001, 42, 548–557. [Google Scholar]
- Friedland, R.P.; Budinger, T.F.; Ganz, E.; Yano, Y.; Mathis, C.A.; Koss, B.; Ober, B.A.; Huesman, R.H.; Derenzo, S.E. Regional cerebral metabolic alterations in dementia of the Alzheimer type: Positron emission tomography with [18F] fluorodeoxyglucose. J. Comput. Assist. Tomogr. 1983, 7, 590–598. [Google Scholar] [CrossRef]
- McGeer, P.L.; Kamo, H.; Harrop, R.; Li, D.K.; Tuokko, H.; McGeer, E.G.; Adam, M.J.; Ammann, W.; Beattie, B.L.; Calne, D.B. Positron emission tomography in patients with clinically diagnosed Alzheimer’s disease. CMAJ Can. Med. Assoc. J. 1986, 134, 597. [Google Scholar]
- Heiss, W.D.; Szelies, B.; Kessler, J.; Herholz, K. Abnormalities of energy metabolism in Alzheimer’s disease studied with PET. Ann. N. Y. Acad. Sci. 1991, 640, 65–71. [Google Scholar] [CrossRef]
- Herholz, K.; Adams, R.; Kessler, J.; Szelies, B.; Grand, M.; Heiss, W.D. Critieria for the diagnosis of Alzheimer’s disease with PET. Dementia 1990, 1, 156–164. [Google Scholar]
- Prestia, A.; Muscio, C.; Caroli, A.; Frisoni, G.B. Computer-aided diagnostic reporting of FDG PET for the diagnosis of Alzheimer’s disease. Clin. Transl. Imaging 2013, 1, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Kajimura, N.; Nishikawa, M.; Uchiyama, M.; Kato, M.; Watanabe, T.; Nakajima, T.; Hori, T.; Nakabayashi, T.; Sekimoto, M.; Ogawa, K.; et al. Deactivation by benzodiazepine of the basal forebrain and amygdala in normal humans during sleep: A placebo-controlled [15O] H2O PET study. Am. J. Psychiatry 2004, 161, 748–751. [Google Scholar] [CrossRef] [Green Version]
- Nayate, A.P.; Dubroff, J.G.; Schmitt, J.E.; Nasrallah, I.; Kishore, R.; Mankoff, D.; Pryma, D.A. Use of standardized uptake value ratios decreases interreader variability of [18F] florbetapir PET brain scan interpretation. Am. J. Neuroradiol. 2015, 36, 1237–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdette, J.H.; Minoshima, S.; Vander Borght, T.; Tran, D.D.; Kuhl, D.E. Alzheimer disease: Improved visual interpretation of PET images by using three-dimensional stereotaxic surface projections. Radiology 1996, 198, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Marcoux, A.A.; Burgos, N.; Bertrand, A.; Teichmann, M.; Routier, A.; Wen, J.; Samper-González, J.; Bottani, S.; Durrleman, S.; Habert, M.-O.; et al. An Automated Pipeline for the Analysis of PET Data on the Cortical Surface. Front. Neuroinform. 2018, 12, 94. [Google Scholar] [CrossRef] [Green Version]
- Iizuka, T.; Morimoto, K.; Sasaki, Y.; Kameyama, M.; Kurashima, A.; Hayasaka, K.; Ogata, H.; Goto, H. Preventive Effect of Rifampicin on Alzheimer Disease Needs at Least 450 mg Daily for 1 Year: An FDG-PET Follow-Up Study. Dement. Geriatr. Cogn. Disord. Extra 2017, 7, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Daerr, S.; Brendel, M.; Zach, C.; Mille, E.; Schilling, D.; Zacherl, M.J.; Bürger, K.; Danek, A.; Pogarell, O.; Schildan, A.; et al. Evaluation of early-phase [18F]-florbetaben PET acquisition in clinical routine cases. NeuroImage Clin. 2016, 14, 77–86. [Google Scholar] [CrossRef]
- Brendel, M.; Wagner, L.; Levin, J.; Zach, C.; Lindner, S.; Bartenstein, P.; Okamura, N.; Rominger, A. Perfusion-Phase [18F]THK5351 Tau-PET Imaging as a Surrogate Marker for Neurodegeneration. J. Alzheimer’s Dis. Rep. 2017, 1, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Beyer, L.; Nitschmann, A.; Barthel, H.; Van Eimeren, T.; Unterrainer, M.; Sauerbeck, J.; Marek, K.; Song, M.; Palleis, C.; Respondek, G.; et al. Early-phase [18F]PI-2620 tau-PET imaging as a surrogate marker of neuronal injury. Eur. J. Pediatr. 2020, 47, 2911–2922. [Google Scholar] [CrossRef]
- Thientunyakit, T.; Sethanandha, C.; Muangpaisan, W.; Minoshima, S. 3D-SSP analysis for amyloid brain PET imaging using 18F-florbetapir in patients with Alzheimer’s dementia and mild cognitive impairment. Med. J. Malays. 2021, 76, 493–501. [Google Scholar]
