
Advanced First-Principle Modeling of Relativistic Ruddlesden—Popper Strontium Iridates


Abstract
:1. Introduction
2. Methodology
2.1. Noncollinear LSDA+U Model
2.2. Constrained Random Phase Approximation
2.3. The Method
2.4. The Bethe–Salpeter Equation
3. Computational Details
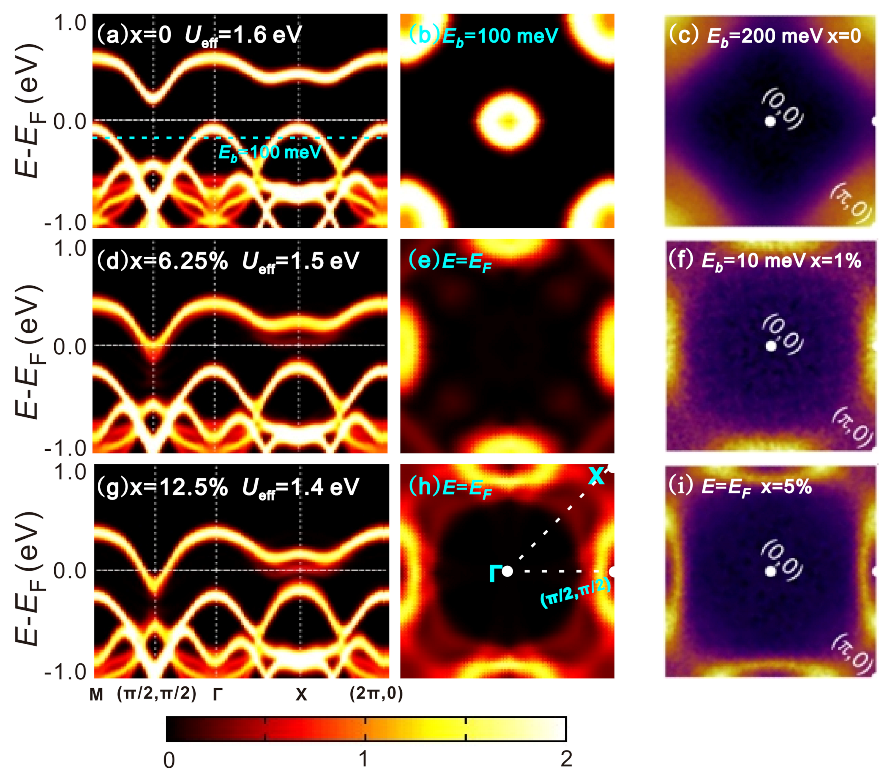
4. Results and Discussions
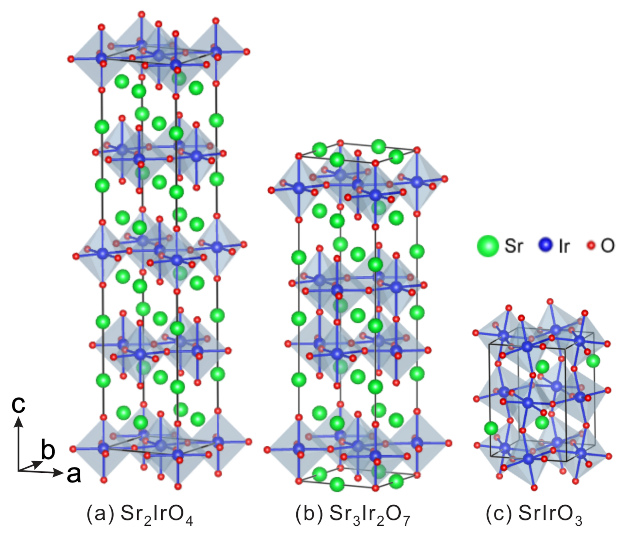
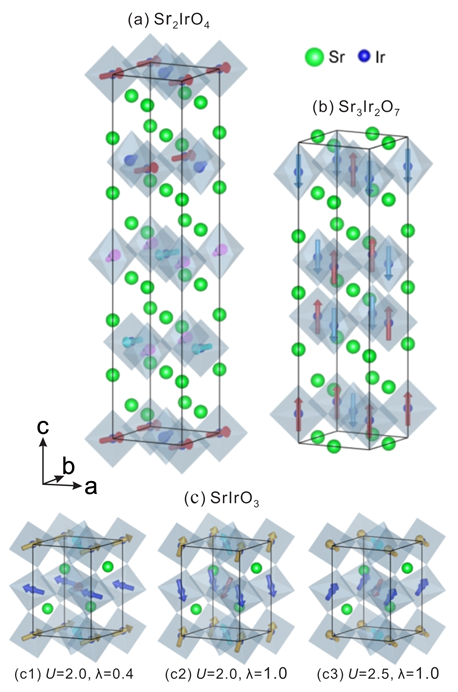
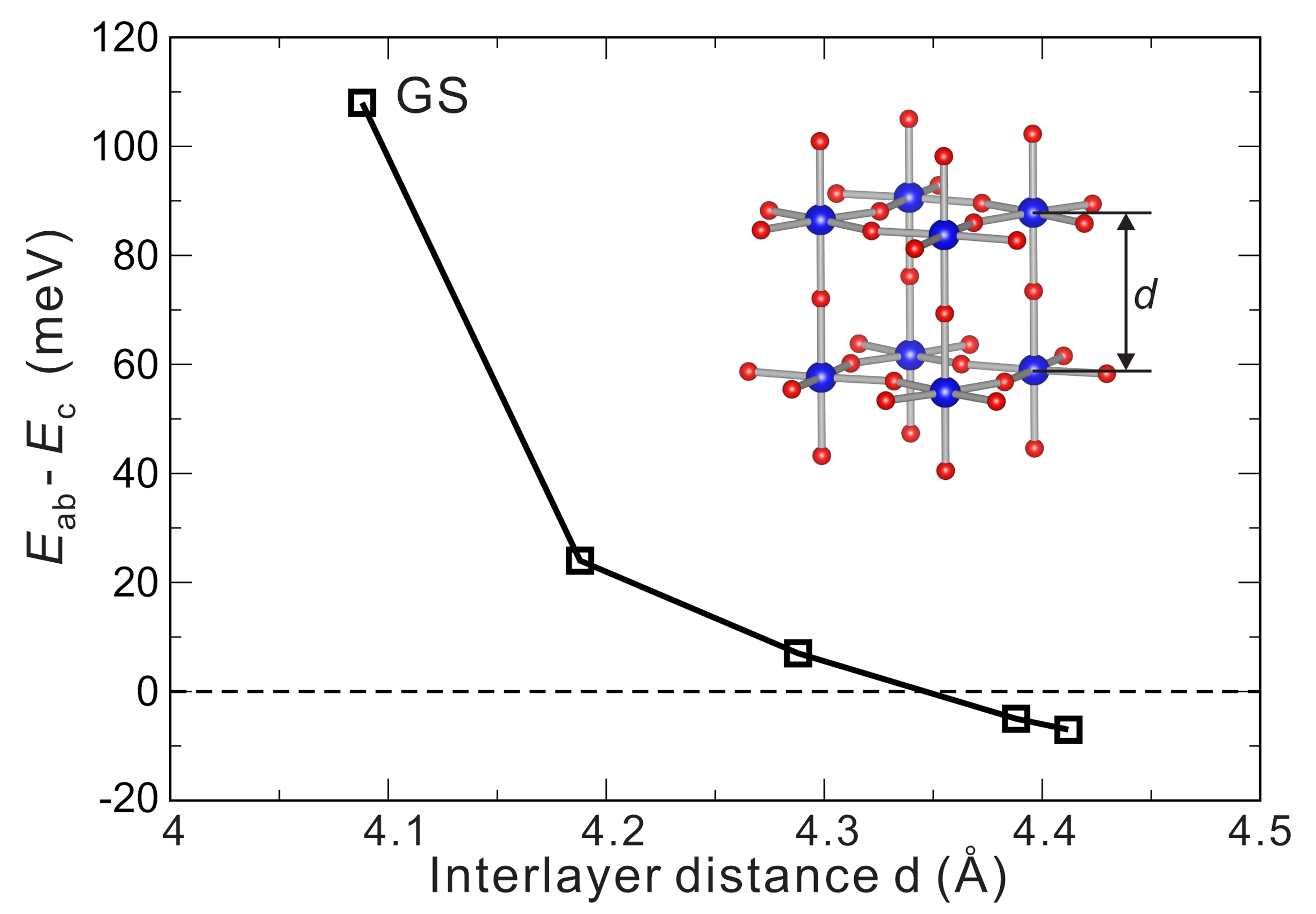
4.1. Crystal Structures
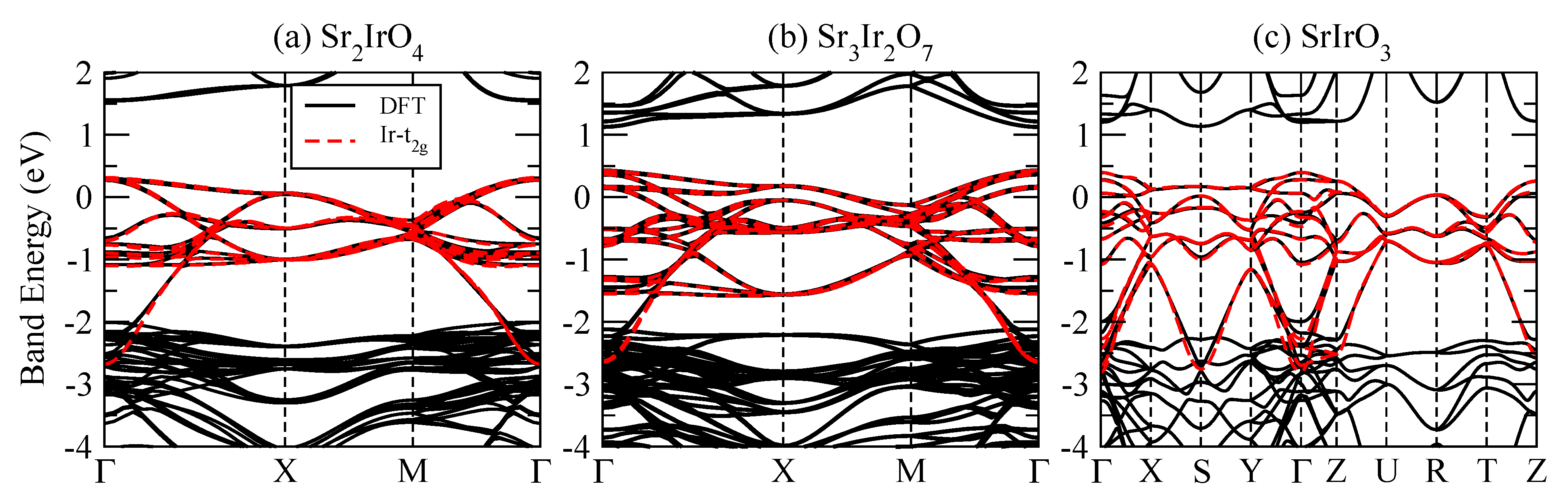
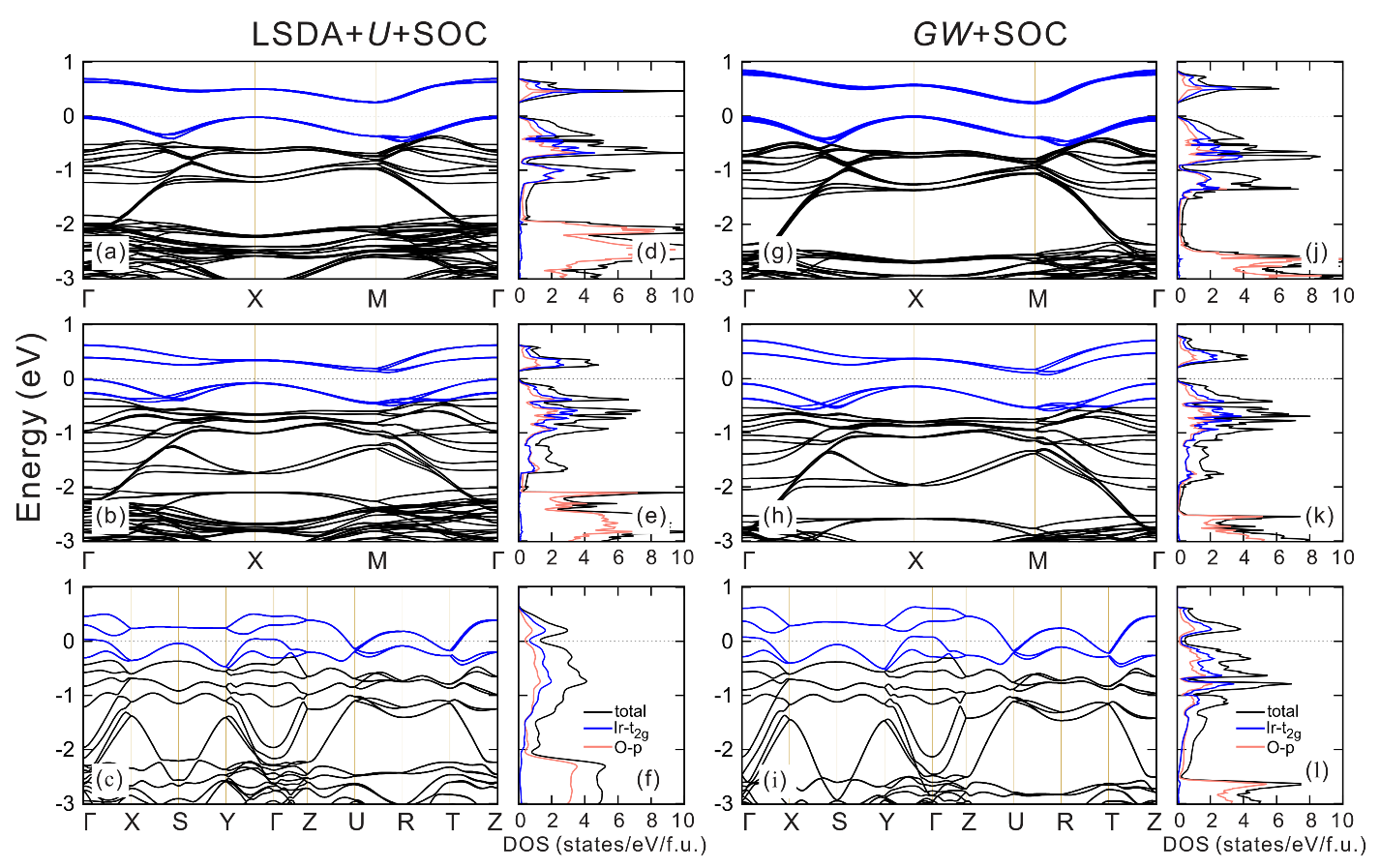
4.2. Electronic Structures
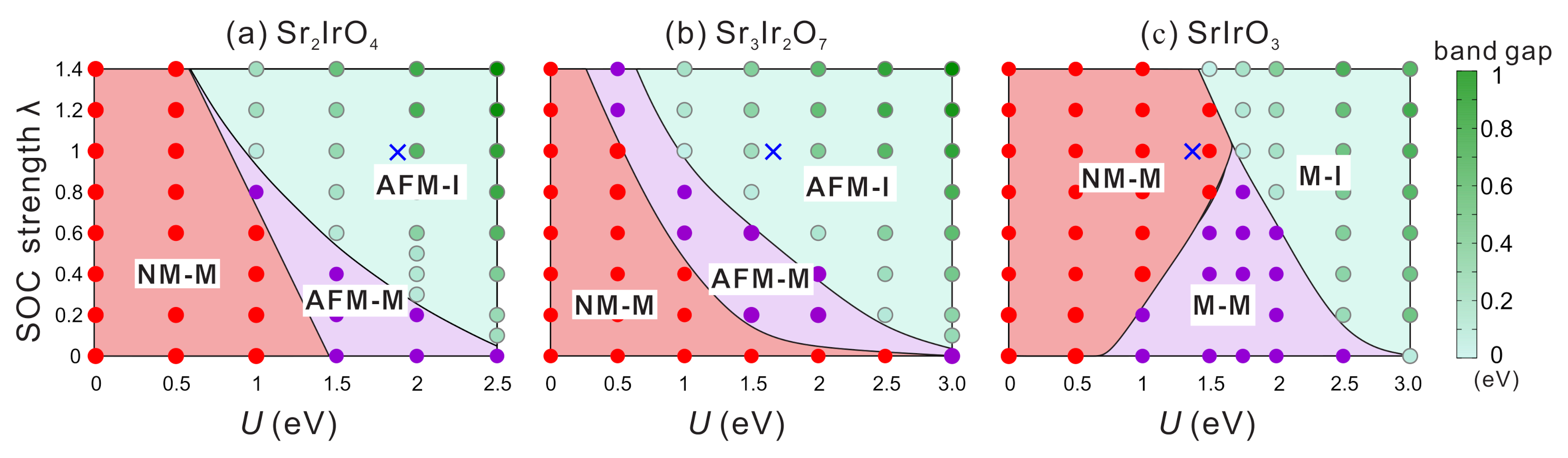
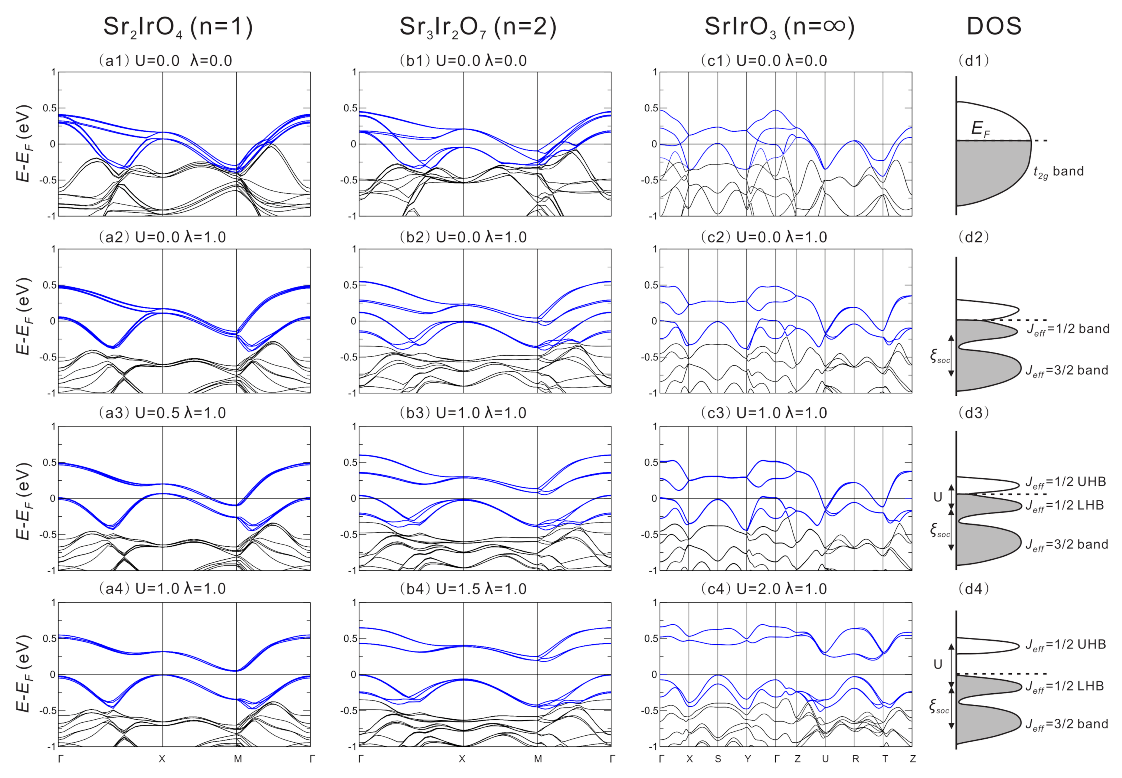
4.2.1. Interplay between U and SOC
4.2.2. Quantification of Relevant Energy Scales
4.2.3. vs. LSDA+U
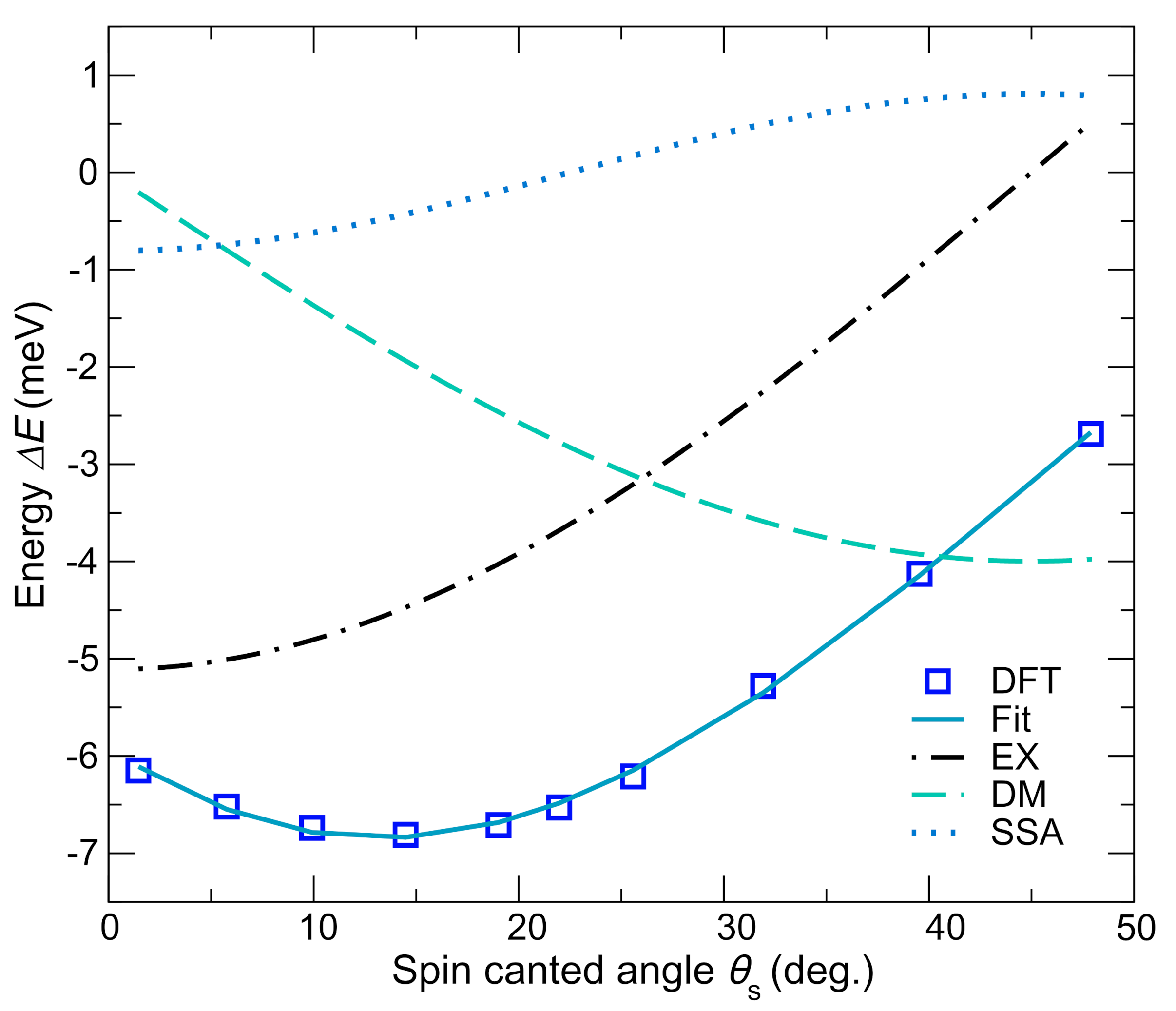
4.3. Magnetic Properties
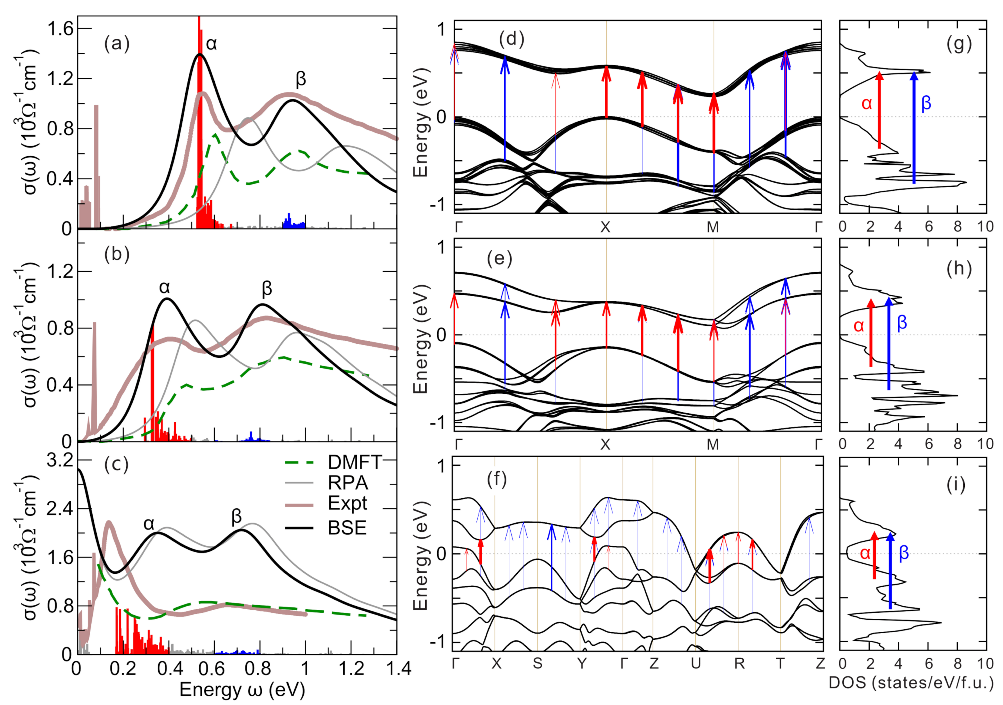
4.4. Optical Spectra
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rau, J.G.; Lee, E.K.H.; Kee, H.Y. Spin-Orbit Physics Giving Rise to Novel Phases in Correlated Systems: Iridates and Related Materials. Annu. Rev. Condens. Matter Phys. 2016, 7, 195–221. [Google Scholar] [CrossRef] [Green Version]
- Witczak-Krempa, W.; Chen, G.; Kim, Y.B.; Balents, L. Correlated Quantum Phenomena in the Strong Spin-Orbit Regime. Annu. Rev. Condens. Matter Phys. 2014, 5, 57–82. [Google Scholar] [CrossRef] [Green Version]
- Cao, G.; Schlottmann, P. The challenge of spin–orbit-tuned ground states in iridates: A key issues review. Rep. Prog. Phys. 2018, 81, 042502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, C.; Aichhorn, M.; Biermann, S. Coulomb correlations in 4d and 5d oxides from first principles—Or how spin–orbit materials choose their effective orbital degeneracies. J. Phys. Condens. Matter 2017, 29, 263001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, L.; Meyers, D.; Dean, M.; Liu, J. Novel spin-orbit coupling driven emergent states in iridate-based heterostructures. J. Phys. Chem. Solids 2019, 128, 39–53. [Google Scholar] [CrossRef]
- Lu, C.; Liu, J.M. The Jeff = 1/2 Antiferromagnet Sr2IrO4: A Golden Avenue toward New Physics and Functions. Adv. Mater. 2020, 32, 1904508. [Google Scholar] [CrossRef]
- Zhang, L.; Pang, B.; Chen, Y.B.; Chen, Y. Review of Spin-orbit Coupled Semimetal SrIrO3 in Thin Film Form. Crit. Rev. Solid State Mater. Sci. 2018, 43, 367–391. [Google Scholar] [CrossRef]
- Kim, B.J.; Jin, H.; Moon, S.J.; Kim, J.Y.; Park, B.G.; Leem, C.S.; Yu, J.; Noh, T.; Kim, C.; Oh, S.J.; et al. Novel Jeff = 1/2 Mott State Induced by Relativistic Spin-Orbit Coupling in Sr2IrO4. Phys. Rev. Lett. 2008, 101, 076402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.J.; Ohsumi, H.; Komesu, T.; Sakai, S.; Morita, T.; Takagi, H.; Arima, T. Phase-Sensitive Observation of a Spin-Orbital Mott State in Sr2IrO4. Science 2009, 323, 1329–1332. [Google Scholar] [CrossRef]
