
Isatin-Hydrazones with Multiple Receptor Tyrosine Kinases (RTKs) Inhibitory Activity and In-Silico Binding Mechanism


Abstract
:1. Introduction
2. Materials and Methods
2.1. General
2.2. General Procedure for the Synthesis of Isatin-Hydrazones
2.3. Calculation of the IC50 Values
2.4. In Vitro EGFR/VEGFR-2 Inhibitory Activity
2.5. In Vitro FLT-3 Inhibitory Activity
2.6. In-Silico Binding Mechanism
3. Results
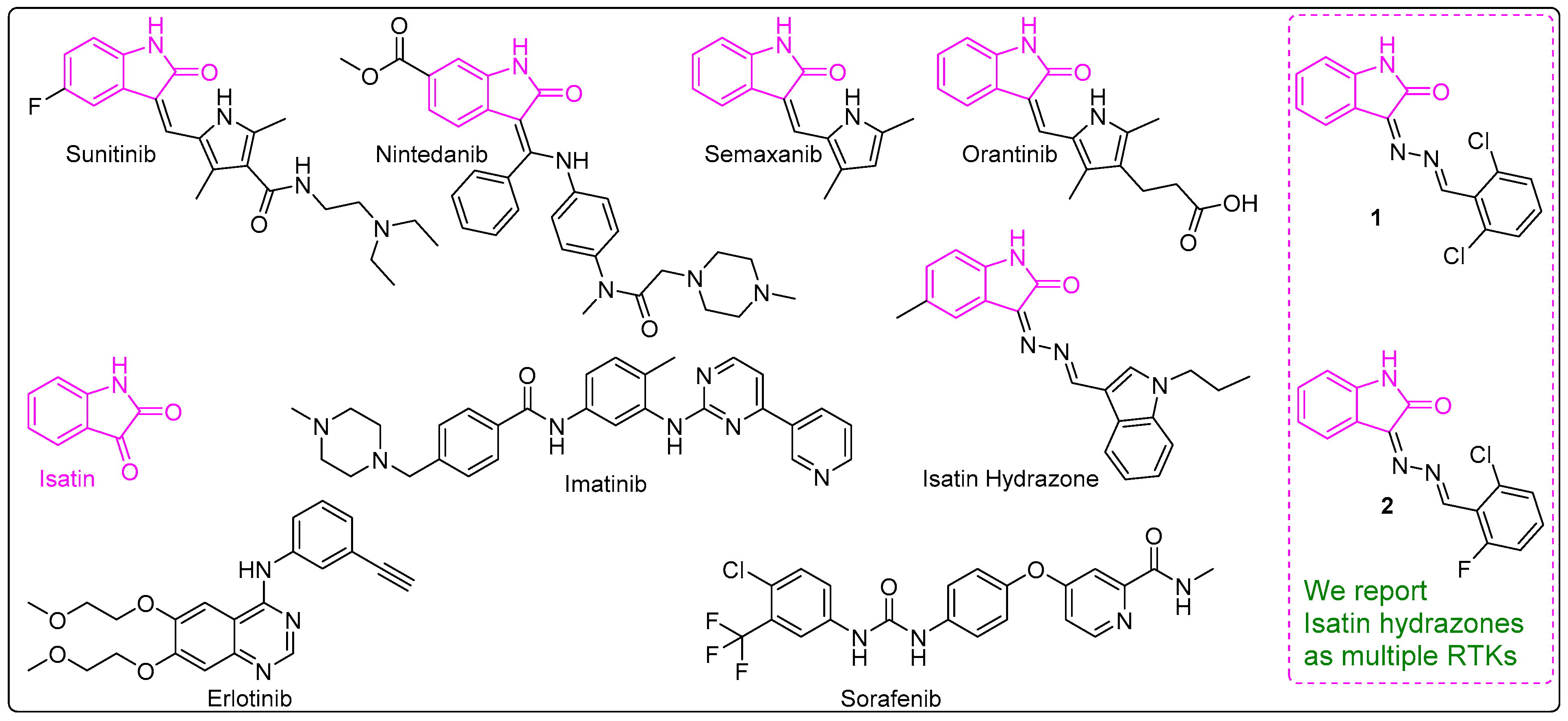
3.1. Synthesis of 1 and 2
3.2. EGFR, VEGFR-2 and FLT-3 Protein Kinase Inhibitory Activities of 1 and 2
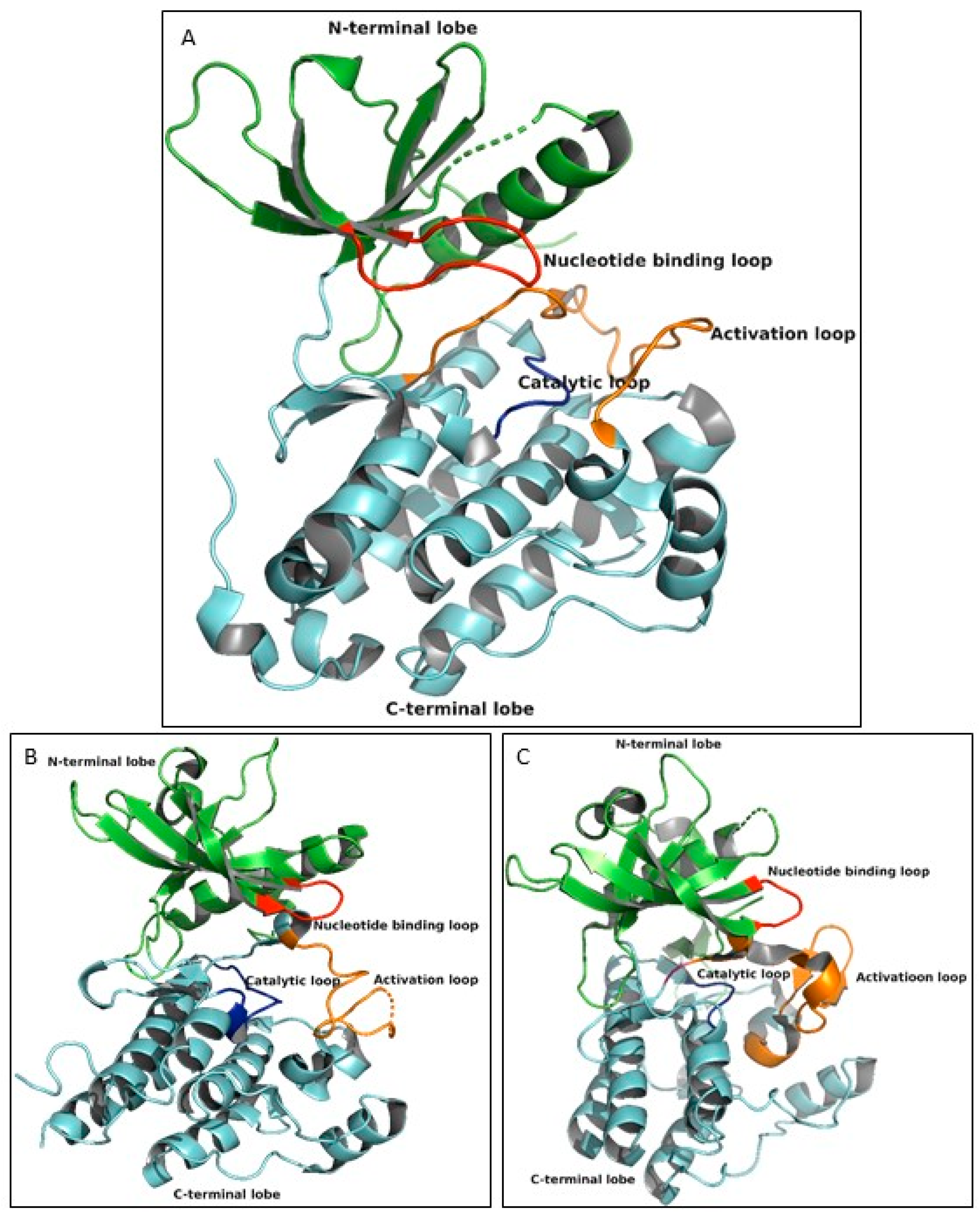
3.3. Overall Structural Arrangement of the Kinase Domain of EGFR, VEGFR-2 and FLT-3 Protein Kinases
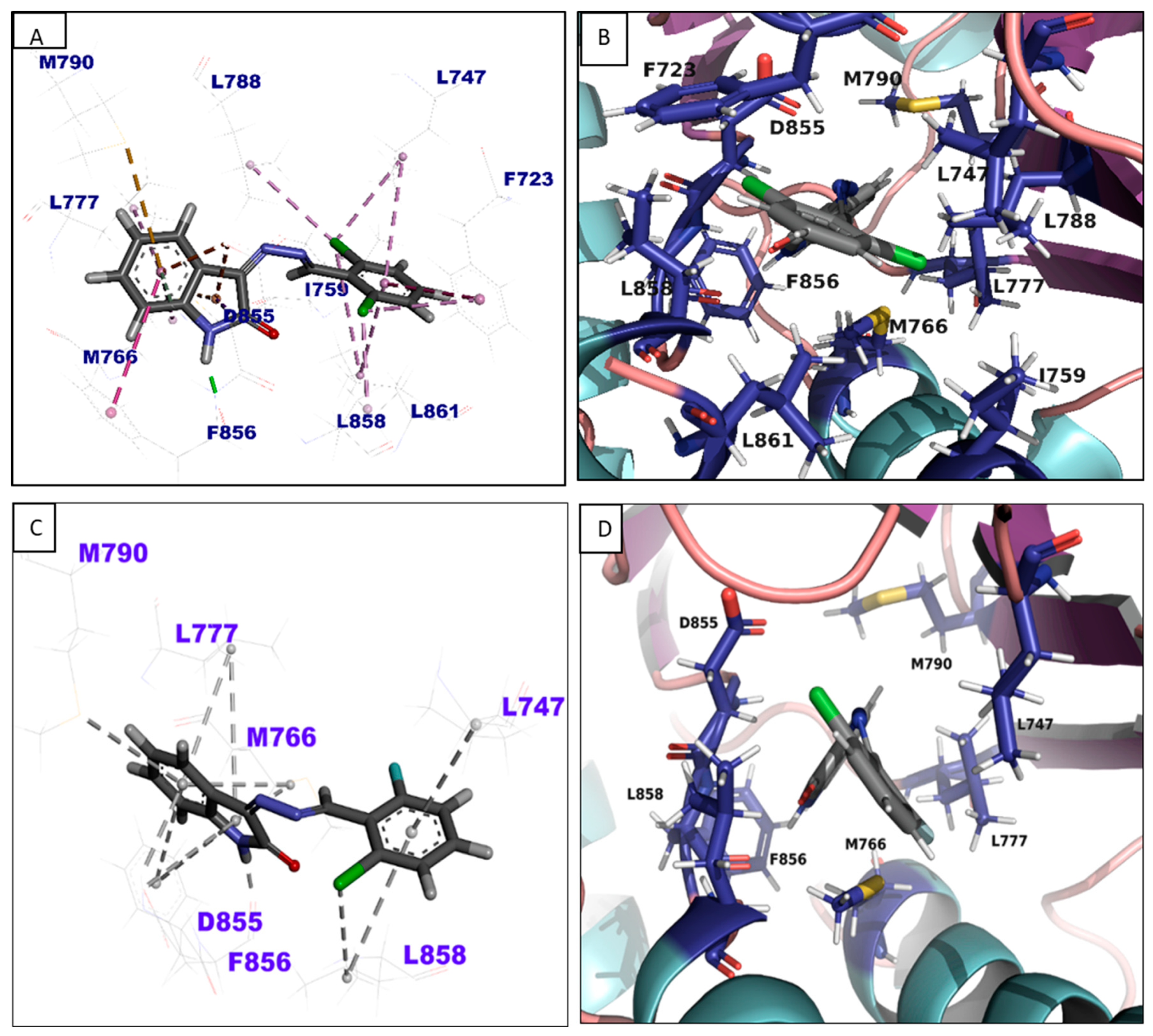
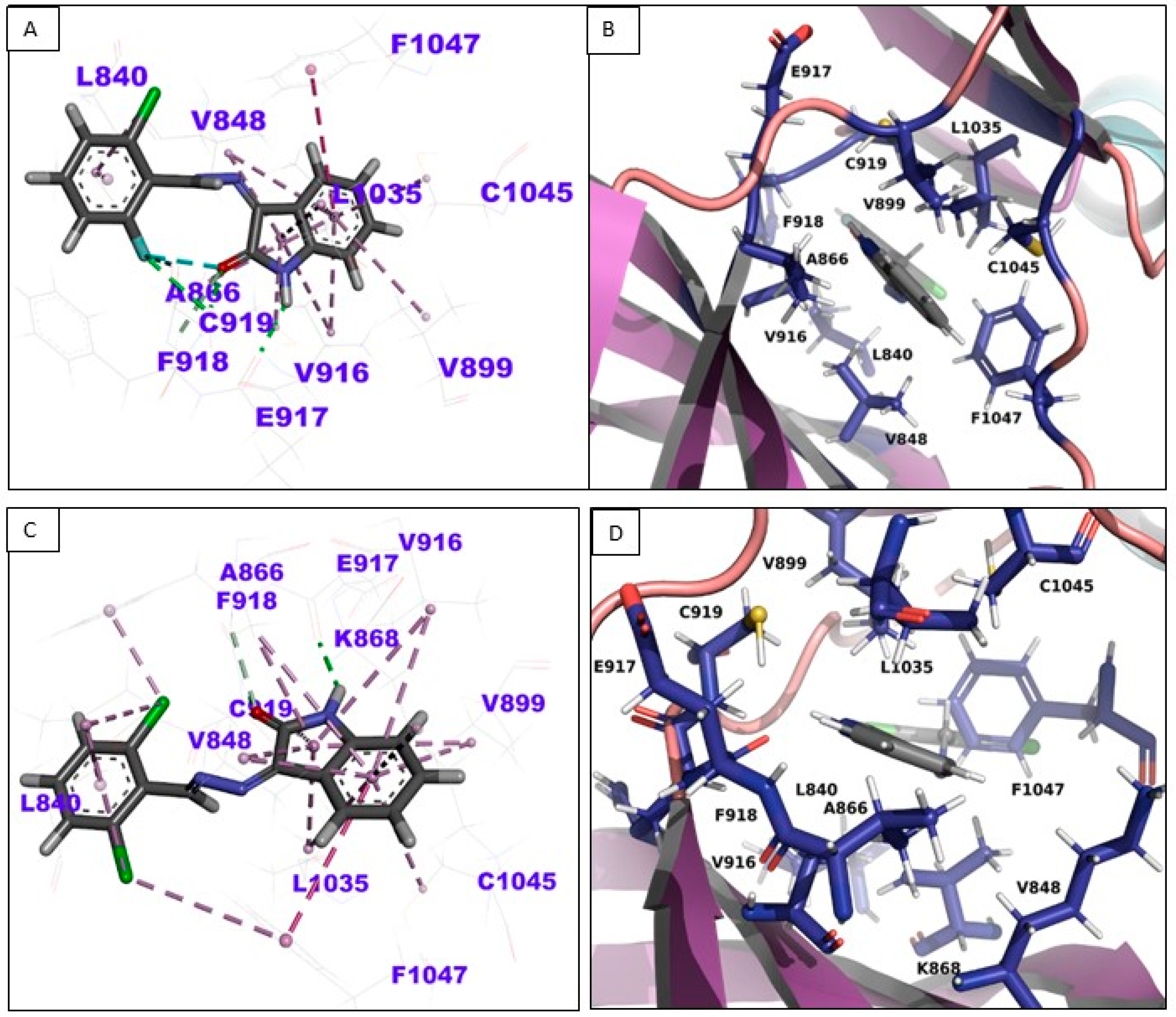
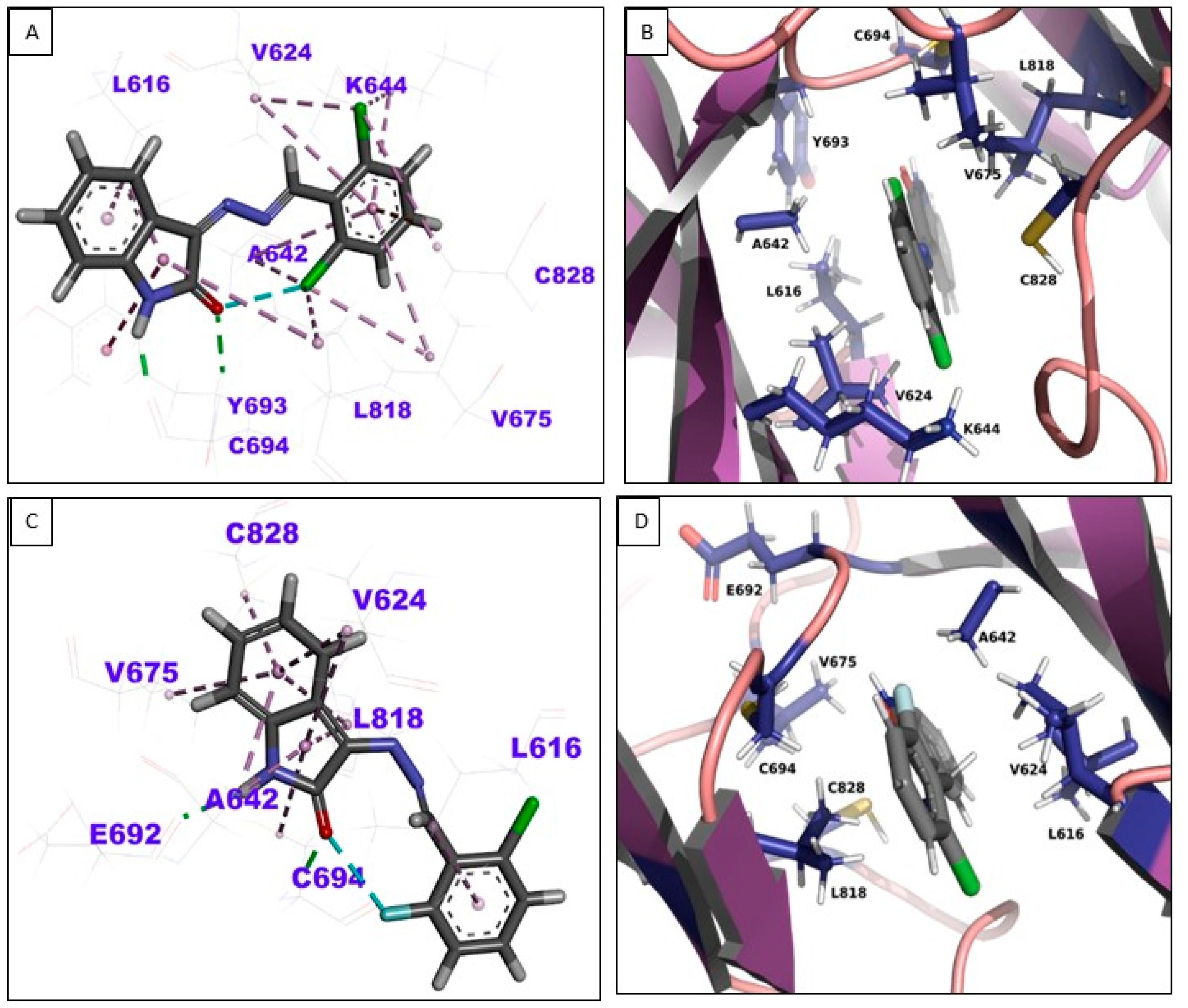
3.4. In-Silico Binding Mechanism Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kozlov, S.; Waters, N.C.; Chavchich, M. Leveraging cell cycle analysis in anticancer drug discovery to identify novel plasmodial drug targets. Infect. Disord. Drug Targets 2010, 10, 165–190. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.F.M.M.; Park, S.-E.; Kadi, A.A.; Kwon, Y. Fluorescein Hydrazones as Novel Nonintercalative Topoisomerase Catalytic Inhibitors with Low DNA Toxicity. J. Med. Chem. 2014, 57, 9139–9151. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, P.; Woo, H.; Jun, K.-Y.; Kadi, A.A.; Abdel-Aziz, H.A.; Kwon, Y.; Rahman, A.F.M.M. Design, synthesis, topoisomerase I & II inhibitory activity, antiproliferative activity, and structure–activity relationship study of pyrazoline derivatives: An ATP-competitive human topoisomerase IIα catalytic inhibitor. Bioorg. Med. Chem. 2016, 24, 1898–1908. [Google Scholar] [CrossRef]
- Islam, M.S.; Park, S.; Song, C.; Kadi, A.A.; Kwon, Y.; Rahman, A.F.M.M. Fluorescein hydrazones: A series of novel non-intercalative topoisomerase IIα catalytic inhibitors induce G1 arrest and apoptosis in breast and colon cancer cells. Eur. J. Med. Chem. 2017, 125, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Al-Salem, H.S.; Arifuzzaman, M.; Alkahtani, H.M.; Abdalla, A.N.; Issa, I.S.; Alqathama, A.; Albalawi, F.S.; Rahman, A.F.M.M. A Series of Isatin-Hydrazones with Cytotoxic Activity and CDK2 Kinase Inhibitory Activity: A Potential Type II ATP Competitive Inhibitor. Molecules 2020, 25, 4400. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S. Review of epidermal growth factor receptor biology. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 21–26. [Google Scholar] [CrossRef]
