1. Introduction
Benzene, toluene, ethylbenzene, and xylene, commonly known as BTEX, are monoaromatic hydrocarbons that are major components of gasolines [
1]. These compounds are said to be the most common contaminant of soils and ground water owing to frequent leakage and accidental spillage [
2]. On the other hand, polycyclic aromatic hydrocarbons (PAHs) are important pollutants which always find their way into the environment through various human activities such as the burning of fossil fuels, oil spillages, and sometimes during vehicle repairs [
3]. The release of PAH and BTEX to surrounding air is worrisome because of the associated hazards on people, animal, and plants [
2]. Exposures to fossil fuels, BTEX, and other hydrocarbon contaminants is known to disrupt agricultural lands, contaminate the surrounding air, and may induce cancer and other mutations in aquatic organisms [
1,
4]. The deleterious effects of the release of BTEX and other hydrocarbons to the environments make remediation processes inevitable. Several remediation processes have been employed for the cleaning of BTEX and PAHs from a contaminated environment. Among such approaches are the use of chemicals and biological methods. Biological approaches are effective measures for the cleaning of sites contaminated by aromatic hydrocarbons [
5,
6]. The degradation of hydrocarbon is hampered by their different molecular structures [
7]. However, some microbes possessed the capacity to degrade both high and low molecular weight hydrocarbons [
8]. Similarly, bacteria and other microbes have been demonstrated as effective tools in the cleaning of BTEX-polluted environments [
6]. These bacteria with good potentials for the degradation of BTEX and PAHs could be tools for the modern bioremediation of areas contaminated by hydrocarbons. Bacteria that have been employed for bioremediation purposes include member of the genera Stenotrophomonas, Pseudomonas, Franconibacter, Bacillus, Enterobacter, and Sphingomonad among others [
9,
10,
11]. Although there were arrays of bacteria with the capacity to grow in and degrade aromatic hydrocarbons, some genera have continuously been reported to be highly important. One of such genera is the genus Sphingomonas [
10]. Sphingomonas and some other genera are commonly referred to as the Sphingomonads.
The Sphingomonads belongs to the family Sphingomonadaceae which was proposed by Yabuuchi et al., 1990 [
12,
13]. The family was later subdivided by Takeuchi et al. [
14,
15] into four genera: Sphingomonas, Sphingobium, Novosphingobium, and Sphingopyxis. Members of these genera are commonly found in nature as endophyte and rhizospheres’ bacteria [
16]. There is an increased attention on these genera in recent times because they play important roles in the remediation of pollutants. They do so by using these pollutants as substrates for growth [
17]. Among the Sphingomonads with promising potential for both biodegradation and bioremediation of pollutants are the Sphingobium and Sphingomonas [
18,
19]. Many Sphingobium species have been reported to have the ability to degrade different pollutants including hydrocarbons [
16]. The significant roles played by Sphingobium in the degradation of hydrocarbon emphasize the importance of continuous studies on them. Such studies can further unravel yet-to-be-identified mechanisms employed by members of this genus in the utilization of hydrocarbons as substrates for growth.
Genome sequence analysis has been used to unravel many underlining factors responsible for bacteria behavior. To understand more about the biology of Sphingobium yanoikuyae S72 and its detailed architecture with respect to the degradation of a hydrocarbon, we sequenced and analyzed the complete genome sequence of Sphingobium yanoikuyae S72. In this study, Sphingobium yanoikuyae S72 was isolated from the rhizosphere of sorghum sample collected from the northeastern part of Mexico (25.983333 latitude, 198.1 longitude, and altitude 120 m a.s.l.). This strain grew effectively in aromatic hydrocarbon supplemented medium. Its potential to degrade aromatic hydrocarbon was evaluated in a hydrocarbon degradation assay. The complete genome was sequenced and analyzed to understand the molecular mechanisms responsible for its survival in the tested hydrocarbons.
2. Materials and Methods
2.1. Bacterial Isolation and Characterization
S72 was isolated from the rhizosphere of a sorghum plant by a colleague in the lab in a previous study [
20]. One gram of soil sample recovered from sorghum plant rhizosphere was added to 10 mL of 0.85% saline solution in a 50 mL falcon tube. The mixture was strongly agitated on a vortex and then serially diluted in sterile distilled water to the factor of 10
−4. Then, 0.1 mL of each diluent was inoculated on an already prepared Tryptic soy agar (TY) and incubated at 30 °C for 24–48 h. The isolates which appeared on the plate were purified by subculturing individual colonies in a freshly prepared TY agar and incubated in the condition previously mentioned. The purified colonies which appeared on plates were identified morphologically with the electron microscope, biochemically using the API
®20NE bacterial identification kit (bioMerieux, 200, Durham, NC, USA), and molecularly by amplifying the 16S rRNA fragment of the bacterial genome. The API 20NE gallery system, used for biochemical characterization, contains a gallery made up of series of microtubes with dehydrated media and substrates. This system is basically for the identification of non-enteric Gram-negative bacilli and some members of the genus Sphingomonads [
21]. The biochemical test was carried out as specified by the manufacturer of the API 2ONE (bioMerieux sa). The galleries were incubated at 30 °C, and the results observed at 24 and 48 h. Selection of the positive and negative tests was made based on the API 20 gallery color chart (apiweb, bioMerieux, 2010). Subsequently, the results were compared with the API and ID 32 identification database (bioMeerieux) and with biochemical characteristics for
Sphingobium yanoikuyae [
13].
The genomic DNA for the bacterial identification was extracted from an overnight broth culture of the suspected colonies with the wizard Promega genomic DNA extraction kit (USA) according to the manufacturer’s instruction. The extracted genomic DNA was amplified with the bacterial 16S rRNA universal primer 27f (5′-GAGAGTTTGATCCTGGCTCAG-3′) and 1495r (5′-CTACGGCTACCTTGTTACGA-3′) [
22] which amplifies about 1500 bp of the 16S rRNA fragment. The amplified region was sequenced and blast check analysis on the NCBI database was carried out to determine the identity of the bacteria.