- Jackeli, G.; Khaliullin, G. Mott Insulators in the Strong Spin-Orbit Coupling Limit: From Heisenberg to a Quantum Compass and Kitaev Models. Phys. Rev. Lett. 2009, 102, 017205. [Google Scholar] [CrossRef] [Green Version]
- Moon, S.J.; Jin, H.; Kim, K.W.; Choi, W.S.; Lee, Y.S.; Yu, J.; Cao, G.; Sumi, A.; Funakubo, H.; Bernhard, C.; et al. Dimensionality-Controlled Insulator-Metal Transition and Correlated Metallic State in 5d Transition Metal Oxides Srn+1IrnO3n+1 (n = 1, 2, and ∞). Phys. Rev. Lett. 2008, 101, 226402. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, A.; Fujiwara, H.; Tachibana, S.; Iwasaki, D.; Higashino, Y.; Yoshimi, C.; Nakagawa, K.; Nakatani, Y.; Yamagami, K.; Aratani, H.; et al. Three-dimensional electronic structures and the metal-insulator transition in Ruddlesden-Popper iridates. Phys. Rev. B 2016, 94, 115103. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Shirakawa, T.; Yunoki, S. Microscopic Study of a Spin-Orbit-Induced Mott Insulator in Ir Oxides. Phys. Rev. Lett. 2010, 105, 216410. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Khmelevskyi, S.; Kim, B.; Marsman, M.; Li, D.; Chen, X.Q.; Sarma, D.D.; Kresse, G.; Franchini, C. Anisotropic magnetic couplings and structure-driven canted to collinear transitions in Sr2IrO4 by magnetically constrained noncollinear DFT. Phys. Rev. B 2015, 92, 054428. [Google Scholar] [CrossRef] [Green Version]
- Fujiyama, S.; Ohsumi, H.; Komesu, T.; Matsuno, J.; Kim, B.J.; Takata, M.; Arima, T.; Takagi, H. Two-Dimensional Heisenberg Behavior of Jeff = 1/2 Isospins in the Paramagnetic State of the Spin-Orbital Mott Insulator Sr2IrO4. Phys. Rev. Lett. 2012, 108, 247212. [Google Scholar] [CrossRef] [Green Version]
- Dhital, C.; Hogan, T.; Yamani, Z.; de la Cruz, C.; Chen, X.; Khadka, S.; Ren, Z.; Wilson, S.D. Neutron scattering study of correlated phase behavior in Sr2IrO4. Phys. Rev. B 2013, 87, 144405. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.; Chi, S.; Chakoumakos, B.C.; Fernandez-Baca, J.A.; Qi, T.; Cao, G. Magnetic and crystal structures of Sr2IrO4: A neutron diffraction study. Phys. Rev. B 2013, 87, 140406. [Google Scholar] [CrossRef] [Green Version]
- Moon, S.J.; Jin, H.; Choi, W.S.; Lee, J.S.; Seo, S.S.A.; Yu, J.; Cao, G.; Noh, T.W.; Lee, Y.S. Temperature dependence of the electronic structure of the Jeff = 1/2 Mott insulator Sr2IrO4 studied by optical spectroscopy. Phys. Rev. B 2009, 80, 195110. [Google Scholar] [CrossRef] [Green Version]
- Arita, R.; Kuneš, J.; Kozhevnikov, A.V.; Eguiluz, A.G.; Imada, M. Ab initio. Phys. Rev. Lett. 2012, 108, 086403. [Google Scholar] [CrossRef] [Green Version]