- Shlens, J. A tutorial on principal component analysis. arXiv 2014, arXiv:1404.1100. [Google Scholar]
- Illán, I.A.; Górriz, J.M.; Ramírez, J.; Salas-Gonzalez, D.; López, M.M.; Segovia, F.; Chaves, R.; Gómez-Rio, M.; Puntonet, C.G.; The Alzheimer’s Disease Neuroimaging Initiative. 18F-FDG PET imaging analysis for computer aided Alzheimer’s diagnosis. Inf. Sci. 2011, 181, 903–916. [Google Scholar] [CrossRef]
- Moeller, J.R.; Strother, S.C.; Sidtis, J.J.; Rottenberg, D.A. Scaled subprofile model: A statistical approach to the analysis of functional patterns in positron emission tomographic data. J. Cereb. Blood Flow Metab. 1987, 7, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Spetsieris, P.G.; Eidelberg, D. Scaled subprofile modeling of resting state imaging data in Parkinson’s disease: Methodological issues. Neuroimage 2011, 54, 2899–2914. [Google Scholar] [CrossRef] [Green Version]
- Hocurscak, L.; Tomanic, T.; Trost, M.; Simoncic, U. Comparison of statistical parametric mapping method and scaled subprofile model for functional neuroimage analysis. Bull. Am. Phys. Soc. 2021, 66, F15-002. [Google Scholar]
- Spetsieris, P.; Ma, Y.; Peng, S.; Ko, J.H.; Dhawan, V.; Tang, C.C.; Eidelberg, D. Identification of disease-related spatial covariance patterns using neuroimaging data. JoVE 2013, 76, e50319. [Google Scholar] [CrossRef] [Green Version]
- Teune, K.L.; Strijkert, F.; Renken, J.R.; Izaks, J.G.; de Vries, J.J.; Segbers, M.; Roerdink, J.; Dierckx, R.; Leenders, L.K. The Alzheimer’s disease-related glucose metabolic brain pattern. Curr. Alzheimer Res. 2014, 11, 725–732. [Google Scholar] [CrossRef]
- Iizuka, T.; Kameyama, M. Spatial metabolic profiles to discriminate dementia with Lewy bodies from Alzheimer disease. J. Neurol. 2020, 267, 1960–1969. [Google Scholar] [CrossRef]
- Meles, S.K.; Pagani, M.; Arnaldi, D.; De Carli, F.; Dessi, B.; Morbelli, S.; Sambuceti, G.; Jonsson, C.; Leenders, K.L.; Nobili, F. Alzheimer’s disease metabolic brain pattern in mild cognitive impairment. J. Cereb. Blood Flow Metab. 2017, 37, 3643–3648. [Google Scholar] [CrossRef]
- Blazhenets, G.G.; Ma, Y.; Sörensen, A.; Schiller, F.; Rücker, G.; Eidelberg, D.; Frings, L.; Meyer, P.T. Predictive Value of 18F-Florbetapir and 18F-FDG PET for Conversion from Mild Cognitive Impairment to Alzheimer Dementia. J. Nucl. Med. 2019, 61, 597–603. [Google Scholar] [CrossRef]
- Blazhenets, G. Clinical Utility of Principal Components Analysis on PET Data in the Prediction of Alzheimer’s Disease Dementia. Ph.D. Thesis, University of Freiburg, Freiburg, Germany, 2021. [Google Scholar]
- Yokoi, T.; Watanabe, H.; Yamaguchi, H.; Bagarinao, E.; Masuda, M.; Imai, K.; Ogura, A.; Ohdake, R.; Kawabata, K.; Hara, K.; et al. Involvement of the precuneus/posterior cingulate cortex is significant for the development of Alzheimer’s disease: A PET (THK5351, PiB) and resting fMRI study. Front. Aging Neurosci. 2018, 10, 304. [Google Scholar] [CrossRef] [Green Version]
- Perovnik, M.; Tomše, P.; Jamšek, J.; Emeršič, A.; Tang, C.; Eidelberg, D.; Trošt, M. Identification and validation of Alzheimer’s disease-related metabolic brain pattern in biomarker confirmed Alzheimer’s dementia patients. Sci. Rep. 2022, 12, 11752. [Google Scholar] [CrossRef]
- Peretti, D.E.; García, D.V.; Renken, R.J.; Reesink, F.E.; Doorduin, J.; de Jong, B.M.; De Deyn, P.P.; Dierckx, R.A.J.O.; Boellaard, R. Alzheimer’s disease pattern derived from relative cerebral flow as an alternative for the metabolic pattern using SSM/PCA. EJNMMI Res. 2022, 12, 37. [Google Scholar] [CrossRef] [PubMed]
- Boyd, S.; Boyd, S.P.; Vandenberghe, L. Convex Optimization; Cambridge University Press, Cambridge, UK, 2004.