- Zhang, X.; Mar, V.; Zhou, W.; Harrington, L.; Robinson, M.O. Telomere shortening and apoptosis in telomerase-inhibited human tumor cells. Genes Dev. 1999, 13, 2388–2399. [Google Scholar] [CrossRef]
- Bishayee, S. Role of conformational alteration in the epidermal growth factor receptor (EGFR) function. Biochem. Pharmacol. 2000, 60, 1217–1223. [Google Scholar] [CrossRef]
- Umekita, Y.; Ohi, Y.; Sagara, Y.; Yoshida, H. Co-expression of epidermal growth factor receptor and transforming growth factor-alpha predicts worse prognosis in breast-cancer patients. Int. J. Cancer 2000, 89, 484–487. [Google Scholar] [CrossRef]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, F.R.; Varella-Garcia, M.; Bunn, P.A., Jr.; Di Maria, M.V.; Veve, R.; Bremmes, R.M.; Barón, A.E.; Zeng, C.; Franklin, W.A. Epidermal growth factor receptor in non-small-cell lung carcinomas: Correlation between gene copy number and protein expression and impact on prognosis. J. Clin. Oncol. 2003, 21, 3798–3807. [Google Scholar] [CrossRef]
- Bazley, L.A.; Gullick, W.J. The epidermal growth factor receptor family. Endocr.-Relat. Cancer 2005, 12, S17–S27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Berezov, A.; Wang, Q.; Zhang, G.; Drebin, J.; Murali, R.; Greene, M.I. ErbB receptors: From oncogenes to targeted cancer therapies. J. Clin. Investig. 2007, 117, 2051–2058. [Google Scholar] [CrossRef] [Green Version]
- Ivanković, M.; Cukusić, A.; Gotić, I.; Skrobot, N.; Matijasić, M.; Polancec, D.; Rubelj, I. Telomerase activity in HeLa cervical carcinoma cell line proliferation. Biogerontology 2007, 8, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Cui, S.; Luan, W.; Wang, S.; Hou, Z.; Liu, Y.; Cheng, M. Natural product-based design, synthesis and biological evaluation of Albiziabioside A derivatives that selectively induce HCT116 cell death. Eur. J. Med. Chem. 2016, 113, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Holmes, K.; Roberts, O.L.; Thomas, A.M.; Cross, M.J. Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cell Signal. 2007, 19, 2003–2012. [Google Scholar] [CrossRef]
- Stuttfeld, E.; Ballmer-Hofer, K. Structure and function of VEGF receptors. IUBMB Life 2009, 61, 915–922. [Google Scholar] [CrossRef]
- Lu, R.-M.; Chiu, C.-Y.; Liu, I.-J.; Chang, Y.-L.; Liu, Y.-J.; Wu, H.-C. Novel human Ab against vascular endothelial growth factor receptor 2 shows therapeutic potential for leukemia and prostate cancer. Cancer Sci. 2019, 110, 3773–3787. [Google Scholar] [CrossRef] [Green Version]
- Rosnet, O.; Matteï, M.G.; Marchetto, S.; Birnbaum, D. Isolation and chromosomal localization of a novel FMS-like tyrosine kinase gene. Genomics 1991, 9, 380–385. [Google Scholar] [CrossRef]