- Katukuri, V.M.; Stoll, H.; van den Brink, J.; Hozoi, L. Ab initio. Phys. Rev. B 2012, 85, 220402. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Casa, D.; Upton, M.H.; Gog, T.; Kim, Y.J.; Mitchell, J.F.; van Veenendaal, M.; Daghofer, M.; van den Brink, J.; Khaliullin, G.; et al. Magnetic Excitation Spectra of Sr2IrO4 Probed by Resonant Inelastic X-Ray Scattering: Establishing Links to Cuprate Superconductors. Phys. Rev. Lett. 2012, 108, 177003. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Senthil, T. Twisted Hubbard Model for Sr2IrO4: Magnetism and Possible High Temperature Superconductivity. Phys. Rev. Lett. 2011, 106, 136402. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Shirakawa, T.; Yunoki, S. Monte Carlo Study of an Unconventional Superconducting Phase in Iridium Oxide Jeff = 1/2 Mott Insulators Induced by Carrier Doping. Phys. Rev. Lett. 2013, 110, 027002. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.K.; Krupin, O.; Denlinger, J.D.; Bostwick, A.; Rotenberg, E.; Zhao, Q.; Mitchell, J.F.; Allen, J.W.; Kim, B.J. Fermi arcs in a doped pseudospin-1/2 Heisenberg antiferromagnet. Science 2014, 345, 187–190. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.K.; Sung, N.H.; Denlinger, J.D.; Kim, B.J. Observation of a d-wave gap in electron-doped Sr2IrO4. Nat. Phys. 2015, 12, 37. [Google Scholar] [CrossRef]
- Lupascu, A.; Clancy, J.P.; Gretarsson, H.; Nie, Z.; Nichols, J.; Terzic, J.; Cao, G.; Seo, S.S.A.; Islam, Z.; Upton, M.H.; et al. Tuning Magnetic Coupling in Sr2IrO4 Thin Films with Epitaxial Strain. Phys. Rev. Lett. 2014, 112, 147201. [Google Scholar] [CrossRef] [Green Version]
- Wojek, B.M.; Berntsen, M.H.; Boseggia, S.; Boothroyd, A.T.; Prabhakaran, D.; McMorrow, D.F.; Ronnow, H.M.; Chang, J.; Tjernberg, O. The Jeff = 1/2 insulator Sr3Ir2O7 studied by means of angle-resolved photoemission spectroscopy. J. Phys. Condens. Matter 2012, 24, 415602. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Cao, Y.; Waugh, J.A.; Park, S.R.; Qi, T.F.; Korneta, O.B.; Cao, G.; Dessau, D.S. Dimensionality-controlled Mott transition and correlation effects in single-layer and bilayer perovskite iridates. Phys. Rev. B 2013, 87, 245109. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Walkup, D.; Lin, H.; Dhital, C.; Chang, T.R.; Khadka, S.; Zhou, W.; Jeng, H.T.; Paranjape, M.; Bansil, A.; et al. Imaging the evolution of metallic states in a correlated iridate. Nat. Mater. 2013, 12, 707. [Google Scholar] [CrossRef] [PubMed]
- Fujiyama, S.; Ohashi, K.; Ohsumi, H.; Sugimoto, K.; Takayama, T.; Komesu, T.; Takata, M.; Arima, T.; Takagi, H. Weak antiferromagnetism of Jeff = 1/2 band in bilayer iridate Sr3Ir2O7. Phys. Rev. B 2012, 86, 174414. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Sohn, C.H.; Jeong, D.W.; Cao, G.; Kim, K.W.; Moon, S.J.; Jin, H.; Cho, D.Y.; Noh, T.W. Phonon-assisted optical excitation in the narrow bandgap Mott insulator Sr3Ir2O7. Phys. Rev. B 2014, 89, 155115. [Google Scholar] [CrossRef] [Green Version]