2.2. Preparation of Hydrocarbon Supplemented Media for Degradation Assay
The hydrocarbon tolerance test was carried out using nutrient broths that had been supplemented with five different concentrations (20, 40, 60, 80, and 100 µg/mL) of hydrocarbons. The broths were then inoculated with isolated bacteria (OD: 0.05) and incubated at 30 °C for 25 days in a rotatory incubator at a revolution of 180 rpm. Prior to adding hydrocarbons to the medium, the hydrocarbons were first dissolved in dimethyl chloride, and then introduced into the culture medium at the desired concentration. Tolerance was checked every day using colony forming unit per mL. The experimental set-ups involved positive control (nutrient broth without hydrocarbon) and negative control (uninoculated nutrient broth).
Survival of hydrocarbons was checked daily by measuring bacterial OD with spectrophotometer at a wavelength of 600 nm. All experimental setups were carried out in 250-mL Erlenmeyer flasks containing 40 mL of Bushnell Haas broth (BH) supplemented with 100 µg/mL of each tested hydrocarbon (naphthalene, phenanthrene, biphenyl, toluene, or xylene). The medium was inoculated with 1 mL of S. yanoikuyae S72, (OD: 0.05). Medium devoid of hydrocarbons served as negative control. Finally, the experimental setups were incubated in a rotatory incubator with revolutions of 180 rpm at 30 °C and monitored daily for bacterial growth until the 25th day of the experiment.
2.3. Bacterial Growth in and Degradation of Hydrocarbons
S. yanoikuyae S72 was inoculated in different Bushnell Haas (BH) agar media (composition: magnesium, 0.20 g; calcium chloride, 0.02; monopotassium phosphate, 1.00 g; dipotassium phosphate, 1.00 g; ammonium nitrate, 1.00 g; ferric chloride, 0.05 g; agar, 20.0 g) already supplemented with either biphenyl, toluene, xylene, naphthalene, or phenanthrene, at different concentrations of 20, 40, 80, 100, and 150 µg/mL for each hydrocarbon. The final bacterial concentration in the medium was approximately 3.0 × 107 CFU/mL as determined in the laboratory. Each culture was incubated at 30 °C for 72 h. The highest concentration of each hydrocarbon tolerated by S72 was selected as the concentration for the degradation studies of each hydrocarbon used in this study.
The hydrocarbon-degrading test was carried out in 5 different BH liquid media with each containing one out of the five hydrocarbons (naphthalene, phenanthrene, biphenyl, toluene, or xylene) as the sole carbon source. A 1 mL inoculum of S. yanoikuyae S72 was added to 40 mL of each of the 5 Bushnell Haas broths (BH) already supplemented with one of the hydrocarbons unless otherwise stated, in a 250 mL flask. The culture was then incubated in a rotatory incubator at a revolution of 180 rpm at 30 °C for 25 days. The bacterial growth was checked every 48 h and growth was determined using the optical density readings obtained from the spectrophotometer. A control containing each of the hydrocarbons tested in liquid medium without a bacteria inoculum was used as the blank for each assay. The cultures were withdrawn on the 12th and 25th day to determine the extent of hydrocarbon degradation by the isolate. The metabolites formed from the degradation of the hydrocarbons were determined with a gas column chromatography–mass spectrophotometry (GC-MS) analysis.
2.4. GC-MS Chromatography Analysis
For the GC-MS analysis, an Agilent Technologies brand gas chromatograph, model 6890N (Net Work GC system), equipped with a DB-5 column, 5%-phenyl-methyl-polysiloxane (Agilent Technologies, Santa Clara, CA, USA), 60 m long, 0.25 mm in internal diameter, and 0.25 µm thick film was used. The starting temperature was 50 °C, which was maintained for 5 min; subsequently the temperature was raised to 280 °C using a heating ramp of 20 °C/min for 20 min. Helium was used as carrier gas at a flow rate of 1 mL/min, the injector temperature was 250 °C, split injection, with a split ratio of 50:1. Once the chromatogram was obtained, the identification of each of the peaks was carried out by mass spectrometry using an Agilent Technologies model 5975 inert XL mass spectrometer. Mass spectra were obtained by electron impact ionization at 70 eV; for identification, the mass spectra obtained for each compound were compared with a database (HP Chemstation-NIST 05 Mass Spectral search program, version 2.0d), in addition to the comparison with a standard, analyzed under the same conditions, which was used as an external standard for quantification.
The GC-MS analysis began with the separation of the organic phase from the aqueous phase of the culture medium in a rotatory funnel. The culture medium (40 mL) was poured in a separating funnel to which 20 mL of HPLC grade hexane (J.T. Baker
® Fisher, Scientific, Waltham, MA, USA) was added. The funnels were manually shaken for 10 min. Funnels were kept at rest until the two phases were clearly separated. From the upper phase containing the hydrocarbon, 1 mL aliquot was transferred to 1.5 mL dark vials. This was used for the analysis of the metabolite formed in a GC-MS machine (Agilent Technologies
® Model 6890 N). Once the chromatograms were obtained, each of the chromatographic peaks was identified by mass spectrometry, using a mass detector (Agilent Technologies
® Mod. 5975 inert XL). The mass spectra observed in the metabolites were confirmed with the HP Chemstation-NIST 05 Mass Spectral Search Program, version 2.0d software. The spectra were compared with the standard for each hydrocarbon and the spectra from the control in which no bacterium was inoculated under the same condition. Each type of hydrocarbon was separately analyzed, and each treatment had five replicates in the experiment. The experiments were repeated three times. Data were analyzed using the SAS statistical program [
23].