- Rosnet, O.; Schiff, C.; Pébusque, M.J.; Marchetto, S.; Tonnelle, C.; Toiron, Y.; Birg, F.; Birnbaum, D. Human FLT3/FLK2 gene: cDNA cloning and expression in hematopoietic cells. Blood 1993, 82, 1110–1119. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Birg, F.; Courcoul, M.; Rosnet, O.; Bardin, F.; Pébusque, M.J.; Marchetto, S.; Tabilio, A.; Mannoni, P.; Birnbaum, D. Expression of the FMS/KIT-like gene FLT3 in human acute leukemias of the myeloid and lymphoid lineages. Blood 1992, 80, 2584–2593. [Google Scholar] [CrossRef] [Green Version]
- Hannum, C.; Culpepper, J.; Campbell, D.; McClanahan, T.; Zurawski, S.; Bazan, J.F.; Kastelein, R.; Hudak, S.; Wagner, J.; Mattson, J.; et al. Ligand for FLT3/FLK2 receptor tyrosine kinase regulates growth of haematopoietic stem cells and is encoded by variant RNAs. Nature 1994, 368, 643–648. [Google Scholar] [CrossRef]
- Kiyoi, H.; Naoe, T.; Nakano, Y.; Yokota, S.; Minami, S.; Miyawaki, S.; Asou, N.; Kuriyama, K.; Jinnai, I.; Shimazaki, C.; et al. Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood 1999, 93, 3074–3080. [Google Scholar] [CrossRef] [PubMed]
- Kiyoi, H.; Kawashima, N.; Ishikawa, Y. FLT3 mutations in acute myeloid leukemia: Therapeutic paradigm beyond inhibitor development. Cancer Sci. 2020, 111, 312–322. [Google Scholar] [CrossRef] [Green Version]
- El-Husseiny, W.M.; El-Sayed, M.A.A.; Abdel-Aziz, N.I.; El-Azab, A.S.; Ahmed, E.R.; Abdel-Aziz, A.A.M. Synthesis, antitumour and antioxidant activities of novel α,β-unsaturated ketones and related heterocyclic analogues: EGFR inhibition and molecular modelling study. J. Enzym. Inhib. Med. Chem. 2018, 33, 507–518. [Google Scholar] [CrossRef] [Green Version]
- Miura, M. Therapeutic drug monitoring of imatinib, nilotinib, and dasatinib for patients with chronic myeloid leukemia. Biol. Pharm. Bull. 2015, 38, 645–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef] [Green Version]
- Rabindran, S.K.; Discafani, C.M.; Rosfjord, E.C.; Baxter, M.; Floyd, M.B.; Golas, J.; Hallett, W.A.; Johnson, B.D.; Nilakantan, R.; Overbeek, E.; et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004, 64, 3958–3965. [Google Scholar] [CrossRef] [Green Version]
- Minami, Y.; Shimamura, T.; Shah, K.; LaFramboise, T.; Glatt, K.A.; Liniker, E.; Borgman, C.L.; Haringsma, H.J.; Feng, W.; Weir, B.A.; et al. The major lung cancer-derived mutants of ERBB2 are oncogenic and are associated with sensitivity to the irreversible EGFR/ERBB2 inhibitor HKI-272. Oncogene 2007, 26, 5023–5027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wissner, A.; Mansour, T.S. The development of HKI-272 and related compounds for the treatment of cancer. Arch. Pharm. 2008, 341, 465–477. [Google Scholar] [CrossRef]