- Pisner, D.A.; Schnyer, D.M. Support vector machine. In Machine Learning; Academic Press: Cambridge, MA, USA, 2020; pp. 101–121. [Google Scholar]
- Illán, I.A.; Górriz, J.M.; López, M.M.; Ramírez, J.; Salas-Gonzalez, D.; Segovia, F.; Chaves, R.; Puntonet, C.G. Computer aided diagnosis of Alzheimer’s disease using component based SVM. Appl. Soft Comput. 2011, 11, 2376–2382. [Google Scholar] [CrossRef]
- Ramírez, J.; Górriz, J.M.; Salas-Gonzalez, D.; Romero, A.; López, M.; Álvarez, I.; Gómez-Río, M. Computer-aided diagnosis of Alzheimer’s type dementia combining support vector machines and discriminant set of features. Inf. Sci. 2013, 237, 59–72. [Google Scholar] [CrossRef]
- Garali, I.; Adel, M.; Bourennane, S.; Guedj, E. Brain region ranking for 18FDG-PET computer-aided diagnosis of Alzheimer’s disease. Biomed. Signal Process. Control 2016, 27, 15–23. [Google Scholar] [CrossRef]
- Hammes, J.; Bischof, G.N.; Bohn, K.P.; Onur, O.; Schneider, A.; Fliessbach, K.; Hoenig, M.C.; Jessen, F.; Neumaier, B.; Drzezga, A.E.; et al. One-Stop Shop: 18F-Flortaucipir PET Differentiates Amyloid-Positive and -Negative Forms of Neurodegenerative Diseases. J. Nucl. Med. 2020, 62, 240–246. [Google Scholar] [CrossRef]
- Damasceno, P.F.; La Joie, R.; Maia, P.D.; Visani, A.; Iaccarino, L.; Strom, A.; Edwards, L.; Tempini, M.L.; Jagust, W.J.; Miller, B.L.; et al. Colocalization of atrophy and tau improves AI classification of Alzheimer phenotypical variants: Tau imaging. Alzheimer’s Dement. 2020, 16, e046258. [Google Scholar] [CrossRef]
- Syaifullah, A.H.; Shiino, A.; Kitahara, H.; Ito, R.; Ishida, M.; Tanigaki, K. Machine learning for diagnosis of AD and prediction of MCI progression from brain MRI using brain anatomical analysis using diffeomorphic deformation. Front. Neurol. 2021, 11, 576029. [Google Scholar] [CrossRef]
- Ding, Y.; Zhao, K.; Che, T.; Du, K.; Sun, H.; Liu, S.; Zheng, Y.; Li, S.; Liu, B.; Liu, Y.; et al. Quantitative Radiomic Features as New Biomarkers for Alzheimer’s Disease: An Amyloid PET Study. Cereb. Cortex 2021, 31, 3950–3961. [Google Scholar] [CrossRef]
- Varatharajah, Y.; Ramanan, V.K.; Iyer, R.; Vemuri, P. Predicting short-term MCI-to-AD progression using imaging, CSF, genetic factors, cognitive resilience, and demographics. Sci. Rep. 2019, 9, 2235. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Yao, Z.; Zheng, W.; Yang, J.; Ding, Z.; Li, M.; Lu, S. Predicting MCI progression with individual metabolic network based on longitudinal FDG-PET. In Proceedings of the 2017 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Kansas City, MO, USA, 13–16 November 2017; pp. 1894–1899. [Google Scholar]
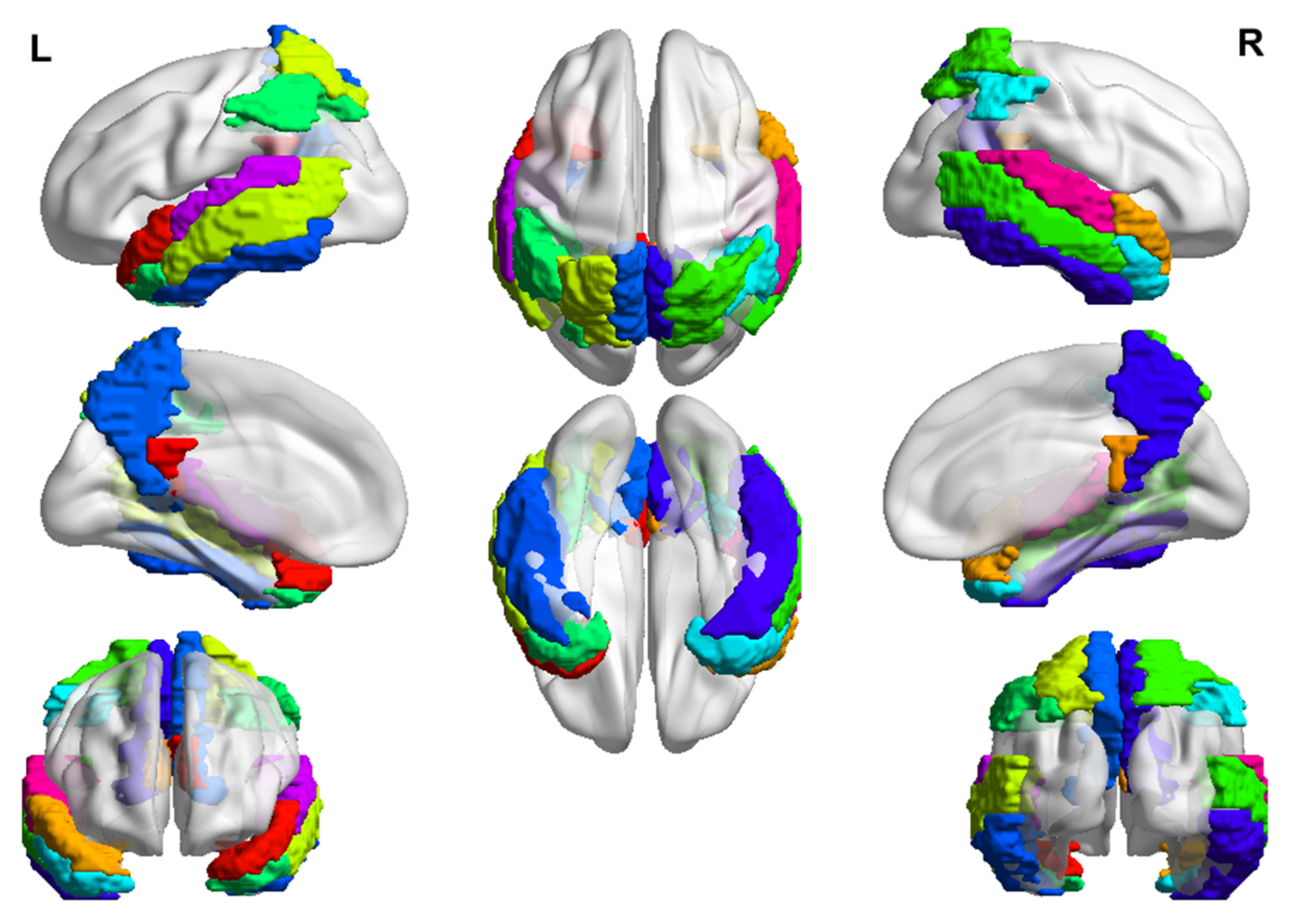