2.5. Genome Sequencing and Analysis of S. yanoikuyae S72
We sequenced the genome of S72 to fully understand the genetic basis underlying its ability to use the tested hydrocarbons as substrate, and the mechanism associated with the observed degradation. As previously stated, genomic DNA was extracted with Promega
® Wizard Genomic DNA purification kit (Promega Corp., Madison, WI, USA) as per the manufacturer’s guideline. The whole genome was sequenced with the Illumina MiSeq™ platform (Illumina Inc., San Diego, CA, USA). The reads obtained from the genome assembly were trimmed with trim-galore, which also checks the quality of the read with its embedded fastqc. The genome assembly was carried out in 2 stages: (i) de novo assembly with the Spades and Velvet Genome Assembler [
24,
25] and (ii) reference-based assembly. The assembled genome obtained from the two softwares (SPAdes and Velvet) were compared using the Metassembler software which suggested the best assembly method among the software used and produced a consensus assembly. The consensus assembly was then used for the reference-based assembly [
26]. The reference-based assembly was used to reduce the genome to 1 contig. This approach employed the alignment of the consensus assembly with Sphingomonas genome strain ATCC512030 retrieved from NCBI database using the Nucmer software and the Consed suite [
27,
28]. The assembled genome was annotated with Prokka (version 1.12) and NCBI prokaryotic genome annotation pipeline (PGAAP) that carried out automatic gene prediction and annotation [
29,
30]. RNAmmer [
31] and tRNAscan [
32] were used for the identification of rRNA and tRNA, respectively. The KEGG and orthologous gene cluster analysis was carried out on the web based PATRIC server [
33]. Alternatively, COGs for unique genes were assigned with BlastP against the COGs database downloaded from NCBI. The bacterial pan genome analysis (BPGA) tool was used for the analysis of the isolate’s pan genome [
34]. The Average Nucleotide Identity based on Mummer (ANIm) was determined with PYANI [
35]. The genomic island in S72 was identified with an online web-based Island viewer4 [
36] and pairwise analysis on Nucmer [
28]. The deep gene sequence analysis for the identification of the genes that were associated with the degradation of hydrocarbon and comparative genomics such as COG abundance was carried out on the JGI-IMG database [
37].
3. Results
S72 is a Gram-negative rod and moves by means of a monotrichous flagellum. It is 1.0–3.5 µm in length and 0.6–1 µm in width (
Figure S1). S72 effectively use
d-glucose,
l-arabinose, mannose,
n-acetyl glucosamine, maltose, potassium gluconate, malic acid, citrate, galactose, citrate, and esculin for its growth. However, it could not use mannitol, adipic acid, capric acid, phenyl acetic acid as a substrate for its growth (
Table 1).
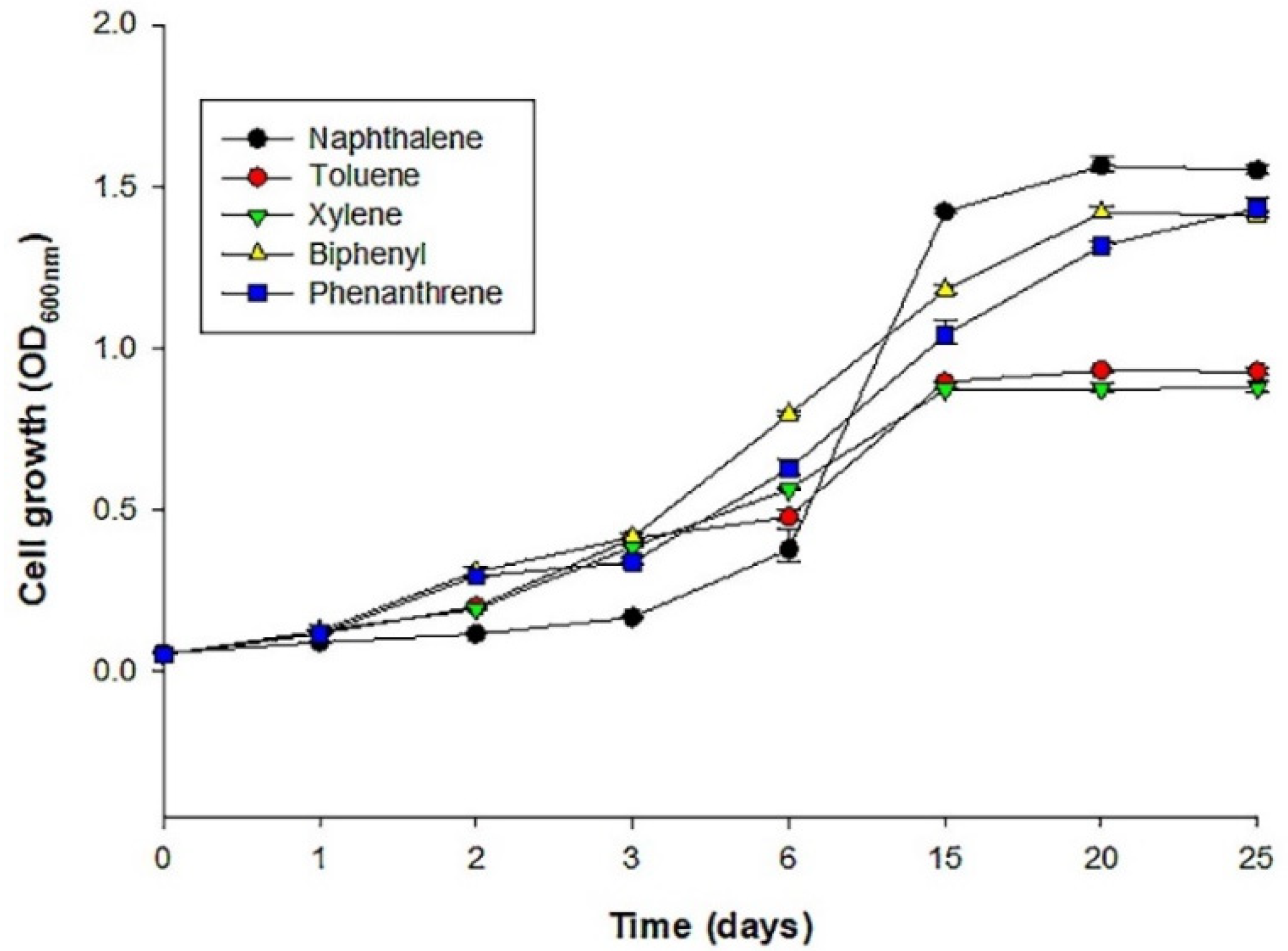
The preliminary analyses showed that S72 can grow in the minimal media (BH) supplemented with either monoaromatic (xylene, toluene) or polycyclic aromatic hydrocarbons (PAH) (biphenyl, phenanthrene, and naphthalene). The number of colonies forming units appearing on the plates decreased as the concentration (0, 20, 40, 80 100, and 150 µg/mL) of the hydrocarbon in the BH medium increased. The preliminary analysis also revealed that S72 has better tolerance for the hydrocarbons at a concentration that is ≤100 µg/mL, and their growth began to decline as the concentrations of the hydrocarbons get higher than 100 µg/mL. S72 did not show any growth when inoculated in BH medium containing 150 µg/mL toluene or xylene and showed limited growth in plate with naphthalene, phenanthrene, or biphenyl at this concentration. Moreover, the rate at which S72 grew in medium containing toluene and xylene is lower than other hydrocarbons tested at 100 µg/mL (
Figure 1). Based on the reported observations 100 µg/mL was selected as the study concentration for the experiment.