- Boseggia, S.; Springell, R.; Walker, H.C.; Boothroyd, A.T.; Prabhakaran, D.; Collins, S.P.; McMorrow, D.F. On the magnetic structure of Sr3Ir2O7: An X-ray resonant scattering study. J. Phys. Condens. Matter 2012, 24, 312202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.W.; Choi, Y.; Kim, J.; Mitchell, J.F.; Jackeli, G.; Daghofer, M.; van den Brink, J.; Khaliullin, G.; Kim, B.J. Dimensionality Driven Spin-Flop Transition in Layered Iridates. Phys. Rev. Lett. 2012, 109, 037204. [Google Scholar] [CrossRef]
- Kim, H.S.; Chen, Y.; Kee, H.Y. Surface states of perovskite iridates AIrO3: Signatures of a topological crystalline metal with nontrivial ℤ2 index. Phys. Rev. B 2015, 91, 235103. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Lu, Y.M.; Kee, H.Y. Topological crystalline metal in orthorhombic perovskite iridates. Nat. Commun. 2015, 6, 6593. [Google Scholar] [CrossRef]
- Fujioka, J.; Okawa, T.; Yamamoto, A.; Tokura, Y. Correlated Dirac semimetallic state with unusual positive magnetoresistance in strain-free perovskite SrIrO3. Phys. Rev. B 2017, 95, 121102. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Liu, P.; Andersson, D.A.; Stanek, C.R.; Ozaki, T.; Franchini, C. Parametrization of LSDA+U for noncollinear magnetic configurations: Multipolar magnetism in UO2. Phys. Rev. Mater. 2019, 3, 083802. [Google Scholar] [CrossRef] [Green Version]
- Wahl, R. The Crystal Lattice: Phonons, Solitons, Dislocations; Wiley-VCH Weinheim: Berlin, Germany, 1999; p. 43. [Google Scholar]
- Coury, M.E.A.; Dudarev, S.L.; Foulkes, W.M.C.; Horsfield, A.P.; Ma, P.W.; Spencer, J.S. Hubbard-like Hamiltonians for interacting electrons in s,p, and d orbitals. Phys. Rev. B 2016, 93, 075101. [Google Scholar] [CrossRef] [Green Version]
- Anisimov, V.I.; Zaanen, J.; Andersen, O.K. Band theory and Mott insulators: Hubbard U instead of Stoner I. Phys. Rev. B 1991, 44, 943–954. [Google Scholar] [CrossRef] [Green Version]
- Liechtenstein, A.I.; Anisimov, V.I.; Zaanen, J. Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators. Phys. Rev. B 1995, 52, R5467–R5470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Cococcioni, M.; de Gironcoli, S. Linear response approach to the calculation of the effective interaction parameters in the LDA+U method. Phys. Rev. B 2005, 71, 035105. [Google Scholar] [CrossRef] [Green Version]
- Biermann, S.; Aryasetiawan, F.; Georges, A. First-Principles Approach to the Electronic Structure of Strongly Correlated Systems: Combining the GW Approximation and Dynamical Mean-Field Theory. Phys. Rev. Lett. 2003, 90, 086402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aryasetiawan, F.; Imada, M.; Georges, A.; Kotliar, G.; Biermann, S.; Lichtenstein, A.I. Frequency-dependent local interactions and low-energy effective models from electronic structure calculations. Phys. Rev. B 2004, 70, 195104. [Google Scholar] [CrossRef] [Green Version]