- Morgillo, F.; Martinelli, E.; Troiani, T.; Orditura, M.; De Vita, F.; Ciardiello, F. Antitumor activity of sorafenib in human cancer cell lines with acquired resistance to EGFR and VEGFR tyrosine kinase inhibitors. PLoS ONE 2011, 6, e28841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, S.H. Crizotinib: A novel and first-in-class multitargeted tyrosine kinase inhibitor for the treatment of anaplastic lymphoma kinase rearranged non-small cell lung cancer and beyond. Drug Des. Dev. Ther. 2011, 5, 471–485. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, H.; de Figueiredo-Pontes, L.L.; Kobayashi, S.; Costa, D.B. Preclinical rationale for use of the clinically available multitargeted tyrosine kinase inhibitor crizotinib in ROS1-translocated lung cancer. J. Thorac. Oncol. 2012, 7, 1086–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, N.; Lucena-Araujo, A.R.; Nakayama, S.; de Figueiredo-Pontes, L.L.; Gonzalez, D.A.; Yasuda, H.; Kobayashi, S.; Costa, D.B. Dual ALK and EGFR inhibition targets a mechanism of acquired resistance to the tyrosine kinase inhibitor crizotinib in ALK rearranged lung cancer. Lung Cancer 2014, 83, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Lee, Y.M.; Chang, G.C.; Yu, S.L.; Hsieh, W.Y.; Chen, J.J.; Chen, H.W.; Yang, P.C. Curcumin induces EGFR degradation in lung adenocarcinoma and modulates p38 activation in intestine: The versatile adjuvant for gefitinib therapy. PLoS ONE 2011, 6, e23756. [Google Scholar] [CrossRef] [Green Version]
- Lv, P.C.; Li, D.D.; Li, Q.S.; Lu, X.; Xiao, Z.P.; Zhu, H.L. Synthesis, molecular docking and evaluation of thiazolyl-pyrazoline derivatives as EGFR TK inhibitors and potential anticancer agents. Bioorg. Med. Chem. Lett. 2011, 21, 5374–5377. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.Y.; Cao, Y.; Ma, H.; Li, H.Q.; Ao, G.Z. Design, synthesis and molecular docking of α,β-unsaturated cyclohexanone analogous of curcumin as potent EGFR inhibitors with antiproliferative activity. Bioorg. Med. Chem. 2013, 21, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.K.; Lee, M.H.; Lim, D.Y.; Kim, J.E.; Singh, P.; Lee, S.Y.; Jeong, C.H.; Lim, T.G.; Chen, H.; Chi, Y.I.; et al. Isoliquiritigenin induces apoptosis and inhibits xenograft tumor growth of human lung cancer cells by targeting both wild type and L858R/T790M mutant EGFR. J. Biol. Chem. 2014, 289, 35839–35848. [Google Scholar] [CrossRef] [Green Version]
