
The Limitations of 5f Delocalization and Dispersion

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
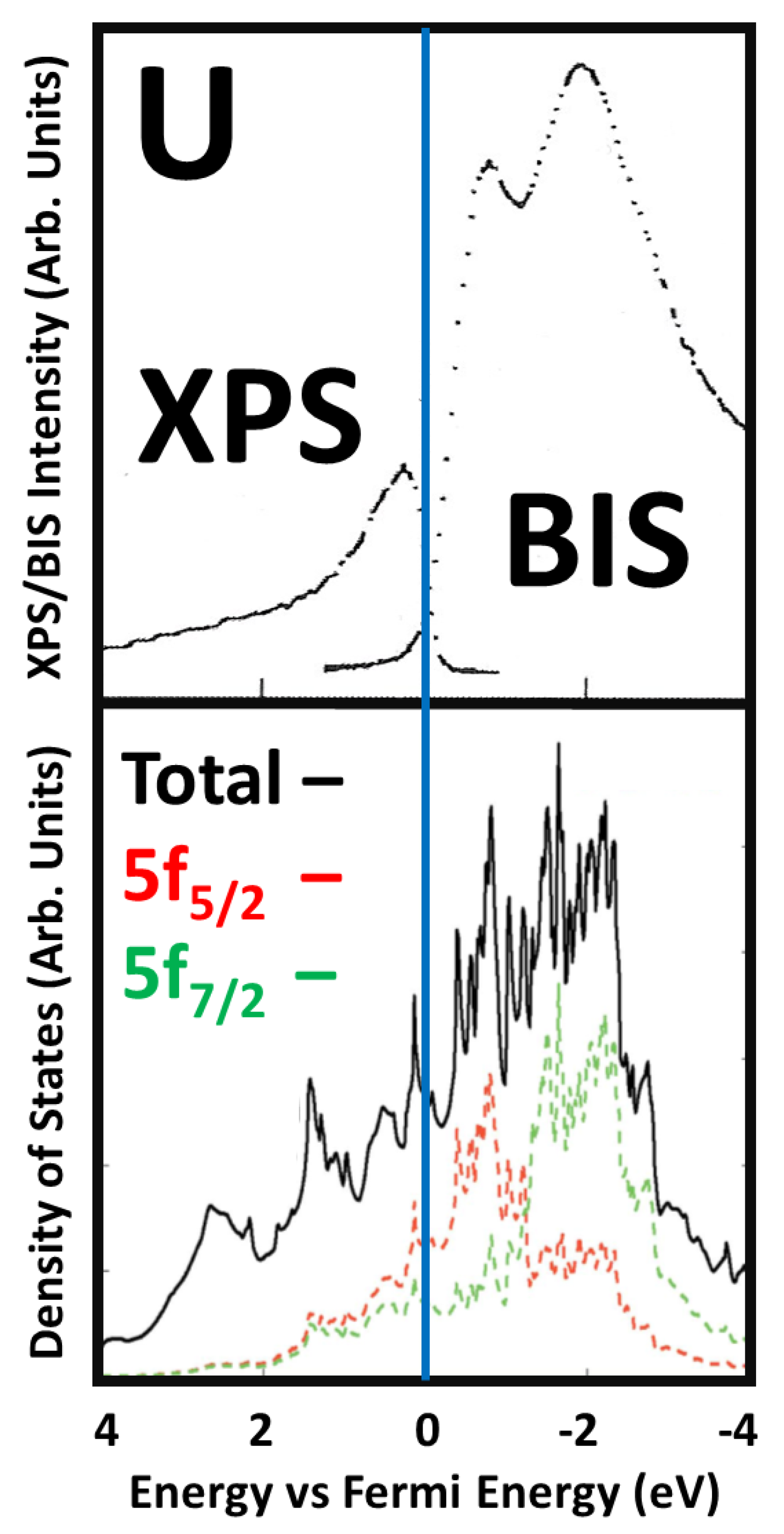
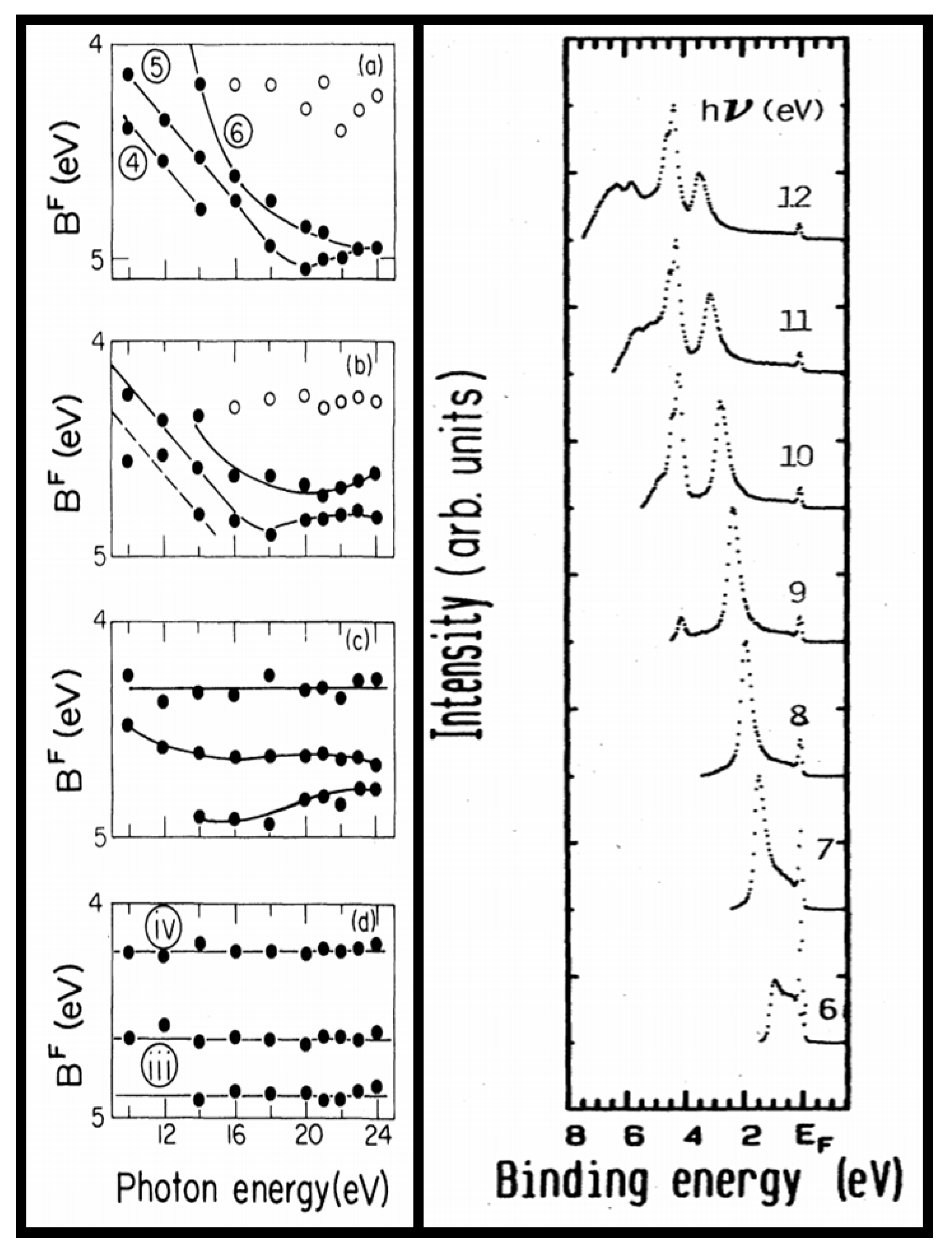
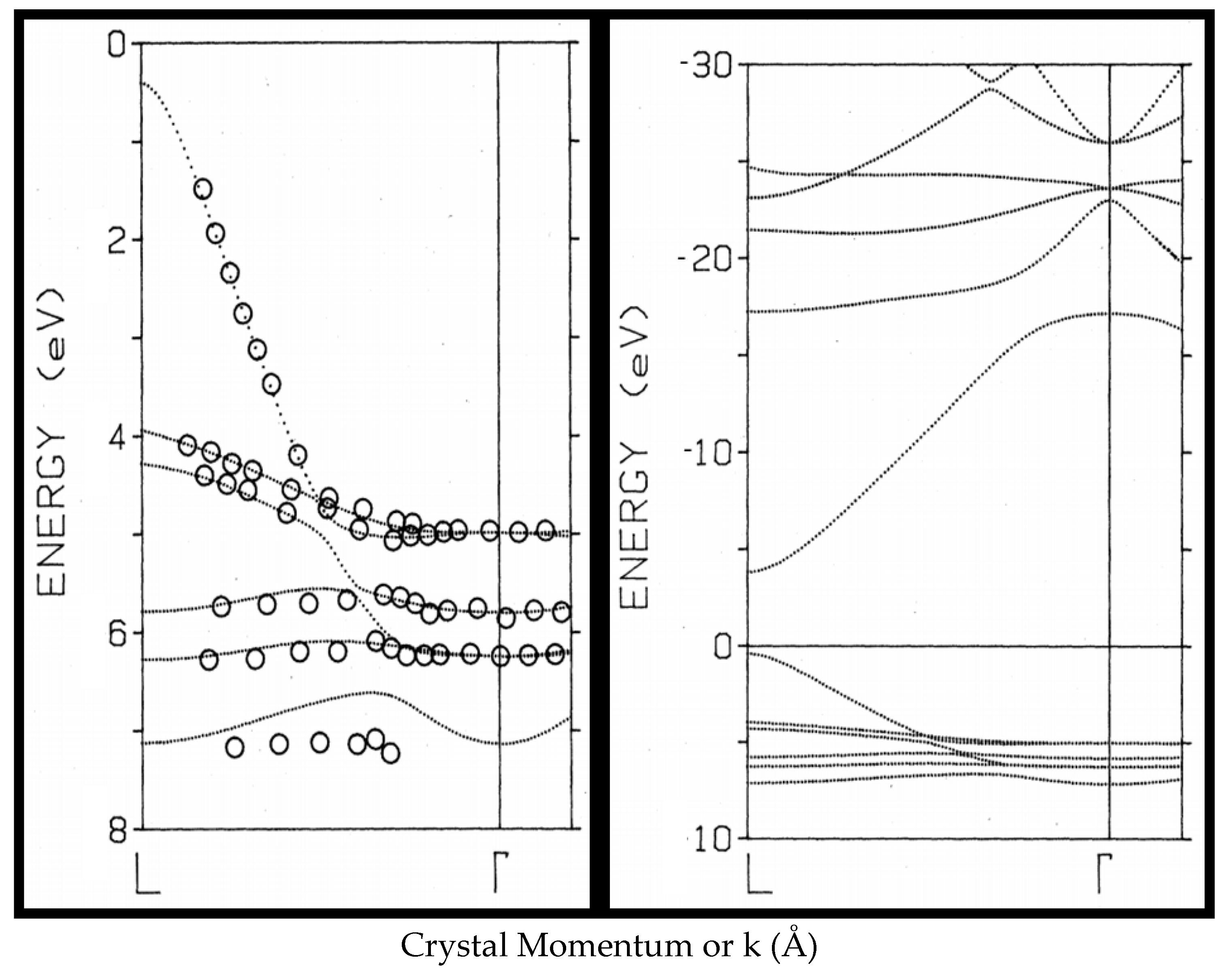
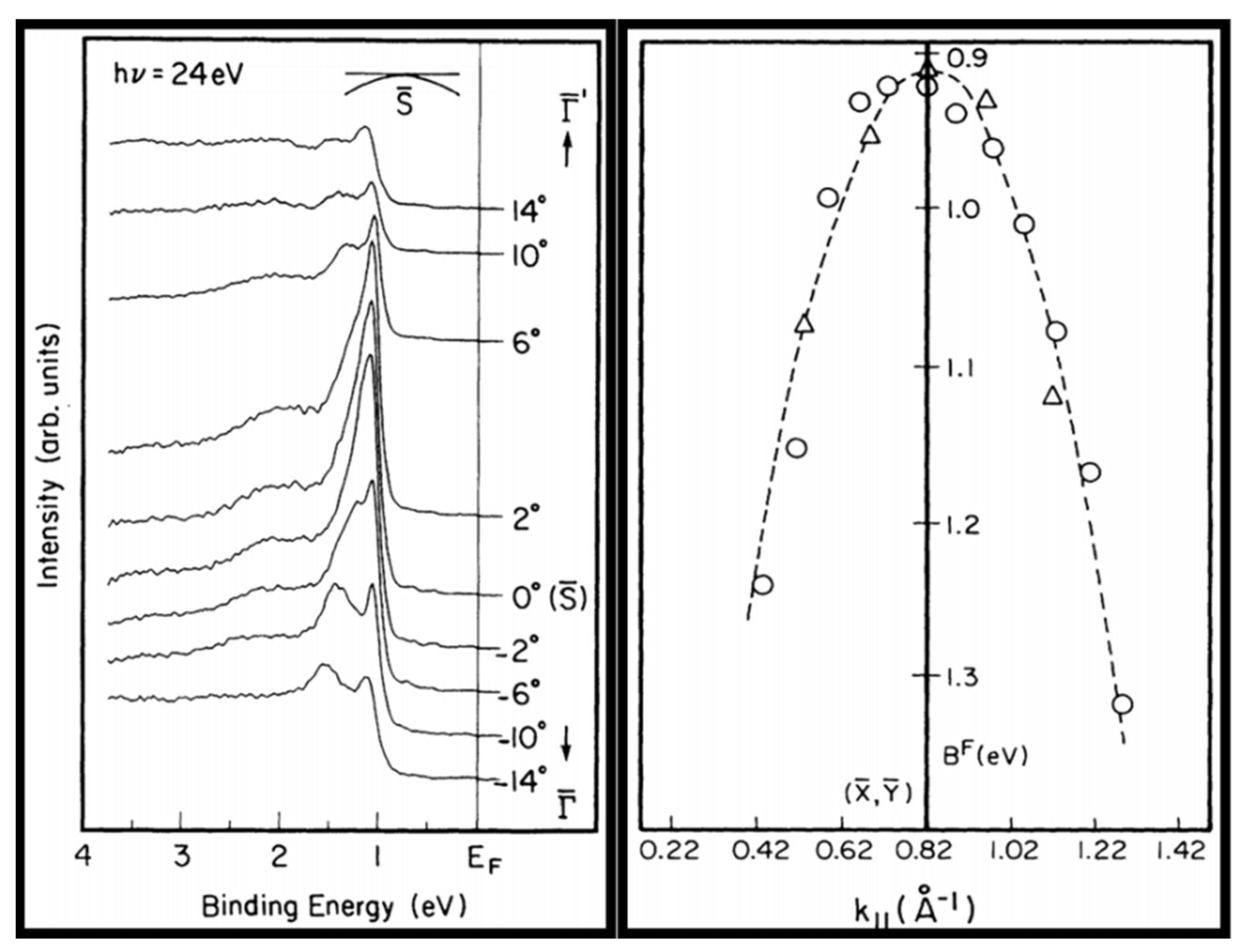
3. Digression on ARPES
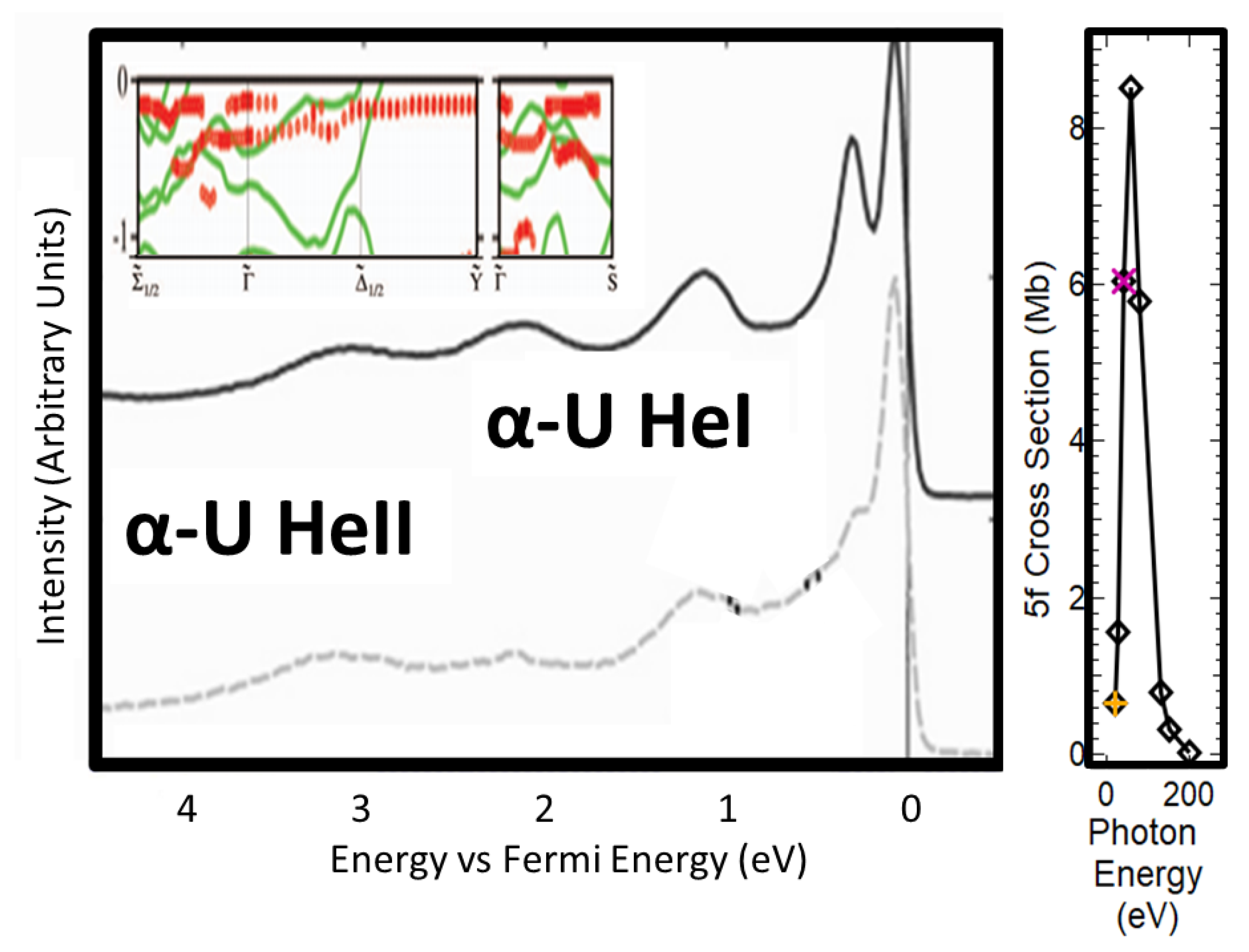
4. ARPES of α-U
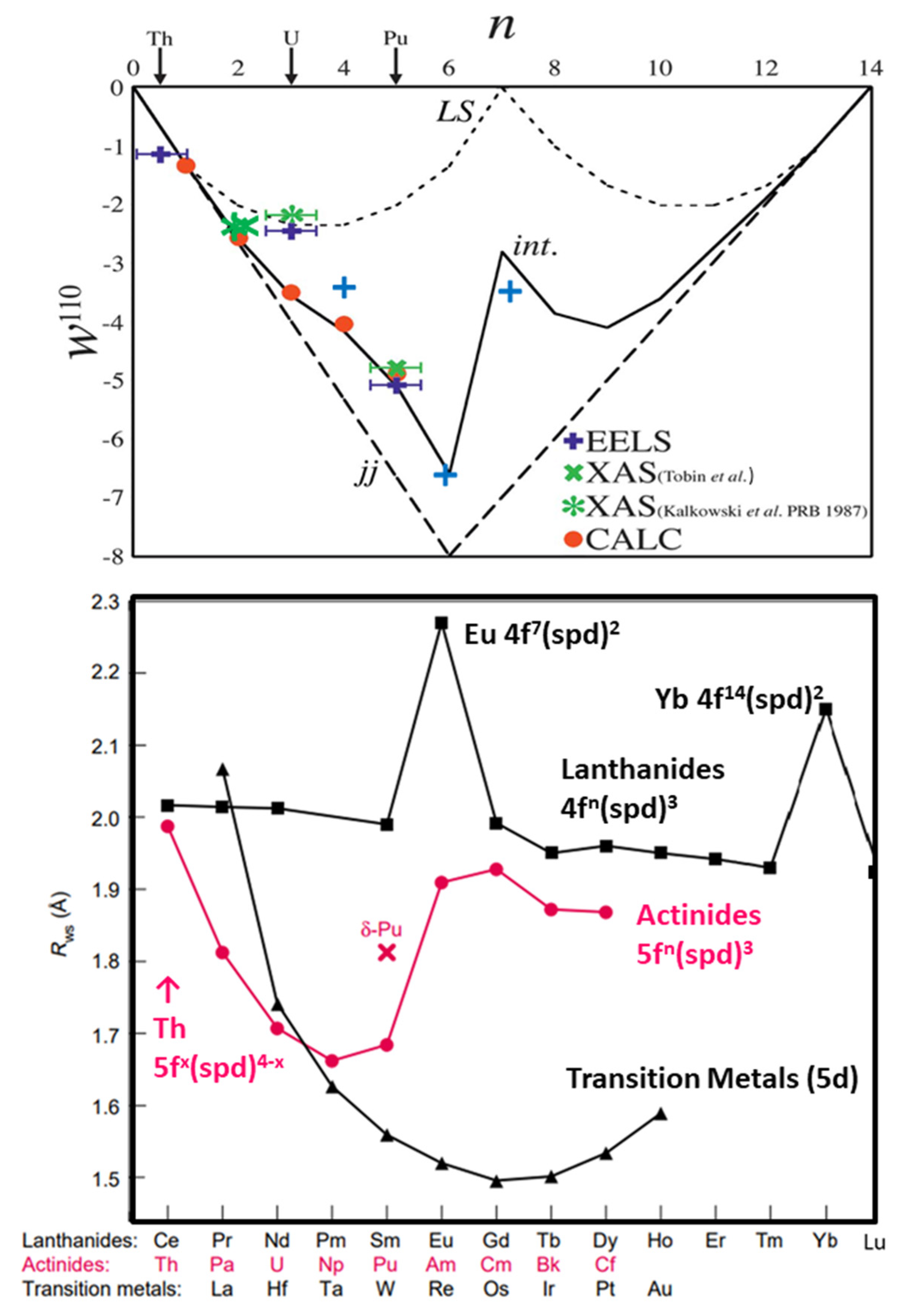
5. X-ray Absorption Spectroscopy: Angular Momentum Coupling and Perturbations
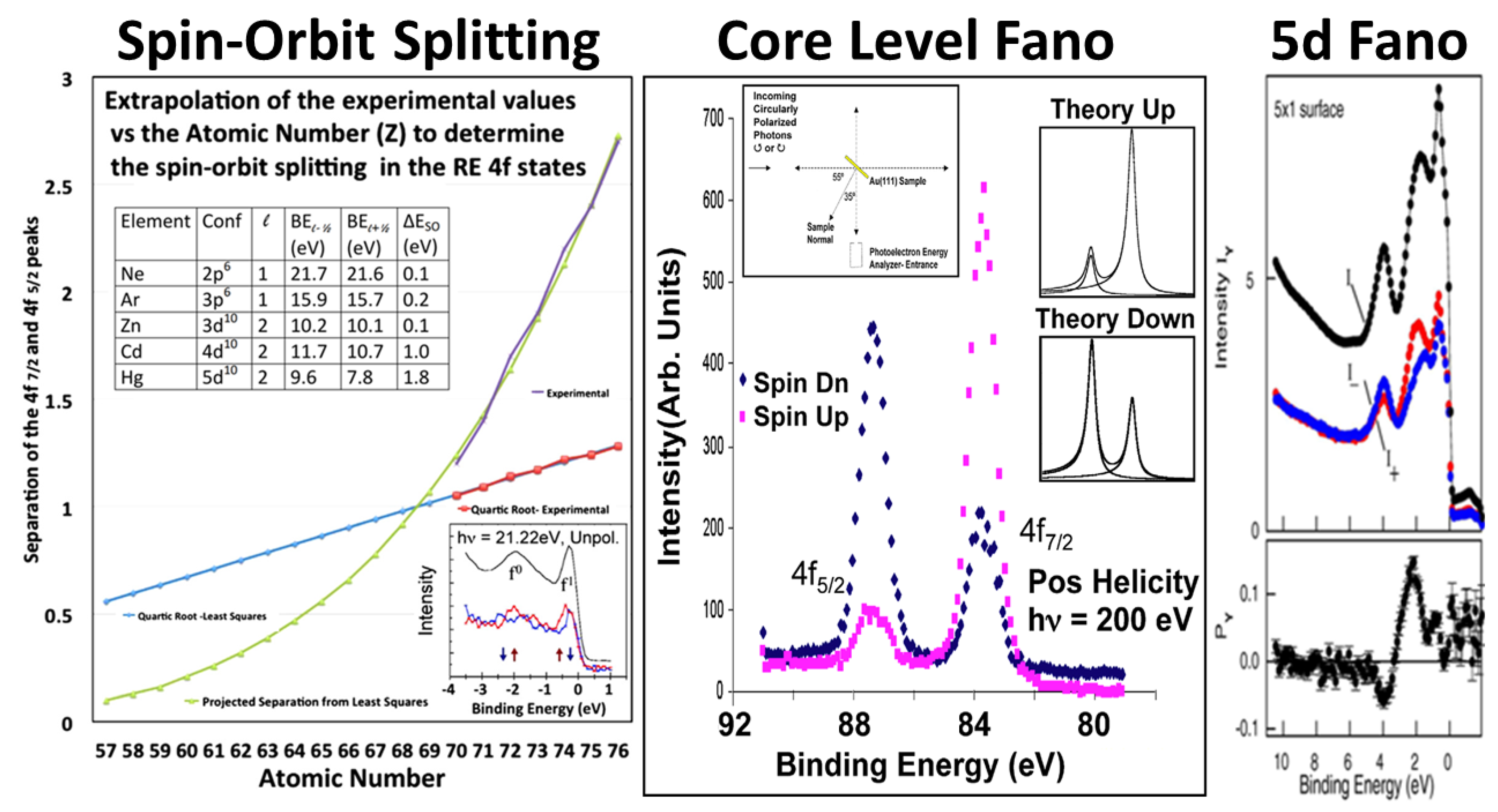
5.1. jj Skewed Intermediate Coupling in the Actinide 5f States
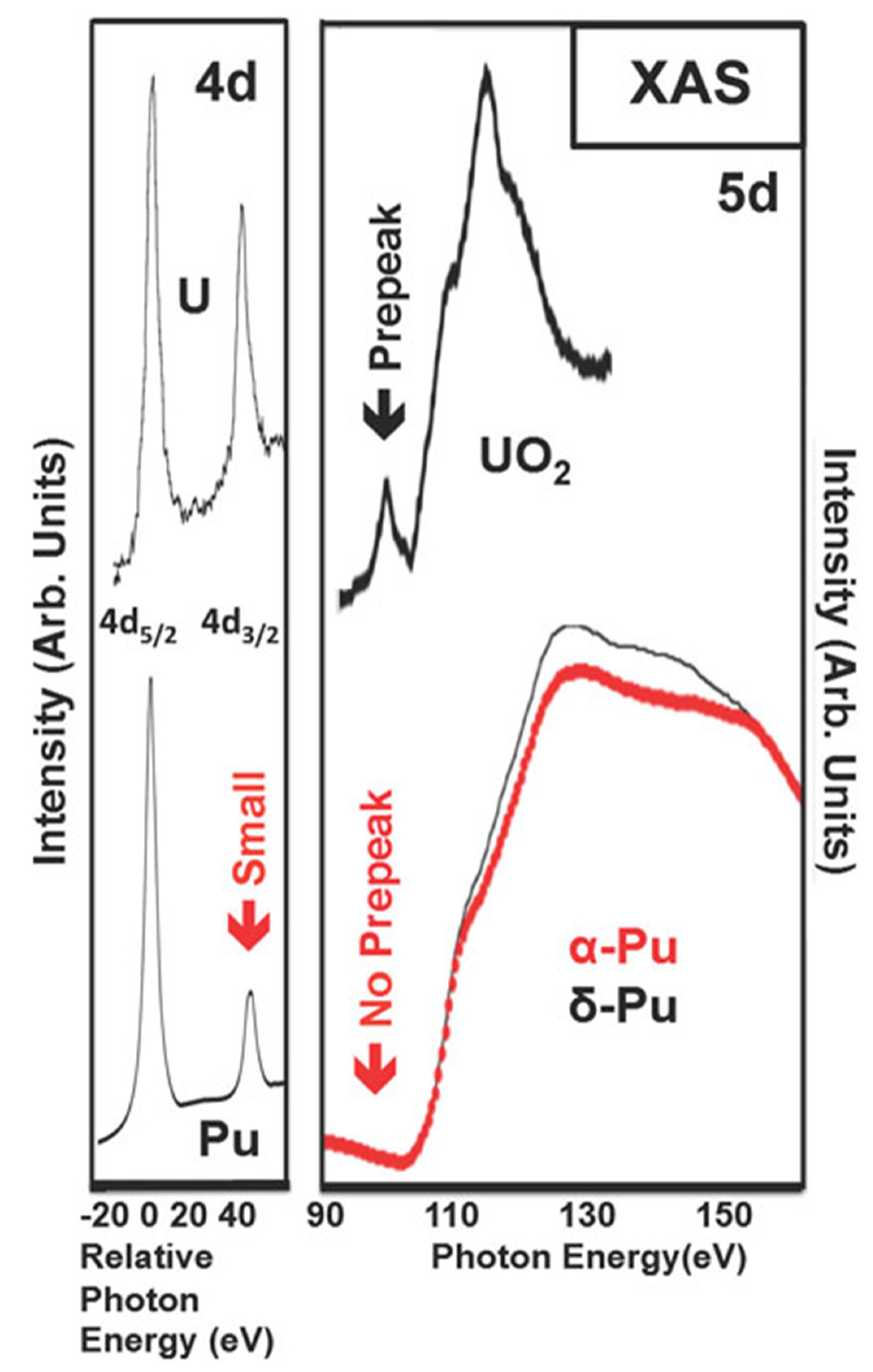
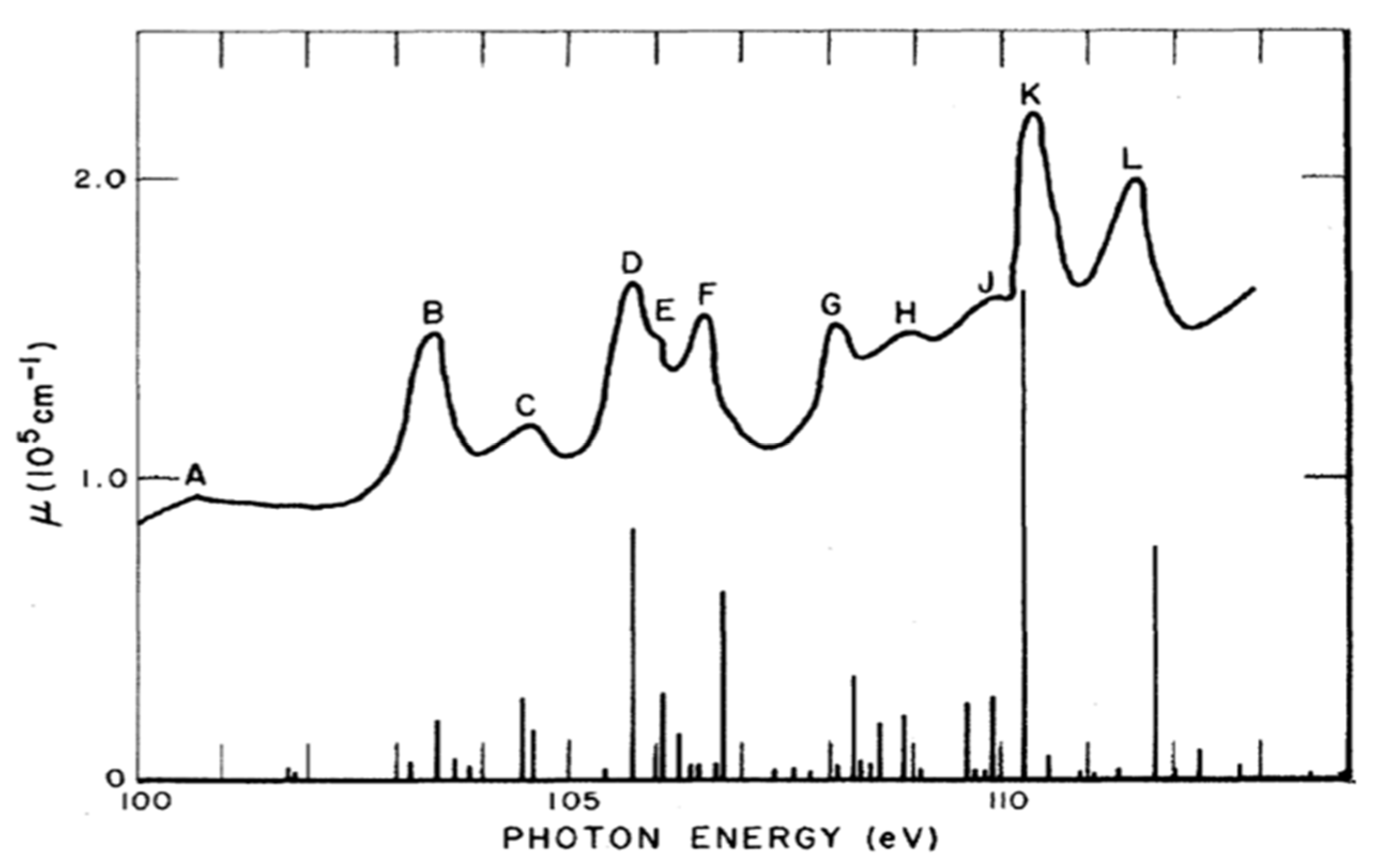
5.2. Prepeak Structures in 4f and 5f Systems
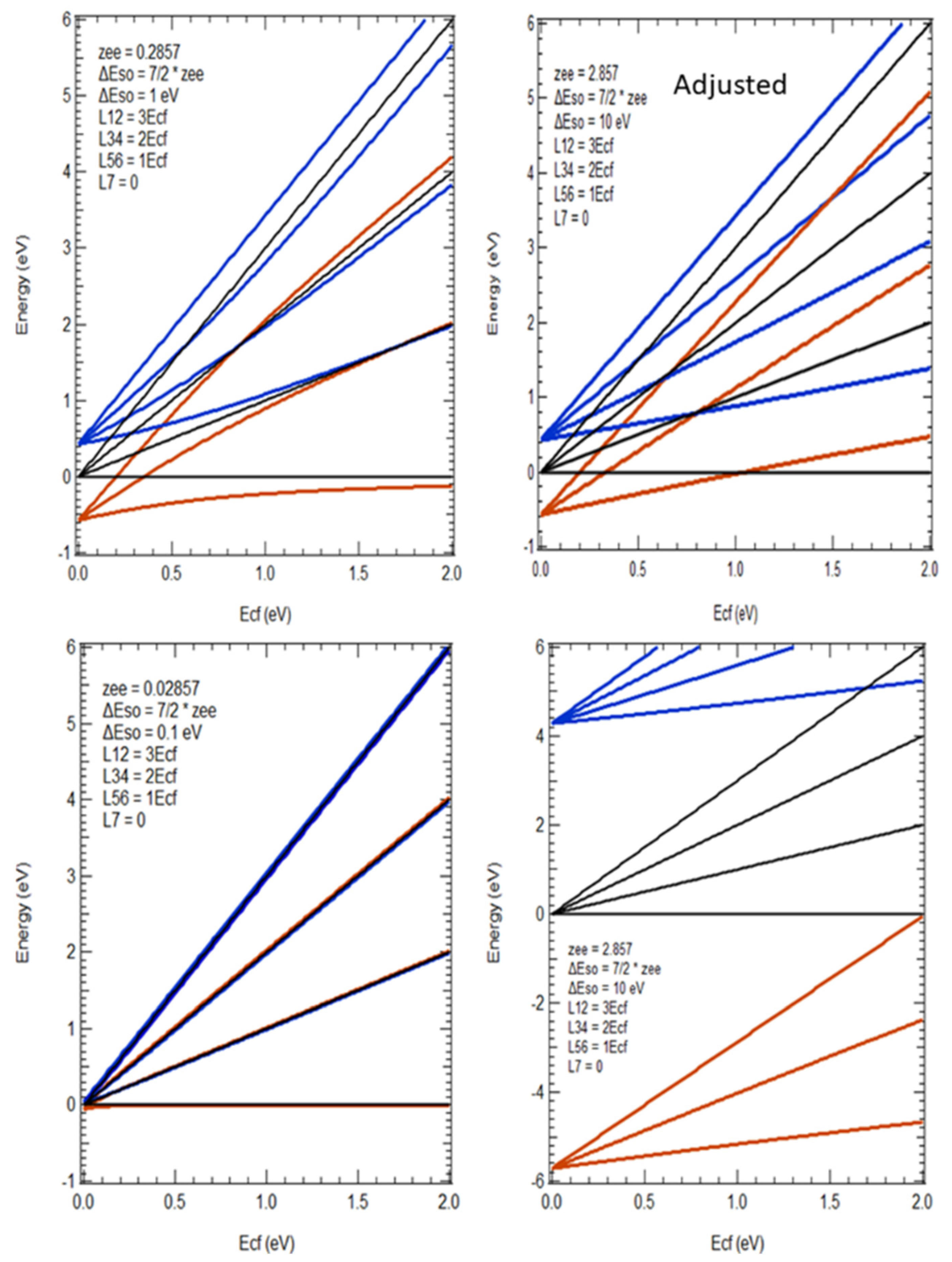
5.3. Crystal Field Modifications of Spherically Symmetric Systems
5.4. Brief Digression on Spin-Orbit Splittings and other Measurements
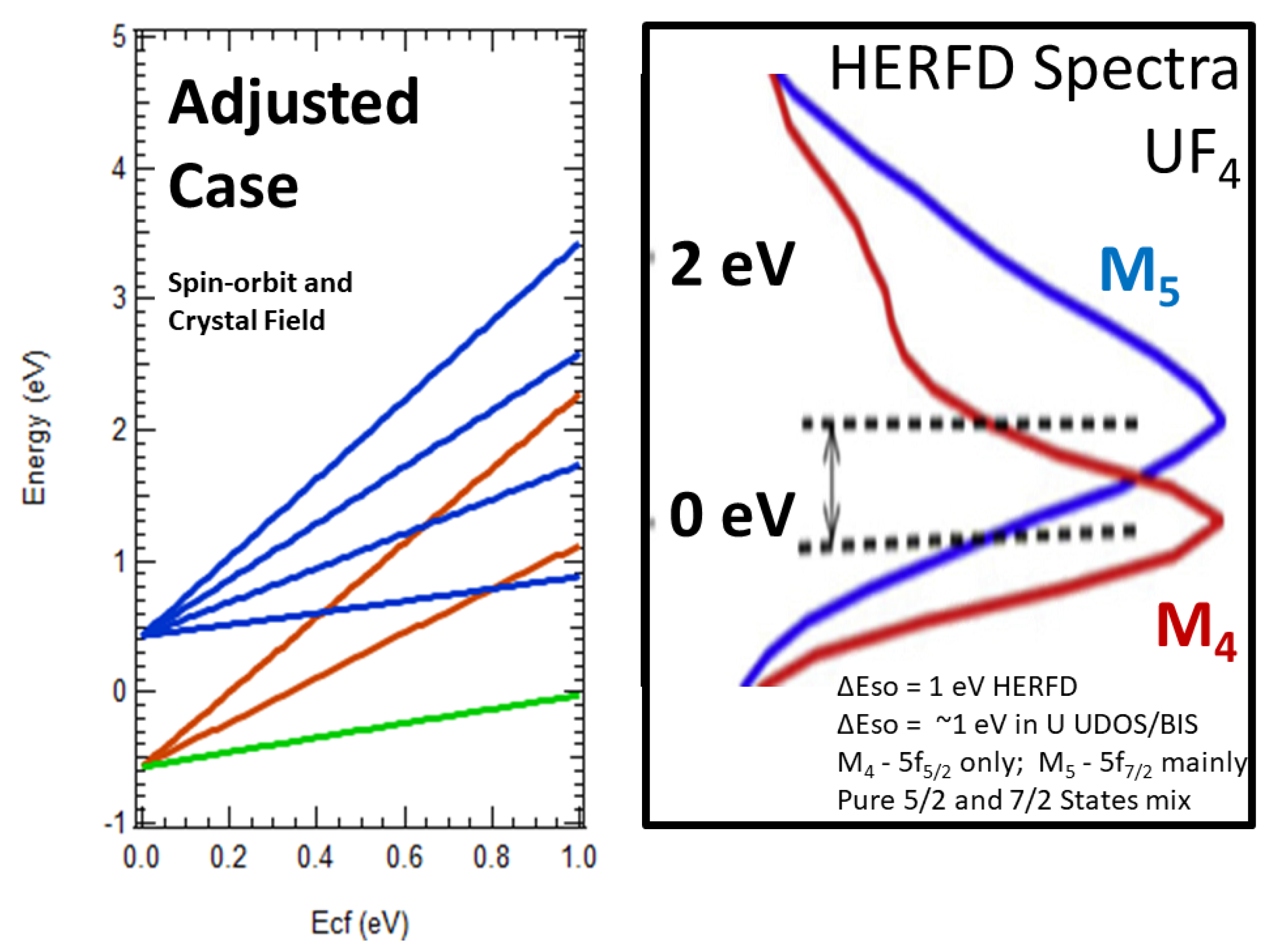
5.5. Evidence of Crystal Field Splittings in HERFD/XAS
5.6. Implications for Cubic Functions
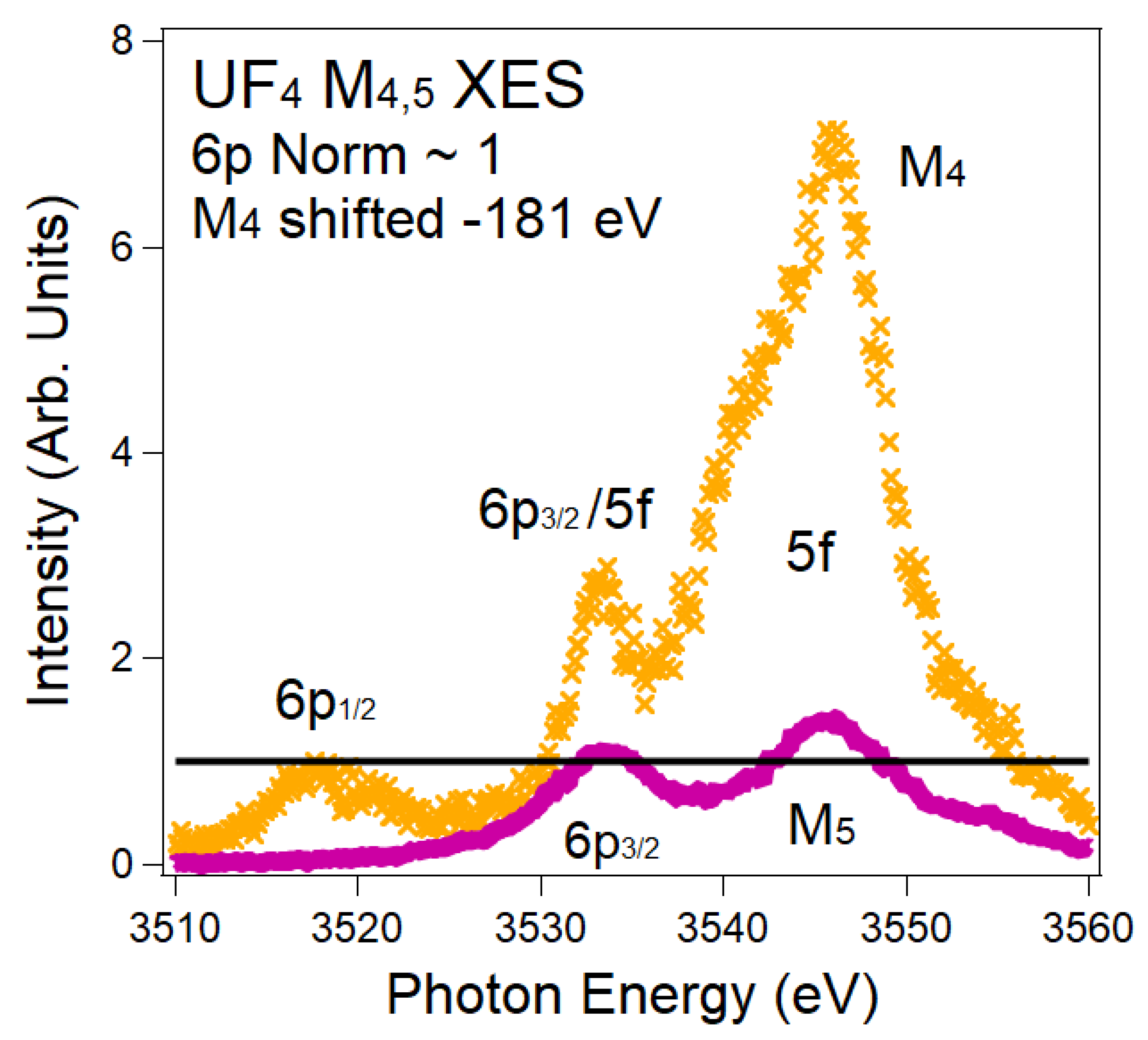
6. X-ray Emission Spectroscopy: A New Approach for the Quantification of Mixing
7. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hecker, S. Plutonium: An element at odds with itself. Alamos Sci. 2000, 26, 16. [Google Scholar]
- Boring, M.; Smith, J.L. Plutonium Condensed-Matter Physics: A survey of theory and experiment. Alamos Sci. 2000, 26, 90. [Google Scholar]
- Wills, J.M.; Eriksson, O. Actinide Ground-State Properties: Theoretical predictions. Alamos Sci. 2000, 26, 128. [Google Scholar]