- Aryasetiawan, F.; Karlsson, K.; Jepsen, O.; Schönberger, U. Calculations of Hubbard U from first-principles. Phys. Rev. B 2006, 74, 125106. [Google Scholar] [CrossRef] [Green Version]
- Metzner, W.; Vollhardt, D. Correlated Lattice Fermions in d=∞ Dimensions. Phys. Rev. Lett. 1989, 62, 324–327. [Google Scholar] [CrossRef]
- Georges, A.; Kotliar, G.; Krauth, W.; Rozenberg, M.J. Dynamical mean-field theory of strongly correlated fermion systems and the limit of infinite dimensions. Rev. Mod. Phys. 1996, 68, 13–125. [Google Scholar] [CrossRef] [Green Version]
- Kotliar, G.; Savrasov, S.Y.; Haule, K.; Oudovenko, V.S.; Parcollet, O.; Marianetti, C.A. Electronic structure calculations with dynamical mean-field theory. Rev. Mod. Phys. 2006, 78, 865–951. [Google Scholar] [CrossRef] [Green Version]
- Held, K.; Nekrasov, I.A.; Keller, G.; Eyert, V.; Blümer, N.; McMahan, A.K.; Scalettar, R.T.; Pruschke, T.; Anisimov, V.I.; Vollhardt, D. Realistic investigations of correlated electron systems with LDA + DMFT. Phys. Status Solidi B 2006, 243, 2599–2631. [Google Scholar] [CrossRef]
- Held, K.; Andersen, O.K.; Feldbacher, M.; Yamasaki, A.; Yang, Y.F. Bandstructure meets many-body theory: The LDA+ DMFT method. J. Phys. Condens. Matter 2008, 20, 064202. [Google Scholar] [CrossRef]
- Pavarini, E.; Koch, E.; Vollhardt, D.; Lichtenstein, A.E. The LDA+DMFT Approach to Strongly Correlated Materials: Lecture Notes of the Autumn School 2011, Hands-on LDA+DMFT; Autumn School Organized by the DFG Research Unit 1346 Dynamical Mean-Field Approach with Predictive Power for Strongly Correlated Materials at Forschungszentrum Jülich 4𠄳7 October 2011; Forschungszentrum: Jülich, Germany, 2011. [Google Scholar]
- Vaugier, L.; Jiang, H.; Biermann, S. Hubbard U and Hund exchange J in transition metal oxides: Screening versus localization trends from constrained random phase approximation. Phys. Rev. B 2012, 86, 165105. [Google Scholar] [CrossRef] [Green Version]
- Marzari, N.; Vanderbilt, D. Maximally localized generalized Wannier functions for composite energy bands. Phys. Rev. B 1997, 56, 12847–12865. [Google Scholar] [CrossRef] [Green Version]
- Mostofi, A.A.; Yates, J.R.; Lee, Y.S.; Souza, I.; Vanderbilt, D.; Marzari, N. wannier90: A tool for obtaining maximally-localised Wannier functions. Comput. Phys. Commun. 2008, 178, 685–699. [Google Scholar] [CrossRef] [Green Version]
- Van Roekeghem, A.; Ayral, T.; Tomczak, J.M.; Casula, M.; Xu, N.; Ding, H.; Ferrero, M.; Parcollet, O.; Jiang, H.; Biermann, S. Dynamical Correlations and Screened Exchange on the Experimental Bench: Spectral Properties of the Cobalt Pnictide BaCo2As2. Phys. Rev. Lett. 2014, 113, 266403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomczak, J.M.; Liu, P.; Toschi, A.; Kresse, G.; Held, K. Merging GW with DMFT and non-local correlations beyond. Eur. Phys. J. Spec. Top. 2017, 226, 2565–2590. [Google Scholar] [CrossRef] [Green Version]