- Wada, K.; Lee, J.Y.; Hung, H.Y.; Shi, Q.; Lin, L.; Zhao, Y.; Goto, M.; Yang, P.C.; Kuo, S.C.; Chen, H.W.; et al. Novel curcumin analogs to overcome EGFR-TKI lung adenocarcinoma drug resistance and reduce EGFR-TKI-induced GI adverse effects. Bioorg. Med. Chem. 2015, 23, 1507–1514. [Google Scholar] [CrossRef] [Green Version]
- Alswah, M.; Bayoumi, A.H.; Elgamal, K.; Elmorsy, A.; Ihmaid, S.; Ahmed, H.E.A. Design, Synthesis and Cytotoxic Evaluation of Novel Chalcone Derivatives Bearing Triazolo [4, 3-a]-quinoxaline Moieties as Potent Anticancer Agents with Dual EGFR Kinase and Tubulin Polymerization Inhibitory Effects. Molecules 2017, 23, 48. [Google Scholar] [CrossRef] [Green Version]
- Whittles, C.E.; Pocock, T.M.; Wedge, S.R.; Kendrew, J.; Hennequin, L.F.; Harper, S.J.; Bates, D.O. ZM323881, a Novel Inhibitor of Vascular Endothelial Growth Factor-Receptor-2 Tyrosine Kinase Activity. Microcirculation 2002, 9, 513–522. [Google Scholar] [CrossRef]
- Jost, L.; Gschwind, H.-P.; Jalava, T.; Wang, Y.; Guenther, C.; Souppart, C.; Rottmann, A.; Denner, K.; Waldmeier, F.; Gross, G.; et al. Metabolism and Disposition of Vatalanib (PTK787/ZK-222584) in Cancer Patients. Drug Metab. Dispos. 2006, 34, 1817–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmange, J.-C.; Weiss, M.M.; Germain, J.; Polverino, A.J.; Borg, G.; Bready, J.; Chen, D.; Choquette, D.; Coxon, A.; DeMelfi, T.; et al. Naphthamides as Novel and Potent Vascular Endothelial Growth Factor Receptor Tyrosine Kinase Inhibitors: Design, Synthesis, and Evaluation. J. Med. Chem. 2008, 51, 1649–1667. [Google Scholar] [CrossRef] [PubMed]
- Nikolinakos, P.; Heymach, J.V. The tyrosine kinase inhibitor cediranib for non-small cell lung cancer and other thoracic malignancies. J. Thorac. Oncol. 2008, 3, S131–S134. [Google Scholar] [CrossRef] [Green Version]
- Kubota, K.; Ichinose, Y.; Scagliotti, G.; Spigel, D.; Kim, J.H.; Shinkai, T.; Takeda, K.; Kim, S.W.; Hsia, T.C.; Li, R.K.; et al. Phase III study (MONET1) of motesanib plus carboplatin/paclitaxel in patients with advanced nonsquamous nonsmall-cell lung cancer (NSCLC): Asian subgroup analysis. Ann. Oncol. 2014, 25, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; DeBraud, F.; Bahleda, R.; Adamo, B.; Andre, F.; Dientsmann, R.; Delmonte, A.; Cereda, R.; Isaacson, J.; Litten, J.; et al. Phase I/IIa study evaluating the safety, efficacy, pharmacokinetics, and pharmacodynamics of lucitanib in advanced solid tumors. Ann. Oncol. 2014, 25, 2244–2251. [Google Scholar] [CrossRef]