- Zachariasen, W. Metallic radii and electron configurations of the 5f−6d metals. J. Inorg. Nucl. Chem. 1973, 35, 3487–3497. [Google Scholar] [CrossRef]
- Skriver, H.L.; Andersen, O.K.; Johansson, B. Calculated Bulk Properties of the Actinide Metals. Phys. Rev. Lett. 1978, 41, 42–45. [Google Scholar] [CrossRef]
- Moore, K.T.; Wall, M.A.; Schwartz, A.J.; Chung, B.W.; Shuh, D.K.; Schulze, R.K.; Tobin, J.G. Failure of Russell-Saunders Coupling in the5fStates of Plutonium. Phys. Rev. Lett. 2003, 90, 196404. [Google Scholar] [CrossRef]
- Van Der Laan, G.; Moore, K.T.; Tobin, J.G.; Chung, B.W.; Wall, M.A.; Schwartz, A.J. Applicability of the Spin-Orbit Sum Rule for the Actinide5fStates. Phys. Rev. Lett. 2004, 93, 097401. [Google Scholar] [CrossRef]
- Tobin, J.G.; Moore, K.T.; Chung, B.W.; Wall, M.A.; Schwartz, A.J.; van der Laan, G.; Kutepov, A.L. Competition between delocalization and spin-orbit splitting in the actinide 5f states. Phys. Rev. B 2005, 72, 085109. [Google Scholar] [CrossRef]
- Van der Laan, G.; Thole, B.T. X-ray-absorption sum rules in jj-coupled operators and ground-state moments of actinide ions. Phys. Rev. B 1996, 53, 14458. [Google Scholar] [CrossRef]
- Tobin, J.G.; Yu, S.-W.; Booth, C.H.; Tyliszczak, T.; Shuh, D.K.; Van Der Laan, G.; Sokaras, D.; Nordlund, D.; Weng, T.-C.; Bagus, P.S. Oxidation and crystal field effects in uranium. Phys. Rev. B 2015, 92, 035111. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.T.; van der Laan, G.; Wall, M.A.; Schwartz, A.J.; Haire, R.G. Rampant changes in 5f5/2 and 5f7/2 filling across the light and middle actinide metals: Electron energy-loss spectroscopy, many-electron atomic spectral calculations, and spin-orbit sum rule. Phys. Rev. B 2007, 76, 073105. [Google Scholar] [CrossRef]