- Kutepov, A.; Haule, K.; Savrasov, S.Y.; Kotliar, G. Self-consistent GW determination of the interaction strength: Application to the iron arsenide superconductors. Phys. Rev. B 2010, 82, 045105. [Google Scholar] [CrossRef] [Green Version]
- Nomura, Y.; Kaltak, M.; Nakamura, K.; Taranto, C.; Sakai, S.; Toschi, A.; Arita, R.; Held, K.; Kresse, G.; Imada, M. Effective on-site interaction for dynamical mean-field theory. Phys. Rev. B 2012, 86, 085117. [Google Scholar] [CrossRef] [Green Version]
- Miyake, T.; Aryasetiawan, F. Screened Coulomb interaction in the maximally localized Wannier basis. Phys. Rev. B 2008, 77, 085122. [Google Scholar] [CrossRef] [Green Version]
- Amadon, B.; Applencourt, T.; Bruneval, F. Screened Coulomb interaction calculations: cRPA implementation and applications to dynamical screening and self-consistency in uranium dioxide and cerium. Phys. Rev. B 2014, 89, 125110. [Google Scholar] [CrossRef] [Green Version]
- Şaşıoğlu, E.; Friedrich, C.; Blügel, S. Effective Coulomb interaction in transition metals from constrained random-phase approximation. Phys. Rev. B 2011, 83, 121101. [Google Scholar] [CrossRef] [Green Version]
- Shih, B.C.; Zhang, Y.; Zhang, W.; Zhang, P. Screened Coulomb interaction of localized electrons in solids from first principles. Phys. Rev. B 2012, 85, 045132. [Google Scholar] [CrossRef] [Green Version]
- Kaltak, M. Merging GW with DMFT. Ph.D. Thesis, University of Vienna, Vienna, Austria, 2015. [Google Scholar]
- Freysoldt, C.; Grabowski, B.; Hickel, T.; Neugebauer, J.; Kresse, G.; Janotti, A.; Van de Walle, C.G. First-principles calculations for point defects in solids. Rev. Mod. Phys. 2014, 86, 253–305. [Google Scholar] [CrossRef]
- Hedin, L. New Method for Calculating the One-Particle Green’s Function with Application to the Electron-Gas Problem. Phys. Rev. 1965, 139, A796–A823. [Google Scholar] [CrossRef]
- Hedin, L.; Lundqvist, S. Solid State Physics; Academic: New York, NY, USA, 1969. [Google Scholar]
- Rojas, H.N.; Godby, R.W.; Needs, R.J. Space-Time Method for Ab Initio Calculations of Self-Energies and Dielectric Response Functions of Solids. Phys. Rev. Lett. 1995, 74, 1827–1830. [Google Scholar] [CrossRef]
- Aryasetiawan, F. Advances in Condensed Matter Science; Gordon and Breach: New York, NY, USA, 2000. [Google Scholar]
- Onida, G.; Reining, L.; Rubio, A. Electronic excitations: Density-functional versus many-body Green’s-function approaches. Rev. Mod. Phys. 2002, 74, 601–659. [Google Scholar] [CrossRef] [Green Version]