- Guffanti, F.; Chilà, R.; Bello, E.; Zucchetti, M.; Zangarini, M.; Ceriani, L.; Ferrari, M.; Lupi, M.; Jacquet-Bescond, A.; Burbridge, M.F.; et al. In Vitro and In Vivo Activity of Lucitanib in FGFR1/2 Amplified or Mutated Cancer Models. Neoplasia 2017, 19, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.-W.; Liu, D.-K.; Zhang, Q.-W.; Xu, Y.-G.; Shi, L. VEGFR-2 inhibitors and the therapeutic applications thereof: A patent review (2012-2016). Expert Opin. Ther. Pat. 2017, 27, 987–1004. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.J.; Kulkarni, V.M. Vascular Endothelial Growth Factor Receptor (VEGFR-2)/KDR Inhibitors: Medicinal Chemistry Perspective. Med. Drug Discov. 2019, 2, 100009. [Google Scholar] [CrossRef]
- Chung, T.-W.; Kim, E.-Y.; Choi, H.-J.; Han, C.W.; Jang, S.B.; Kim, K.-J.; Jin, L.; Koh, Y.J.; Ha, K.-T. 6′-Sialylgalactose inhibits vascular endothelial growth factor receptor 2-mediated angiogenesis. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Li, C.; Zhu, X. FLT3 inhibitors in acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 133. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Kantarjian, H.; Ravandi, F.; Daver, N. Emerging treatment paradigms with FLT3 inhibitors in acute myeloid leukemia. Ther. Adv. Hematol. 2019, 10, 204062071982731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Song, Y.; Liu, D. Gilteritinib: A novel FLT3 inhibitor for acute myeloid leukemia. Biomark. Res. 2019, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Antar, A.I.; Otrock, Z.K.; Jabbour, E.; Mohty, M.; Bazarbachi, A. FLT3 inhibitors in acute myeloid leukemia: Ten frequently asked questions. Leukemia 2020, 34, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Li, X.; Hu, Y.; Liu, T. Recent advances in FLT3 inhibitors for acute myeloid leukemia. Future Med. Chem. 2020, 12, 961–981. [Google Scholar] [CrossRef] [PubMed]
- Tarver, T.C.; Hill, J.E.; Rahmat, L.; Perl, A.E.; Bahceci, E.; Mori, K.; Smith, C.C. Gilteritinib is a clinically active FLT3 inhibitor with broad activity against FLT3 kinase domain mutations. Blood Adv. 2020, 4, 514–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manetti, F.; Locatelli, G.A.; Maga, G.; Schenone, S.; Modugno, M.; Forli, S.; Corelli, F.; Botta, M. A Combination of Docking/Dynamics Simulations and Pharmacophoric Modeling to Discover New Dual c-Src/Abl Kinase Inhibitors. J. Med. Chem. 2006, 49, 3278–3286. [Google Scholar] [CrossRef]
- Abbas, H.-A.S.; Abd El-Karim, S.S. Design, synthesis and anticervical cancer activity of new benzofuran–pyrazol-hydrazono-thiazolidin-4-one hybrids as potential EGFR inhibitors and apoptosis inducing agents. Bioorg. Chem. 2019, 89, 103035. [Google Scholar] [CrossRef]