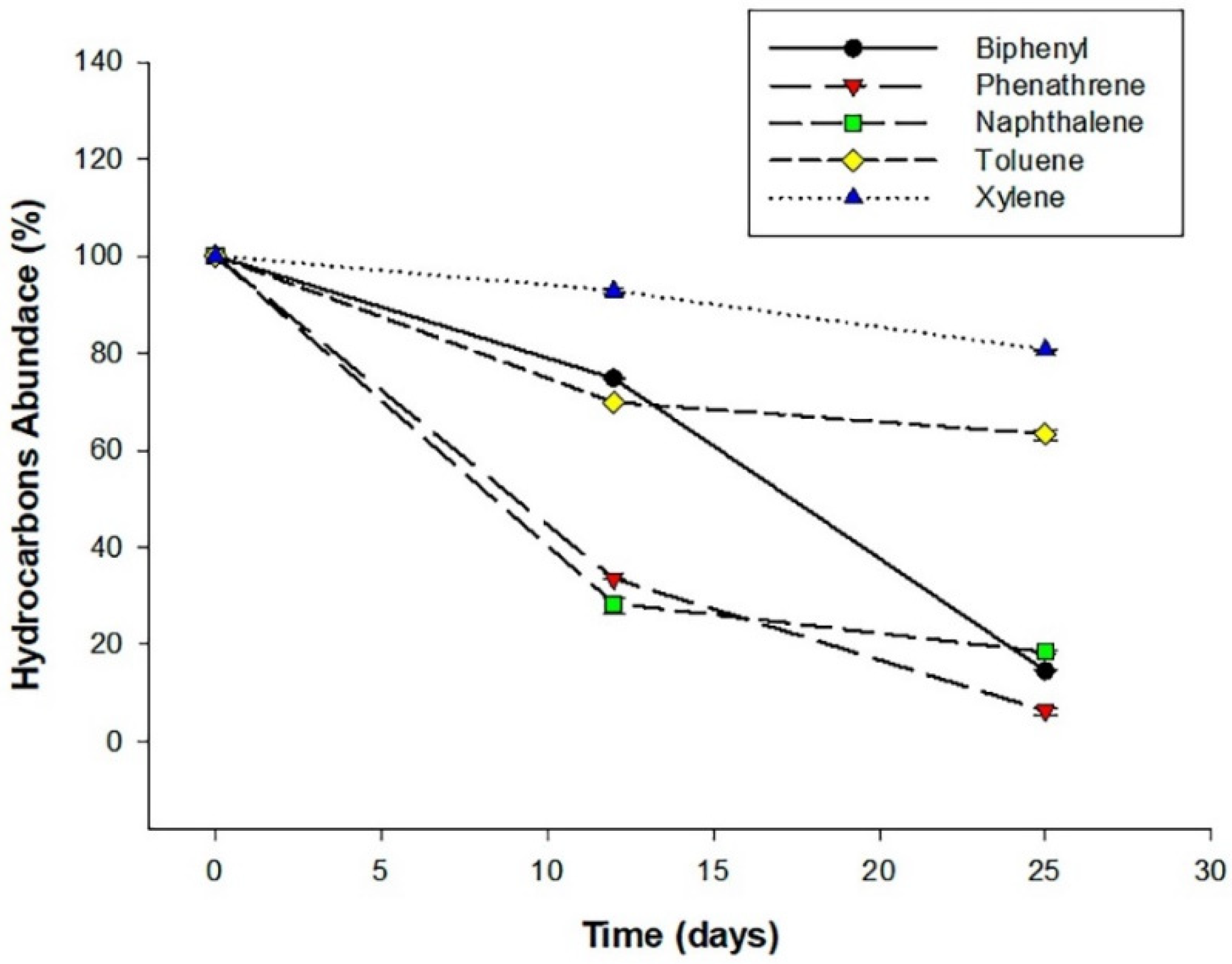
S72 degraded all the hydrocarbons tested at 100 µg/mL with varying efficiencies. S72 showed the best activity in phenanthrene, resulting in 93% degradation. The degradation of biphenyl and naphthalene was up to 85 and 81% reduction, respectively (
Figure 2).
However, a low degradation efficiency was observed in the media containing xylene (19% and 31%). The degradation activity was confirmed by the reduction in the peak for the hydrocarbons as compared to the control from the GC-MS analysis (
Figure S2).