- Golze, D.; Dvorak, M.; Rinke, P. The GW Compendium: A Practical Guide to Theoretical Photoemission Spectroscopy. Front. Chem. 2019, 7, 377. [Google Scholar] [CrossRef] [PubMed]
- Strinati, G.; Mattausch, H.J.; Hanke, W. Dynamical Correlation Effects on the Quasiparticle Bloch States of a Covalent Crystal. Phys. Rev. Lett. 1980, 45, 290–294. [Google Scholar] [CrossRef]
- Strinati, G.; Mattausch, H.J.; Hanke, W. Dynamical aspects of correlation corrections in a covalent crystal. Phys. Rev. B 1982, 25, 2867–2888. [Google Scholar] [CrossRef]
- Hybertsen, M.S.; Louie, S.G. First-Principles Theory of Quasiparticles: Calculation of Band Gaps in Semiconductors and Insulators. Phys. Rev. Lett. 1985, 55, 1418–1421. [Google Scholar] [CrossRef] [PubMed]
- Hybertsen, M.S.; Louie, S.G. Electron correlation in semiconductors and insulators: Band gaps and quasiparticle energies. Phys. Rev. B 1986, 34, 5390–5413. [Google Scholar] [CrossRef] [PubMed]
- Aulbur, W.G.; Jonsson, L.; Wilkins, J.W. Quasiparticle Calculations in Solids. Solid State Phys. 1999, 54, 1–218. [Google Scholar] [CrossRef]
- Van Schilfgaarde, M.; Kotani, T.; Faleev, S. Quasiparticle Self-Consistent GW Theory. Phys. Rev. Lett. 2006, 96, 226402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shishkin, M.; Kresse, G. Self-consistent GW calculations for semiconductors and insulators. Phys. Rev. B 2007, 75, 235102. [Google Scholar] [CrossRef]
- Ergönenc, Z.; Kim, B.; Liu, P.; Kresse, G.; Franchini, C. Converged GW quasiparticle energies for transition metal oxide perovskites. Phys. Rev. Mater. 2018, 2, 024601. [Google Scholar] [CrossRef] [Green Version]
- Shishkin, M.; Marsman, M.; Kresse, G. Accurate Quasiparticle Spectra from Self-Consistent GW Calculations with Vertex Corrections. Phys. Rev. Lett. 2007, 99, 246403. [Google Scholar] [CrossRef] [Green Version]
- Holm, B.; von Barth, U. Fully self-consistent GW self-energy of the electron gas. Phys. Rev. B 1998, 57, 2108–2117. [Google Scholar] [CrossRef] [Green Version]
- Stan, A.; Dahlen, N.E.; van Leeuwen, R. Levels of self-consistency in the GW approximation. J. Chem. Phys. 2009, 130, 114105. [Google Scholar] [CrossRef] [Green Version]
- Kutepov, A.L. Self-consistent solution of Hedin’s equations: Semiconductors and insulators. Phys. Rev. B 2017, 95, 195120. [Google Scholar] [CrossRef] [Green Version]
- Grumet, M.; Liu, P.; Kaltak, M.; Klimeš, J.; Kresse, G. Beyond the quasiparticle approximation: Fully self-consistent GW calculations. Phys. Rev. B 2018, 98, 155143. [Google Scholar] [CrossRef] [Green Version]
- Kutepov, A.L. Electronic structure of Na, K, Si, and LiF from self-consistent solution of Hedin’s equations including vertex corrections. Phys. Rev. B 2016, 94, 155101. [Google Scholar] [CrossRef] [Green Version]