- Fan, L.; Li, H.; Zhuo, J.; Zhang, Y.; Wang, J.; Chen, L.; Yang, Z.; Chu, C.; Xie, S.; Laird, A.R.; et al. The Human Brainnetome Atlas: A New Brain Atlas Based on Connectional Architecture. Cereb. Cortex 2016, 26, 3508–3526. [Google Scholar] [CrossRef] [Green Version]
- Yakushev, I.; Hammers, A.; Fellgiebel, A.; Schmidtmann, I.; Scheurich, A.; Buchholz, H.-G.; Peters, J.; Bartenstein, P.; Lieb, K.; Schreckenberger, M. SPM-based count normalization provides excellent discrimination of mild Alzheimer’s disease and amnestic mild cognitive impairment from healthy aging. NeuroImage 2009, 44, 43–50. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Goodfellow, I.; Bengio, Y.; Courville, A. Deep Learning; MIT Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Sharma, S.; Sharma, S.; Athaiya, A. Activation functions in neural networks. Towards Data Sci. 2017, 6, 310–316. [Google Scholar] [CrossRef]
- Ruder, S. An overview of gradient descent optimization algorithms. arXiv 2016, arXiv:1609.04747. [Google Scholar]
- Medsker, L.R.; Jain, L.C. Recurrent neural networks. Des. Appl. 2001, 5, 64–67. [Google Scholar]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative adversarial networks. Commun. ACM 2020, 63, 139–144. [Google Scholar] [CrossRef]
- Liu, M.; Cheng, D.; Yan, W.; Alzheimer’s Disease Neuroimaging Initiative. Classification of Alzheimer’s disease by combination of convolutional and recurrent neural networks using FDG-PET images. Front. Neuroinform. 2018, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Ruwanpathirana, G.P.; Williams, R.C.; Masters, C.L.; Rowe, C.C.; Johnston, L.A.; Davey, C.E. Mapping the association between tau-PET and Aβ-amyloid-PET using deep learning. Sci. Rep. 2022, 12, 14797. [Google Scholar] [CrossRef]
- Ding, Y.; Sohn, J.H.; Kawczynski, M.G.; Trivedi, H.; Harnish, R.; Jenkins, N.W.; Lituiev, D.; Copeland, T.P.; Aboian, M.S.; Mari Aparici, C.; et al. A deep learning model to predict a diagnosis of Alzheimer disease by using 18F-FDG PET of the brain. Radiology 2019, 290, 456–464. [Google Scholar] [CrossRef]
- Guo, J.; Qiu, W.; Li, X.; Zhao, X.; Guo, N.; Li, Q. Predicting Alzheimer’s disease by hierarchical graph convolution from positron emission tomography imaging. In Proceedings of the 2019 IEEE International Conference on Big Data (Big Data), Los Angeles, CA, USA, 9–12 December 2019; pp. 5359–5363. [Google Scholar]
- Choi, H.; Jin, K.H.; Alzheimer’s Disease Neuroimaging Initiative. Predicting cognitive decline with deep learning of brain metabolism and amyloid imaging. Behav. Brain Res. 2018, 344, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Yee, E.; Popuri, K.; Beg, M.F.; Alzheimer’s Disease Neuroimaging Initiative. Quantifying brain metabolism from FDG-PET images into a probability of Alzheimer’s dementia score. Hum. Brain