- Kalkowski, G.; Kaindl, G.; Brewer, W.D.; Krone, W. Near-edge x-ray-absorption fine structure in uranium compounds. Phys. Rev. B 1987, 35, 2667–2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksson, O.; Uppsalla University, Uppsala, Sweden. Private Communication, September 2020.
- Baer, Y.; Lang, J.K. High-energy spectroscopic study of the occupied and unoccupied 5f and valence states in Th and U metals. Phys. Rev. B 1980, 21, 2060. [Google Scholar] [CrossRef]
- Naegele, J.R. Actinides and some of their alloys and compounds. In Electronic Structure of Solids: Photoemission Spectra and Related Data, Landolt-Bornstein Numerical Data and Functional Relationships in Science and Technology; A Goldmann Group III: Toronto, ON, Canada, 1994; pp. 183–327. [Google Scholar]
- Butterfield, M.T.; Tobin, J.G.; Teslich, N.E., Jr.; Bliss, R.A.; Wall, M.A.; McMahan, A.K.; Chung, B.W.; Schwartz, A.J.; Kutepov, A.L. Utilizing Nano-focussed Bremstrahlung Isochromat Spectroscopy (nBIS) to Determine the Unoccupied Electronic Structure of Pu. Matl. Res. Soc. Symp. Proc. 2006, 893, 95. [Google Scholar]
- Tobin, J.G.; Nowak, S.; Yu, S.-W.; Alonso-Mori, R.; Kroll, T.; Nordlund, D.; Weng, T.-C.; Sokaras, D. Towards the Quantification of 5f Delocalization. Appl. Sci. 2020, 10, 2918. [Google Scholar] [CrossRef] [Green Version]