Genomic analysis has been used often to unravel several details about bacterial behavior. As a result of this, the genome of S72 was sequenced and analyzed to relate its function with its genetic constituent. The sequenced genome of
S. yanoikuyae S72 was reduced to 1 contig, consisting of 5,532,633 bp, 5005 CDS, and 4 identical copies of the rRNA gene operon (23S, 16S, and 5S). The overall G + C content of the assembled genome is 64.23%. The genome has 5231 putative genes and 75 RNAs (67 tRNA and 12 rRNA). A total of 1140 genes are in operons, 2515 are parts of the clusters of orthologous gene (COGs) (
Table 2).
S72 has some COG categories more in abundance than some other Sphingobium species, which has been previously reported. These COG categories include energy production and conversion (COG C, 6.35), carbohydrate transport and metabolism (COG G, 6.20%), lipid transport and metabolism (COG I, 6.99), secondary metabolites biosynthesis (COG Q, 4.52%), general function prediction only in transport and catabolism (COG R, 9.93) (
Table S1).
In a related analysis S72 was found to have higher COG abundance than 17 other hydrocarbon-degrading bacteria retrieved from the IMG database (
Table 3).
There are more genes in abundance in the COG categories mobilome: prophages, transposons (COG X, 2.03%), secondary metabolites biosynthesis, transport and catabolism (COG Q, 4.52%), carbohydrate transport and metabolism (COG G, 6.20%), lipid transport and metabolism (COG I, 6.99%), cell wall/membrane/envelope biogenesis (COG M, 6.58%), replication, recombination, and repair (COG L, 3.33%), inorganic ion transport and metabolism (COG P, 6.22%), transcription (COG K, 7.8%), posttranslational modification, protein turnover, chaperones (COG O, 4.27%), and general function prediction only (COG R, 9.93%). The deep sequence analysis of S72 showed that it possesses 37 genes in different COG categories which are associated with the degradation of xenobiotics and polycyclic aromatic hydrocarbons. These genes include 2 pyrone-4-6-decarboxylase hydrolase (A6768_17510, COG R) and 4 carboxy-2-hydroxy muconic semialdehyde dehydrogenase (A6768_17470, COG R). Others include S-(hydroxymethyl) glutathione dehydrogenase/alcohol dehydrogenase (A6768_05785, COG R) that is associated with the degradation of naphthalene and chloroalkane). Alcohol dehydrogenase (cytochrome c) (A6768_00755, COG G), aldehyde dehydrogenase (NAD+) (A6768_10925, COG E), aldo/keto oxidoreductase (A6768_11370, COG R), phenylacetaldehyde dehydrogenase (A6768_11850, COG C) known to participate in the degradation of fluorobenzoate toluene and naphthalene are also present in S72 [
3,
8]. Other hydrocarbon degrading genes identified in S72 include propanol-preferring alcohol dehydrogenase (A6768_17370, EC:1.1.1.1), carboxymethylenebutenolidase (A6768_23915), catechol 1,2-dioxygenase (A6768_16795), muconate cycloisomerase (A6768_16805), aryl-alcohol dehydrogenase (A6768_11850), 2-keto-4-pentenoate hydratase (A6768_14055), oxalocrotonate tautomerase (A6768_04785) benzoate 1,2-dioxygenase alpha subunit (A6768_16790), benzoate/toluate 1,2-dioxygenase beta subunit (Ben B), dihydroxy cyclohexadiene carboxylate dehydrogenase (Ben D). Similarly, a gene encoding the lactoylglutathione lyase was found in S72. The lactoylglutathione lyase enzyme is known to be actively involved in the cleavage of aromatic bond in many aromatic hydrocarbons [
38]. Among the observed genes in S72 are genes essential for the degradation of toluene. The genes include four genes encoding 3-hydroxyacyl-CoA dehydrogenase (EC:1.1.1.35), one gene for aryl-alcohol dehydrogenase (EC:1.1.1.90), and a gene for catechol 1,2-dioxygenase (EC:1.13.11.1). Others include two genes encoding the enzyme oxidoreductases (EC:1.14.13) which often act on paired donors, with incorporation or reduction of molecular oxygen. Three copies of the genes encoding ferredoxin—NAD (+) reductase (EC:1.18.1.3), seven copies of acyltransferases (EC:2.3.1), three copies of carboxymethylenebutenolidase, and one copy of muconate cycloisomerase are present in S72’s genome.