Mapp. 2020, 41, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Phan, T.L.; Adel, M.; Fossati, C.; Gaidon, T.; Wojak, J.; Guedj, E. Multi-view separable pyramid network for AD prediction at MCI stage by 18 F-FDG brain PET imaging. IEEE Trans. Med. Imaging 2020, 40, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Etminani, K.; Soliman, A.; Davidsson, A.; Chang, J.R.; Martínez-Sanchis, B.; Byttner, S.; Camacho, V.; Bauckneht, M.; Stegeran, R.; Ressner, M.; et al. A 3D deep learning model to predict the diagnosis of dementia with Lewy bodies, Alzheimer’s disease, and mild cognitive impairment using brain 18F-FDG PET. Eur. J. Pediatr. 2021, 49, 563–584. [Google Scholar] [CrossRef] [PubMed]
- Hojjati, S.H.; Babajani-Feremi, A.; Alzheimer’s Disease Neuroimaging Initiative. Prediction and Modeling of Neuropsychological Scores in Alzheimer’s Disease Using Multimodal Neuroimaging Data and Artificial Neural Networks. Front. Comput. Neurosci. 2021, 15, 769982. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, H.G.; Choi, H.; Lee, D.S. Distinct subtypes of spatial brain metabolism patterns in Alzheimer’s disease identified by deep learning based FDG PET clusters. Alzheimer’s Res. Ther. 2021, 13, 49. [Google Scholar] [CrossRef]
- Jo, T.; Nho, K.; Risacher, S.L.; Saykin, A.J. Deep learning detection of informative features in tau PET for Alzheimer’s disease classification. BMC Bioinform. 2020, 21, 496. [Google Scholar] [CrossRef]
- Bach, S.; Binder, A.; Montavon, G.; Klauschen, F.; Müller, K.R.; Samek, W. On pixel-wise explanations for non-linear classifier decisions by layer-wise relevance propagation. PLoS ONE 2015, 10, e0130140. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Popuri, K.; Ding, G.W.; Balachandar, R.; Beg, M.F.; Alzheimer’s Disease Neuroimaging Initiative. Multiscale deep neural network-based analysis of FDG-PET images for the early diagnosis of Alzheimer’s disease. Med. Image Anal. 2018, 46, 26–34. [Google Scholar] [CrossRef]
- Shen, T.; Jiang, J.; Lu, J.; Wang, M.; Zuo, C.; Yu, Z.; Yan, Z. Predicting Alzheimer disease from mild cognitive impairment with a deep belief network based on 18F-FDG-PET images. Mol. Imaging 2019, 18, 1536012119877285. [Google Scholar] [CrossRef]
- Zhang, Z.; Beck, M.W.; Winkler, D.A.; Huang, B.; Sibanda, W.; Goyal, H. Opening the black box of neural networks: Methods for interpreting neural network models in clinical applications. Ann. Transl. Med. 2018, 6, 216. [Google Scholar] [CrossRef]
- Selvaraju, R.R.; Cogswell, M.; Das, A.; Vedantam, R.; Parikh, D.; Batra, D. Grad-cam: Visual explanations from deep networks via gradient-based localization. In Proceedings of the IEEE International Conference on Computer Vision, Venice, Italy, 22–29 October 2017; pp. 618–626. [Google Scholar]