- Larrosa-Garcia, M.; Baer, M.R. FLT3 Inhibitors in Acute Myeloid Leukemia: Current Status and Future Directions. Mol. Cancer Ther. 2017, 16, 991–1001. [Google Scholar] [CrossRef] [Green Version]
- Pradeepkiran, J.A.; Kumar, K.K.; Kumar, Y.N.; Bhaskar, M. Modeling, molecular dynamics, and docking assessment of transcription factor rho: A potential drug target in Brucella melitensis 16M. Drug Des. Dev. Ther. 2015, 9, 1897–1912. [Google Scholar] [CrossRef] [Green Version]
- Pradeepkiran, J.A.; Sainath, S.B.; Kumar, K.K.; Bhaskar, M. Complete genome-wide screening and subtractive genomic approach revealed new virulence factors, potential drug targets against bio-war pathogen Brucella melitensis 16M. Drug Des. Dev. Ther. 2015, 9, 1691–1706. [Google Scholar] [CrossRef] [Green Version]
- Sudhana, S.M.; Adi, P.J. Synthesis, Biological Evaluation and Molecular Docking Studies of Novel Di-hydropyridine Analogs as Potent Antioxidants. Curr. Top. Med. Chem. 2019, 19, 2676–2686. [Google Scholar] [CrossRef]
- Pradeepkiran, J.A.; Reddy, P.H. Structure Based Design and Molecular Docking Studies for Phosphorylated Tau Inhibitors in Alzheimer’s Disease. Cells 2019, 8, 260. [Google Scholar] [CrossRef] [Green Version]
- Pradeepkiran, J.A.; Reddy, A.P.; Reddy, P.H. Pharmacophore-based models for therapeutic drugs against phosphorylated tau in Alzheimer’s disease. Drug Discov. Today 2019, 24, 616–623. [Google Scholar] [CrossRef]
- Pradeepkiran, J.A.; Reddy, A.P.; Yin, X.; Manczak, M.; Reddy, P.H. Protective effects of BACE1 inhibitory ligand molecules against amyloid beta-induced synaptic and mitochondrial toxicities in Alzheimer’s disease. Hum. Mol. Genet. 2020, 29, 49–69. [Google Scholar] [CrossRef] [PubMed]
- McTigue, M.A.; Wickersham, J.A.; Pinko, C.; Showalter, R.E.; Parast, C.V.; Tempczyk-Russell, A.; Gehring, M.R.; Mroczkowski, B.; Kan, C.C.; Villafranca, J.E.; et al. Crystal structure of the kinase domain of human vascular endothelial growth factor receptor 2: A key enzyme in angiogenesis. Structure 1999, 7, 319–330. [Google Scholar] [CrossRef] [Green Version]
- Griffith, J.; Black, J.; Faerman, C.; Swenson, L.; Wynn, M.; Lu, F.; Lippke, J.; Saxena, K. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol. Cell 2004, 13, 169–178. [Google Scholar] [CrossRef]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef] [Green Version]
- Fiedler, W.; Kayser, S.; Kebenko, M.; Janning, M.; Krauter, J.; Schittenhelm, M.; Götze, K.; Weber, D.; Göhring, G.; Teleanu, V.; et al. A phase I/II study of sunitinib and intensive chemotherapy in patients over 60 years of age with acute myeloid leukaemia and activating FLT3 mutations. Br. J. Haematol. 2015, 169, 694–700. [Google Scholar] [CrossRef]