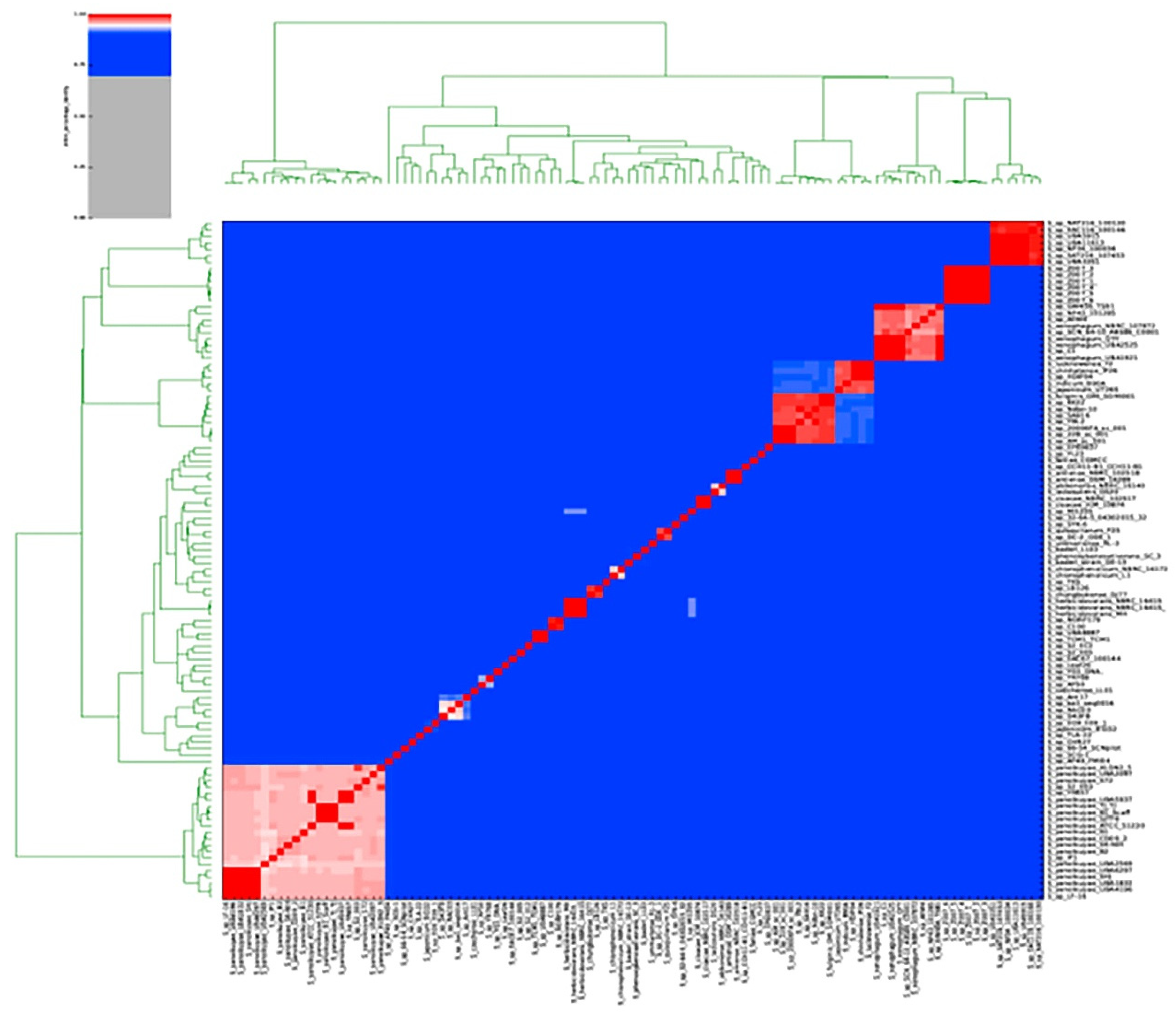
S72 relatedness to other
Sphingobium strain (105) was evaluated on PYANI (Pritchard et al. 2016) using the average nucleotide identity mummer (ANIm) comparison measure. The ANIm result showed that S72 is closely related to
S. yanoikuyae strain UBA2097 sharing 97% average identity with it (
Figure 3). The analysis of S72 pan genome showed that it shared 1734 core genome with other
Sphingobium species and possesses 403 unique genes. We noted that 126 of the unique genes are associated with the catabolism of xenobiotics. Twenty of the unique genes have been previously reported to be involved in the degradation of toluene, xylene, ethylbenzene, biphenyl, benzoate, naphthalene, anthracene, tetrachloroethene, 1,4-dichlorobenzene, bisphenol, and trinitrotoluene in bacteria. These genes are involved in both the catabolism of central aromatic intermediate and the peripheral catabolic pathway for aromatic hydrocarbon (
Table S1).
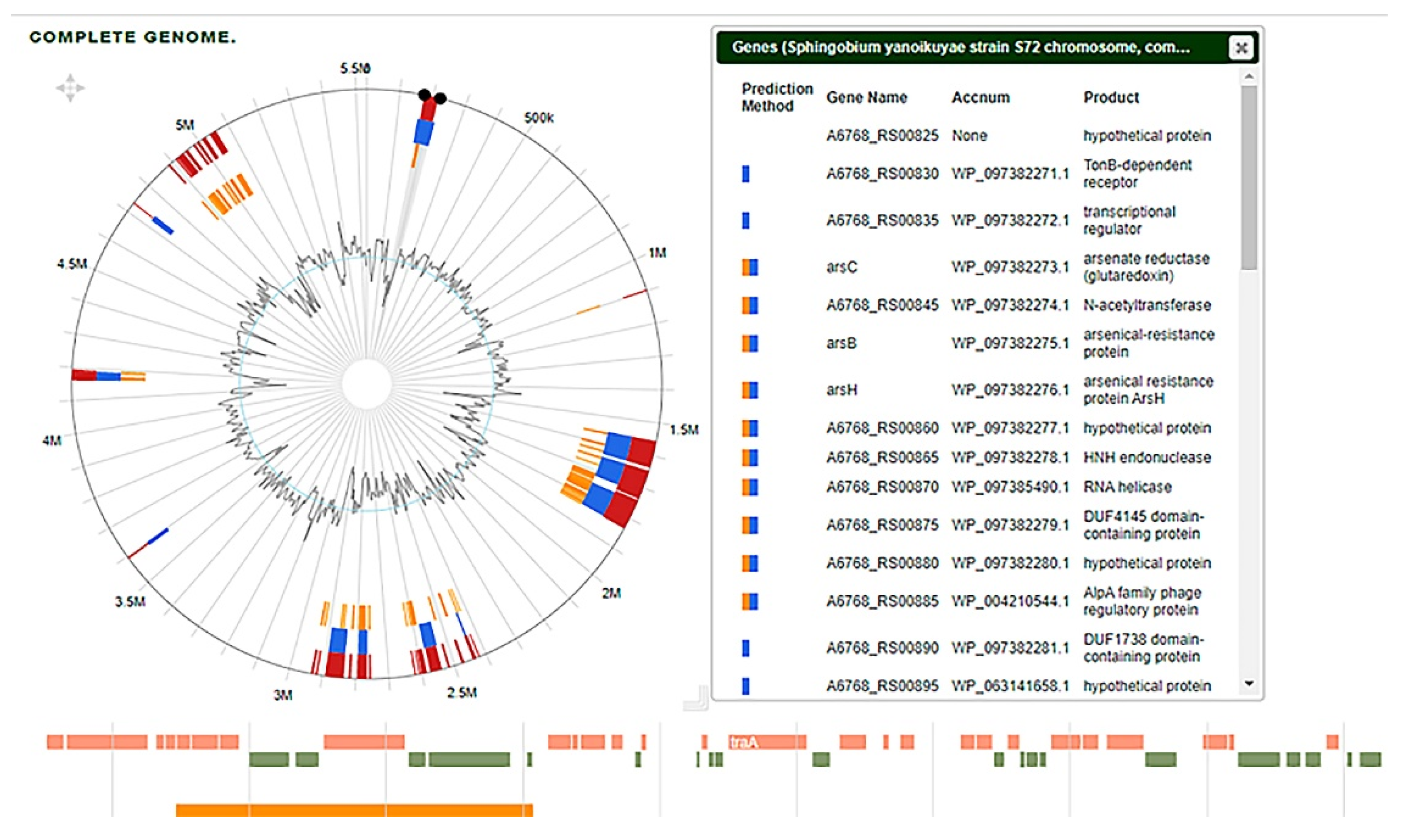
Horizontal gene transfer is a common phenomenon through which bacteria often acquire some genes that are essential for their survival [
3]. The acquired genes are commonly found on the genomic islands in bacteria or as mobile genetic elements. The analysis of the S72’s genome with the online based Island viewer 4.0 revealed that it has 37 genomic islands (
Figure 4). Five large regions were found in the genomic island with regions II and III being the largest consisting of 317,308 kb and 579,060 kb in length, respectively. An in-depth look into the genomic island showed the presence of many genes that are associated with the degradation of xenobiotics and hydrocarbons. Among such genes are SDR family NAD(P)-dependent oxidoreductase (A6768_07840), aldo/keto reductase (A6768_07825), cytochrome P450 (A6768_11830), 4-hydroxybenzoate 3-monooxygenase (A6768_12975), and aromatic alcohol reductase (A6768_RS13080). The other hydrocarbon degrading genes found on the genomic island can be seen in the
Supplementary File.
4. Discussion
The biochemical characteristics and the molecular identification of S72 confirmed that it is a
S. yanoikuyae strain. It grew effectively in the tested hydrocarbon concentration until 100 µg/mL, implying that S72 can use the tested hydrocarbon as substrates for growth. S72’s growth rate, however, decreased as the hydrocarbon’s concentration increased. The reduction in growth as shown in