- Ghazal, T.M.; Issa, G. Alzheimer disease detection empowered with transfer learning. Comput. Mater. Contin. 2022, 70, 5005–5019. [Google Scholar] [CrossRef]
- Ashraf, A.; Naz, S.; Shirazi, S.H.; Razzak, I.; Parsad, M. Deep transfer learning for Alzheimer neurological disorder detection. Multimed. Tools Appl. 2021, 80, 30117–30142. [Google Scholar] [CrossRef]
- Aderghal, K.; Khvostikov, A.; Krylov, A.; Benois-Pineau, J.; Afdel, K.; Catheline, G. Classification of Alzheimer disease on imaging modalities with deep CNNs using cross-modal transfer learning. In Proceedings of the 2018 IEEE 31st International Symposium on Computer-Based Medical Systems (CBMS), Karlstad, Sweden, 18–21 June 2018; pp. 345–350. [Google Scholar]
- Aderghal, K.; Afdel, K.; Benois-Pineau, J.; Catheline, G.; Alzheimer’s Disease Neuroimaging Initiative. Improving Alzheimer’s stage categorization with Convolutional Neural Network using transfer learning and different magnetic resonance imaging modalities. Heliyon 2020, 6, e05652. [Google Scholar] [CrossRef]
- Doersch, C. Tutorial on variational autoencoders. arXiv 2016, arXiv:1606.05908. [Google Scholar]
- Penning, J.; John, R.; Chandler, H.; Fielding, P.; Marshall, C.; Smith, R. Generative Adversarial Network” Steerability” for Brain PET Image Generation. In Proceedings of the 2021 IEEE Nuclear Science Symposium and Medical Imaging Conference (NSS/MIC), Piscataway, NJ, USA, 16–23 October 2021; pp. 1–4. [Google Scholar]
- John, R.; Penning, J.; Chandler, H.; Fielding, P.; Marshall, C.; Smith, R. Quantitative Evaluation of Synthesized Brain PET Using a Variational Autoencoder. In Proceedings of the 2021 IEEE Nuclear Science Symposium and Medical Imaging Conference (NSS/MIC), Piscataway, NJ, USA, 16–23 October 2021; pp. 1–4. [Google Scholar]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Ioffe, S.; Szegedy, C. Batch normalization: Accelerating deep network training by reducing internal covariate shift. In Proceedings of the International Conference on Machine Learning (PMLR), Lille, France, 6–11 July 2015; pp. 448–456. [Google Scholar]
- Wan, L.; Zeiler, M.; Zhang, S.; Le Cun, Y.; Fergus, R. Regularization of neural networks using dropconnect. In Proceedings of the International Conference on Machine Learning (PMLR), Atlanta, GA, USA, 16–21 June 2013; pp. 1058–1066. [Google Scholar]
- Lee, C.; Landgrebe, D.A. Decision boundary feature extraction for neural networks. IEEE Trans. Neural Netw. 1997, 8, 75–83. [Google Scholar]
- Mao, J.; Jain, A.K. Artificial neural networks for feature extraction and multivariate data projection. IEEE Trans. Neural Netw. 1995, 6, 296–317. [Google Scholar]