- Liao, Q.H.; Gao, Q.Z.; Wei, J.; Chou, K.C. Docking and molecular dynamics study on the inhibitory activity of novel inhibitors on epidermal growth factor receptor (EGFR). Med. Chem. 2011, 7, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Belal, A. Synthesis, molecular docking and antitumor activity of novel pyrrolizines with potential as EGFR-TK inhibitors. Bioorg. Chem. 2015, 59, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Kunz, R.K.; Rumfelt, S.; Chen, N.; Zhang, D.; Tasker, A.S.; Bürli, R.; Hungate, R.; Yu, V.; Nguyen, Y.; Whittington, D.A.; et al. Discovery of amido-benzisoxazoles as potent c-Kit inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 5115–5117. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.A.; Serya, R.A.T.; Lasheen, D.S.; Abdel-Aziz, A.K.; Esmat, A.; Mansour, A.M.; Singab, A.N.B.; Abouzid, K.A.M. Discovery of Potent VEGFR-2 Inhibitors based on Furopyrimidine and Thienopyrimidne Scaffolds as Cancer Targeting Agents. Sci. Rep. 2016, 6, 24460. [Google Scholar] [CrossRef] [Green Version]
- Zorn, J.A.; Wang, Q.; Fujimura, E.; Barros, T.; Kuriyan, J. Crystal Structure of the FLT3 Kinase Domain Bound to the Inhibitor Quizartinib (AC220). PLoS ONE 2015, 10, e0121177. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | IC50 (µM) | Kinase IC50 (µM) a | |||
|---|---|---|---|---|---|
| MCF7 [5] | CDK2 [5] | EGFR | VEGFR-2 | FLT-3 | |
| 1 | 1.51 ± 0.09 | 0.246 ± 0.05 | 0.269 ± 0.08 | 0.232 ± 0.01 | 1.535 ± 0.03 |
| 2 | 3.56 ± 0.31 | 0.301 ± 0.02 | 0.369 ± 0.32 | 0.266 ± 0.04 | 0.546 ± 0.28 |
| Control b–f | 3.10 ± 0.29 b | 0.131 ± 0.24 c | 0.056 ± 0.02 d | 0.091 ± 0.03 e | 0.262 ± 0.01 f |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Salem, H.S.; Arifuzzaman, M.; Issa, I.S.; Rahman, A.F.M.M. Isatin-Hydrazones with Multiple Receptor Tyrosine Kinases (RTKs) Inhibitory Activity and In-Silico Binding Mechanism. Appl. Sci. 2021, 11, 3746. https://doi.org/10.3390/app11093746
Al-Salem HS, Arifuzzaman M, Issa IS, Rahman AFMM. Isatin-Hydrazones with Multiple Receptor Tyrosine Kinases (RTKs) Inhibitory Activity and In-Silico Binding Mechanism. Applied Sciences. 2021; 11(9):3746. https://doi.org/10.3390/app11093746
Chicago/Turabian StyleAl-Salem, Huda S., Md Arifuzzaman, Iman S. Issa, and A. F. M. Motiur Rahman. 2021. "Isatin-Hydrazones with Multiple Receptor Tyrosine Kinases (RTKs) Inhibitory Activity and In-Silico Binding Mechanism" Applied Sciences 11, no. 9: 3746. https://doi.org/10.3390/app11093746