- Smith, N.V. Inverse photoemission. Rep. Prog. Phys. 1988, 51, 1227–1294. [Google Scholar] [CrossRef]
- Tobin, J.G.; Robey, S.W.; Klebanoff, L.E.; Shirley, D.A. Ag/Cu(001): Observation of the development of the electronic structure in metal overlayers from two to three dimensionality. Phys. Rev. B 1983, 28, 6169. [Google Scholar] [CrossRef] [Green Version]
- Nelson, J.G.; Kim, S.; Gignac, W.J.; Williams, R.S.; Tobin, J.G.; Robey, S.W.; Shirley, D.A. High-resolution angle-resolved photoemission study of the Ag band structure along. Phys. Rev. B 1985, 32, 3465. [Google Scholar] [CrossRef]
- Tobin, J.G.; Robey, S.W.; Shirley, D.A. Two-dimensional valence-electronic structure of a monolayer of Ag on Cu(001). Phys. Rev. B 1986, 33, 2270. [Google Scholar] [CrossRef] [PubMed]
- Tobin, J.G.; Robey, S.W.; Klebanoff, L.E.; Shirley, D.A. Development of a three-dimensional valence-band structure in Ag overlayers on Cu(001). Phys. Rev. B 1987, 35, 9056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobin, J.G.; Olson, C.G.; Gu, C.; Liu, J.Z.; Solal, F.R.; Fluss, M.J.; Howell, R.H.; O’Brien, J.C.; Radousky, H.B.; Sterne, P.A. Valence bands and Fermi-surface topology of untwinned single-crystalYBa2Cu3O6.9. Phys. Rev. B 1992, 45, 5563–5576. [Google Scholar] [CrossRef] [Green Version]
- Opeil, C.P.; Schulze, R.K.; Manley, M.E.; Lashley, J.C.; Hults, W.L.; Hanrahan, R.J., Jr.; Smith, J.L.; Mihaila, B.; Blagoev, K.B.; Albers, R.C.; et al. Valence-band UPS, 6p core-level XPS, and LEED of a uranium (001) single crystal. Phys. Rev. B 2006, 73, 165109. [Google Scholar] [CrossRef] [Green Version]