- Chen, Y.; Jiang, H.; Li, C.; Jia, X.; Ghamisi, P. Deep feature extraction and classification of hyperspectral images based on convolutional neural networks. IEEE Trans. Geosci. Remote Sens. 2016, 54, 6232–6251. [Google Scholar] [CrossRef] [Green Version]
- Nixon, M.; Aguado, A. Feature Extraction and Image Processing for Computer Vision; Academic Press: Cambridge, MA, USA, 2019. [Google Scholar]
- Egmont-Petersen, M.; de Ridder, D.; Handels, H. Image processing with neural networks—A review. Pattern Recognit. 2002, 35, 2279–2301. [Google Scholar] [CrossRef]
- Wachinger, C.; Reuter, M.; Klein, T. DeepNAT: Deep convolutional neural network for segmenting neuroanatomy. NeuroImage 2018, 170, 434–445. [Google Scholar] [CrossRef] [PubMed]
- De Brebisson, A.; Montana, G. Deep neural networks for anatomical brain segmentation. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition (CVPR) Workshops, Boston, MA, USA, 7–12 June 2015; pp. 20–28. [Google Scholar]
- Hunter, C.A.; Kirson, N.Y.; Desai, U.; Cummings, A.K.G.; Faries, D.E.; Birnbaum, H.G. Medical costs of Alzheimer’s disease misdiagnosis among US Medicare beneficiaries. Alzheimer’s Dement. 2015, 11, 887–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, R.C.; Aisen, P.S.; Beckett, L.A.; Donohue, M.C.; Gamst, A.C.; Harvey, D.J.; Jack, C.R., Jr.; Jagust, W.J.; Shaw, L.M.; Toga, A.W.; et al. Alzheimer’s disease neuroimaging initiative (ADNI): Clinical characterization. Neurology 2010, 74, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Iwatsubo, T. Japanese Alzheimer’s Disease Neuroimaging Initiative: Present status and future. Alzheimer’s Dement. 2010, 6, 297–299. [Google Scholar] [CrossRef]
- Carrillo, M.C.; Bain, L.J.; Frisoni, G.B.; Weiner, M.W. Worldwide Alzheimer’s disease neuroimaging initiative. Alzheimer’s Dement. 2012, 8, 337–342. [Google Scholar] [CrossRef]
- Frisoni, G.B. Alzheimer’s disease neuroimaging initiative in Europe. Alzheimer’s Dement. 2010, 6, 280–285. [Google Scholar] [CrossRef]
- Cavedo, E.; Redolfi, A.; Angeloni, F.; Babiloni, C.; Lizio, R.; Chiapparini, L.; Bruzzone, M.G.; Aquino, D.; Sabatini, U.; Alesiani, M.; et al. The Italian Alzheimer’s Disease Neuroimaging Initiative (I-ADNI): Validation of Structural MR Imaging. J. Alzheimer’s Dis. 2014, 40, 941–952. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author/Reference | Methodology | Task | Performance |
|---|---|---|---|
| Katako et al. [85] | GLM | AD vs. HC | SENS = 85.6% SPEC = 86.2% AUC = 0.922 |
| Ottoy et al. [87] | GLM | MCI to AD conversion | SENS = 92% SPEC = 96% |
| Katako et al. [85] | PCA/SSM—single PC PCA/SSM—many PCs | AD vs. HC AD vs. HC | SENS = 80.2% SPEC = 78.7% SENS = 86.5% SPEC = 81.9% |
| Teune et al. [114] | PCA/SSM | AD vs. HC | SENS = 93% SPEC = 94% |
| Meles et al. [116] | PCA/SSM | pMCI vs. HC | SENS = 82.4% SPEC = 85.7% |