Figure 1 could be due to increased toxicity that is associated with increased hydrocarbon concentration. This toxicity may be due to the reduction in rate of solubility that may occur as hydrocarbon concentration increases. Previous study has shown that as concentration of hydrocarbon increases, the rate of solubility decreases resulting in the reduction in growth for bacteria [
39]. Low solubility could make it difficult for bacteria to utilize certain compounds as a nutrient for growth [
40].
S72 successfully degraded the tested hydrocarbons during the 25-day degradation study. S72 was able to degrade phenanthrene by 93% at a concentration of 100 µg/mL (
Figure 3). This result is similar to a previous report where
Sphingobium chlorophenolicum C3R degraded phenanthrene by 65% [
19]. Hence, S72 can degrade phenanthrene but has a lower efficacy compared to
Sphingobium chlorophenolicum C3R because of S72 inactivity in phenanthrene at a concentration higher than 100 µg/mL. S72’s ability to degrade hydrocarbon is not limited to phenanthrene (PHE), it also degraded biphenyl and naphthalene by 85 and 81%, respectively. This may imply that S72 had learnt to survive in and use different hydrocarbon as substrates for growth. S72 degradation efficiency is slightly weaker than what was reported for
Burkholderia fungorum FM-2 which degraded PHE by 65% after 3 days [
19] because the effective degradation of phenanthrene by S72 took more than 3 days. However, it has an advantage of surviving in PHE and ensuring its degradation after 25 days. On the other hand, S72 did not effectively degrade xylene and toluene. The in-depth analysis of the genome of S72 showed that it lacked some genes that are essential for the complete metabolism of toluene. The absence of these genes may be responsible for S72 inability to effectively degrade toluene or xylene. It has only four genes that that are directly associated with the degradation of toluene (aryl-alcohol dehydrogenase [EC:1.1.1.90], oxidoreductases acting on the CH-OH group of donors with NAD+ or NADP+ as acceptor, and muconate cycloisomerase [EC:5.5.1.1] a lyase which isomerizes 3 methyl
cis muconate to 3 methyl muconolactone. The other two genes encode catechol 1, 2 dioxygenase and carboxymethylenebutenolidase [EC:3.1.1.45]. Catechol 1, 2 dioxygenase catalyzes the decyclizing of the aromatic ring of methyl catechol to generate 3-methyl-cis, cis-hexadienedioate, while carboxymethylenebutenolidase [EC:3.1.1.45] is acting as dienelactone hydrolase converting 3-chloro-2-methyldienelactone to 3-Chloro-2-methylmaleylacetate in the breakdown of toluene). This observation is same for the genes required for the degradation of xylene as reported by the KEGG database.