- Opeil, C.P.; Schulze, R.K.; Volz, H.M.; Lashley, J.C.; Manley, M.E.; Hults, W.L.; Hanrahan, R.J., Jr.; Smith, J.L.; Mihaila, B.; Blagoev, K.B.; et al. Angle-resolved photoemission and first-principles electronic structure of single-crystalline α-U(001). Phys. Rev. B 2007, 75, 045120. [Google Scholar] [CrossRef] [Green Version]
- Tobin, J.G.; Nowak, S.; Yu, S.W.; Mori, R.A.; Kroll, T.; Nordlund, D.; Weng, T.-C.; Sokaras, D. Observation of 5f intermediate coupling in uranium x-ray emission spectroscopy. J. Phys. Commun. 2020, 4, 015013. [Google Scholar] [CrossRef]
- Nowak, S.H.; Armenta, R.; Schwartz, C.P.; Gallo, A.; Abraham, B.; Garcia-Esparza, A.T.; Biasin, E.; Prado, A.; Maciel, A.; Zhang, D.; et al. A versatile Johansson-type tender x-ray emission spectrometer. Rev. Sci. Instrum. 2020, 91, 033101. [Google Scholar] [CrossRef] [PubMed]
- Trelenberg, T.W.; Glade, S.C.; Tobin, J.G.; Hamza, A.V. The production and oxidation of uranium nanoparticles produced via pulsed laser ablation. Surf. Sci. 2006, 600, 2338. [Google Scholar] [CrossRef]
- Tobin, J.; Nowak, S.; Yu, S.-W.; Alonso-Mori, R.; Kroll, T.; Nordlund, D.; Weng, T.-C.; Sokaras, D. EXAFS as a probe of actinide oxide formation in the tender X-ray regime. Surf. Sci. 2020, 698, 121607. [Google Scholar] [CrossRef]
- Cohen-Tannoudji, C.; Diu, B.; Laloë, F. Quantum Mechanics; Wiley: New York, NY, USA, 1973; Volumes 1 and 2. [Google Scholar]
- Kittel, C. Introduction to Solid State Physics; John Wiley and Sons: New York, NY, USA, 1976. [Google Scholar]