| Yokoi et al. [119] | PCA/SSM | AD vs. HC | SENS = 79.1% SPEC = 82.6% |
| Perovnik et al. [120] | PCA/SSM | AD vs. HC AD vs. MCI & bvFTD AD vs. DLB | AUC = 0.95 AUC = 0.76–0.85 AUC = 0.87 |
| Katako et al. [85] | SVM—ISDA | AD vs. HC | SENS = 0.84 SPEC = 0.955 AUC = 0.945 |
| Ilan et al. [124] | PCA-SVM | AD vs. HC | SENS = 88.64% SPEC = 87.70% ACC = 88.24% |
| Ramirez et al. [125] | SVM | AD vs. HC | ACC = 90.38% |
| Garali et al. [126] | SVM | AD vs. HC | ACC = 95.07% |
| Damasceno et al. [128] | SVM | AD vs. MCI vs. non-AD | AUC = 0.9 |
| Svaifullah et al. [129] | SVM | MCI to AD conversion | SENS = 81.7% SPEC = 90.1% ACC = 87.2% AUC = 0.94 |
| Ding et al. [130] | SVM | AD vs. HC pMCI vs. sMCI | AUC = 0.93 AUC = 0.83 |
| Varatharajah et al. [131] | SVM—linear kernel | MCI to AD conversion | SENS = 93% SPEC = 77% ACC = 81% AUC = 0.93 |
| Zhao et al. [132] | SVM | sMCI vs. pMCI | ACC = 89.9% AUC = 0.892 |
| Liu et al. [141] | NN—RNN | AD vs. HC MCI vs. HC | SENS = 91.4% SPEC = 91% ACC = 91.2% AUC = 0.953 SENS = 78.1% SPEC = 80% ACC = 78.9% AUC = 0.839 |
| Ding et al. [143] | NN—CNN | AD identification MCI identification non-AD/non-MCI identification | AUC = 0.93 AUC = 0.63 AUC = 0.73 |
| Guo et al. [144] | NN—Graph CNN | AD vs. HC AD vs. MCI vs. HC | ACC = 93% ACC = 77% |
| Choi & Jin [145] | NN—3D CNN | AD vs. HC pMCI vs. sMCI | ACC = 93% ACC = 84.2% |
| Yee et al. [146] | NN—Residual CNN | AD vs. HC pMCI vs. sMCI | ACC = 93.5% AUC = 0.976 ACC = 74.7% AUC = 0.811 |
| Pan et al. [147] | NN—Pyramidal CNN | MCI to AD conversion | ACC = 83.05% |
| Etmani et al. [148] | NN—3D CNN | DLB identification AD identification MCI identification | AUC = 0.962 AUC = 0.964 AUC = 0.714 |
| Choi et al. [150] | NN—3D CNN | MCI to AD conversion | AUC = 0.89 |
| Jo et al. [152] | NN—3D CNN | AD vs. HC MCI vs. HC | SENS = 95.4% SPEC = 96.9% ACC = 96.2% SENS = 48.2% SPEC = 82.4% ACC = 64.2% |
| Lu et al. [154] | NN—Multi-scale CNN | pMCI vs. sMCI | ACC = 82.51% |
| Shen et al. [155] | NN—SVM-CNN | MCI to AD conversion | ACC = 86.6% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perron, J.; Ko, J.H. Review of Quantitative Methods for the Detection of Alzheimer’s Disease with Positron Emission Tomography. Appl. Sci. 2022, 12, 11463. https://doi.org/10.3390/app122211463
Perron J, Ko JH. Review of Quantitative Methods for the Detection of Alzheimer’s Disease with Positron Emission Tomography. Applied Sciences. 2022; 12(22):11463. https://doi.org/10.3390/app122211463
Chicago/Turabian StylePerron, Jarrad, and Ji Hyun Ko. 2022. "Review of Quantitative Methods for the Detection of Alzheimer’s Disease with Positron Emission Tomography" Applied Sciences 12, no. 22: 11463. https://doi.org/10.3390/app122211463