The molecular mechanisms responsible for the survival of S72 in and degradation of hydrocarbons was evaluated by the sequencing of its genome. The genomic analysis of the S72 genome show the presence of some genes which are essential for the degradation of hydrocarbons. Genes such as S-(hydroxymethyl) glutathione dehydrogenase/alcohol dehydrogenase (A6768_05785, COG R) which is known to enhance the degradation of hydrocarbon by catalyzing the oxidation of long chain primary alcohol (Uniprot). S-(hydroxymethyl) glutathione dehydrogenase/alcohol dehydrogenase has been previously reported to be associated with the degradation of naphthalene [
8,
40]. Thus, the presence of S-(hydroxymethyl) glutathione dehydrogenase/alcohol dehydrogenase in S72 could be associated with its need to degrade naphthalene as a substrate for growth. We also noted several copies of other essential genes for the degradation of hydrocarbon. For example, there were 18 genes that are associated with the cytochrome P450 superfamily. The cytochrome P450 is a monooxygenase, which is involved in the degradation of hydrocarbon. A recent report showed that cytochrome P450 can play important role in the degradation of naphthalene and pyrene [
41]. The presence of cytochrome P450 monooxygenase (CP450) in S72 may be due to its role in helping S72 in the degradation of hydrocarbons. CP450′s importance in S72 is evident with the presence of many of its copies in S72. Eighteen genes which are member of CP450 superfamily were found in S72. The role of the unique CP450 gene could not be fully explained here but it is likely that they are involved in some pathways that are associated with the degradation of hydrocarbon.
Similarly, several genes which have been linked with the degradation of tetrachloroethane were seen in the genome of S72. These genes include nitrilotriacetate monooxygenase (A6768_04895, EC 1.14.13.), aldehyde dehydrogenase (A6768_04870, EC 1.2.1.3), and alcohol dehydrogenase (A6768_22375, EC 1.1.1.1). There are 22 genes in these three categories that have been associated with the degradation of tetrachloroethane. Tetrachloroethane (CCl4) and trichloroethanes are organic solvents commonly used for laundries and metal cleaning; as a result, they are often found as environmental pollutants [
42]. No aerobic bacteria have been reported to possess the ability to degrade CCl4 [
43]. The presence of genes needed to degrade CCl4 may suggest that S72 may be effective for the decontamination of an area polluted by CCl4. S72’s ability to grow in and degrade naphthalene is associated with the presence of some genes in its genome. The genome has six genes which are involved in the pathway for the degradation of naphthalene (alcohol dehydrogenase, aldehyde dehydrogenase, and nitrilotriacetate monooxygenase). Although these genes are not the only ones that are needed in the pathway for the degradation of naphthalene, their presence in S72’s genome may be the reason for its naphthalene degradation. More so, there are several hypothetical genes in S72 which are in the same operon with some genes that take part in the degradation of hydrocarbons (
Figure S3) and, as such, may be complementing the roles of the identified genes in the degradation of naphthalene (
Figure S3). Most genes in this region had been previously reported to play an essential role in the adaptation to hydrocarbon environment, uptake of hydrocarbon, utilization, and degradation of hydrocarbon. They include the lysR family protein gene which is known to play significant role in the regulation of bacterial adaptation to stress and the use of hydrocarbon. Pal et al. (2017) reported that lysR are part of the two-component system in bacteria through which they sense and respond to the presence of PAH in their environment [
8].
Comparative genomic analysis of S72 showed that it is more closely related to the
S. yanoikuyae strain UBA2097 because it shares 97% average nucleotide identity with it. Further analysis on its genome showed that it has higher abundance in some cog categories (mobilome: prophages, transposons (COG X), secondary metabolites biosynthesis, transport and catabolism (COG Q), carbohydrate transport and metabolism (COG G), lipid transport and metabolism (COG I), cell wall/membrane/envelope biogenesis (COG M), replication, recombination, and repair (COG L), inorganic ion transport and metabolism (COG P), transcription (COG K), posttranslational modification, protein turnover, chaperones (COG O), and general function prediction only (COG R) when compared with previously described hydrocarbon degrading bacteria retrieved from the JGI_IMG database. Many enzymes that are essential for the degradation of hydrocarbon by bacteria have been reported to be encoded by genes in some of the COG categories that were found in abundance in S72 [
40]. For example, COG category K, 307 genes belonging to this category were present in S72. The genes encoding the LTTR family are part of the genes in this category, LTTR are important regulators in bacteria helping them to cope effectively in their environment. A total of 49 LTTR encoding genes were identified in S72; this observation is similar to the findings from the study of Pal et al. on Franconibacter, another hydrocarbon-degrading gene which possess a higher number of LTTR family proteins in its genome [
8]. Binnewies et al. had previously reported that the LTTR family plays important role in regulating genes responsible for aromatic compound catabolism, motility of cell, and quorum sensing [
44]. The abundance of the LTTR gene family in S72 implies that it is highly metabolically active and may employ these genes in the detection of and regulation of its response to hydrocarbons. Twenty-one proteins of the OmpR family transcriptional regulator of the two components system, six Mer family regulator protein, twenty-one proteins belonged to the multiple antibiotic resistance regulator (MarR) family (COG1846) [
45]. S72 has 41 TetR regulation protein (COG1309 and COG3226), and 14 GntR family transcriptional regulators protein (COG1167, COG1802, COG2186 and COG2188). These transcription regulators are known to play essential roles in the degradation of aromatic hydrocarbon [
38]. Other transcriptional regulating protein in S72 include 23 AraC family (COG, 2207, COG4977, COG1609) known to be involved in the metabolism of different sugars [
46], 8 proteins of the IclR family transcriptional regulator (COG1414), 2 proteins in the AsnC transcriptional regulator, and 8 proteins of the ArsR family transcriptional regulator. S72 also has genes in the two-component system which are responding to the limitation of phosphate in the environment. It has a Pho R and Pho B genes which are phosphate regulon regulator in the OmpR family which can play an essential role in the phosphate transport system [
8].
S72 also has in abundance genes in the COG category for mobilome: prophages, transposons (COG X). This COG category comprises genes that were acquired via horizontal gene transfer. Some studies have shown that bacterial adaptation to polycyclic aromatic hydrocarbon could be mediated by an acquired mobile genetic element such as transposons, integron, plasmids, or even prophages [
47]. There are about 80 genes in this COG category. The high abundance of genes from the COG X category could be an indication that certain genes, which are essential for the degradation of hydrocarbons, may be part of the mobilome, prophage, or transposon.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}