- Tobin, J.G.; Chung, B.W.; Schulze, R.K.; Terry, J.; Farr, J.D.; Shuh, D.K.; Heinzelman, K.; Rotenberg, E.; Waddill, G.D.; Van Der Laan, G. Resonant Photoemission in f-electron Systems: Pu and Gd. Phys. Rev. 2003, 68, 155109. [Google Scholar] [CrossRef]
- Naegele, J.R.; Ghijsen, J.; Manes, L. Localization and hybridization of 5f states in the metallic and ionic bond as investigated by photoelectron spectroscopy. Actin. Chem. Phys. Prop. 2007, 59, 197–262. [Google Scholar] [CrossRef]
- Evans, S. Determination of the valence electronic configuration of uranium dioxide by photoelectron spectroscopy. J. Chem. Soc. Faraday Trans. 2 1977, 73, 1341–1343. [Google Scholar] [CrossRef]
- Greuter, F.; Hauser, E.; Oelhafen, P.; Güntherodt, H.-J.; Reihl, B.; Vogt, O. Core level and valence band photoemission from UAs. Phys. B+C 1980, 102, 117–121. [Google Scholar] [CrossRef]
- Yeh, J.; Lindau, I. Atomic subshell photoionization cross sections and asymmetry parameters: 1 ⩽ Z ⩽ 103. At. Data Nucl. Data Tables 1985, 32, 1–155. [Google Scholar] [CrossRef]
- Tobin, J. The apparent absence of chemical sensitivity in the 4d and 5d X-ray absorption spectroscopy of uranium compounds. J. Electron Spectrosc. Relat. Phenom. 2014, 194, 14–22. [Google Scholar] [CrossRef]
- Tobin, J.G.; Yu, S.-W.; Chung, B.W. Splittings, Satellites and Fine Structure in the Soft X-ray Spectroscopy of the Actinides. Top. Catal. 2013, 56, 1104. [Google Scholar] [CrossRef]
- Dehmer, J.L.; Starace, A.F.; Fano, U.; Sugar, J.; Cooper, J.W. Raising of Discrete Levels into the Far Continuum. Phys. Rev. Lett. 1971, 26, 1521–1525. [Google Scholar] [CrossRef] [Green Version]
- Tobin, J.G.; Söderlind, P.; Landa, A.; Moore, K.T.; Schwartz, A.J.; Chung, B.W.; A Wall, M.; Wills, J.M.; Haire, R.G.; Kutepov, A.L. On the electronic configuration in Pu: Spectroscopy and theory. J. Phys. Condens. Matter 2008, 20, 125204. [Google Scholar] [CrossRef]
- Moore, K.T.; Chung, B.W.; Morton, S.A.; Schwartz, A.J.; Tobin, J.G.; Lazar, S.; Tichelaar, F.D.; Zandbergen, H.W.; Söderlind, P.; Van Der Laan, G. Changes in the electronic structure of cerium due to variations in close packing. Phys. Rev. B 2004, 69, 193104. [Google Scholar] [CrossRef] [Green Version]
- Tobin, J.G. 5f states with spin-orbit and crystal field splittings. J. Vac. Sci. Technol. A 2019, 37, 031201. [Google Scholar] [CrossRef]
- Tobin, J.G.; Nowak, S.; Booth, C.H.; Bauer, E.D.; Yu, S.-W.; Alonso-Mori, R.; Kroll, T.; Nordlung, D.; Weng, T.-C.; Sokaras, D. Separate Measurement of the 5f5/2 and 5f7/2 Unoccupied Density of States of UO2. J. Electron. Spectrosc. Relat. Phenom. 2019, 232, 100. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.; Vaughan, D. (Eds.) X-ray Data Booklet; Lawrence Berkeley National Laboratory, University of California: Berkley, CA, USA, 2001. [Google Scholar]
- Tobin, J.G.; Yu, S.W.; Komesu, T.; Chung, B.W.; A Morton, S.; Waddill, G.D. Evidence of dynamical spin shielding in Ce from spin-resolved photoelectron spectroscopy. EPL Europhys. Lett. 2007, 77, 17004. [Google Scholar] [CrossRef]
- Tobin, J.; Morton, S.; Chung, B.; Yu, S.; Waddill, G. Spin-resolved electronic structure studies of non-magnetic systems: Possible observation of the Fano Effect in polycrystal Ce. Phys. B Condens. Matter 2006, 378–380, 925–928. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.-W.; Tobin, J. Observation of an underlying relativistic effect in the valence bands of Pt. Surf. Sci. 2007, 601, L127–L131. [Google Scholar] [CrossRef]
- Burdge, J. Chemistry, 3rd ed.; McGraw Hill: New York, NY, USA, 2014. [Google Scholar]
- Cotton, F.A. Chemical Applications of Group Theory, 2nd ed.; Wiley Interscience, John Wiley and Sons: Baffins Lane, UK, 1971. [Google Scholar]
- Orbitron, Gallery of Atomic Orbitals. Available online: https://winter.group.shef.ac.uk/orbitron/AOs/2p/index.html (accessed on 24 April 2021).













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tobin, J.G.; Nowak, S.; Yu, S.W.; Alonso-Mori, R.; Kroll, T.; Nordlund, D.; Weng, T.C.; Sokaras, D. The Limitations of 5f Delocalization and Dispersion. Appl. Sci. 2021, 11, 3882. https://doi.org/10.3390/app11093882
Tobin JG, Nowak S, Yu SW, Alonso-Mori R, Kroll T, Nordlund D, Weng TC, Sokaras D. The Limitations of 5f Delocalization and Dispersion. Applied Sciences. 2021; 11(9):3882. https://doi.org/10.3390/app11093882
Chicago/Turabian StyleTobin, J. G., S. Nowak, S. W. Yu, R. Alonso-Mori, T. Kroll, D. Nordlund, T. C. Weng, and D. Sokaras. 2021. "The Limitations of 5f Delocalization and Dispersion" Applied Sciences 11, no. 9: 3882. https://doi.org/10.3390/app11093882