- Maggio, E.; Kresse, G. GW Vertex Corrected Calculations for Molecular Systems. J. Chem. Theory Comput. 2017, 13, 4765–4778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruneval, F.; Gonze, X. Accurate GW self-energies in a plane-wave basis using only a few empty states: Towards large systems. Phys. Rev. B 2008, 78, 085125. [Google Scholar] [CrossRef]
- Giustino, F.; Cohen, M.L.; Louie, S.G. GW method with the self-consistent Sternheimer equation. Phys. Rev. B 2010, 81, 115105. [Google Scholar] [CrossRef] [Green Version]
- Govoni, M.; Galli, G. Large Scale GW Calculations. J. Chem. Theory Comput. 2015, 11, 2680–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umari, P.; Stenuit, G.; Baroni, S. GW quasiparticle spectra from occupied states only. Phys. Rev. B 2010, 81, 115104. [Google Scholar] [CrossRef] [Green Version]
- Foerster, D.; Koval, P.; Sánchez-Portal, D. An O(N3) implementation of Hedin’s GW approximation for molecules. J. Chem. Phys. 2011, 135, 074105. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Kaltak, M.; Klimeš, J.; Kresse, G. Cubic scaling GW: Towards fast quasiparticle calculations. Phys. Rev. B 2016, 94, 165109. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, J.; Golze, D.; Talirz, L.; Hutter, J.; Pignedoli, C.A. Toward GW Calculations on Thousands of Atoms. J. Phys. Chem. Lett. 2018, 9, 306–312. [Google Scholar] [CrossRef]
- Kim, M.; Martyna, G.J.; Ismail-Beigi, S. Complex-time shredded propagator method for large-scale GW calculations. Phys. Rev. B 2020, 101, 035139. [Google Scholar] [CrossRef] [Green Version]
- Ben, M.D.; Yang, C.; Li, Z.; da Jornada, F.H.; Louie, S.G.; Deslippe, J. SC ’20: Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, Atlanta, GA, USA, 9–19 November 2020; IEEE Press: New York, NY, USA, 2020. [Google Scholar]
- Bruneval, F.; Marques, M.A.L. Benchmarking the Starting Points of the GW Approximation for Molecules. J. Chem. Theory Comput. 2013, 9, 324–329. [Google Scholar] [CrossRef]
- Fuchs, F.; Furthmüller, J.; Bechstedt, F.; Shishkin, M.; Kresse, G. Quasiparticle band structure based on a generalized Kohn-Sham scheme. Phys. Rev. B 2007, 76, 115109. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Gomez-Abal, R.I.; Rinke, P.; Scheffler, M. Localized and Itinerant States in Lanthanide Oxides United by GW@LDA + U. Phys. Rev. Lett. 2009, 102, 126403. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Gomez-Abal, R.I.; Rinke, P.; Scheffler, M. First-principles modeling of localized d states with the GW@LDA + U approach. Phys. Rev. B 2010, 82, 045108. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Kim, B.; Chen, X.Q.; Sarma, D.D.; Kresse, G.; Franchini, C. Relativistic GW+BSE study of the optical properties of Ruddlesden-Popper iridates. Phys. Rev. Mater. 2018, 2, 075003. [Google Scholar] [CrossRef] [Green Version]
- Held, K. Electronic structure calculations using dynamical mean field theory. Adv. Phys. 2007, 56, 829–926. [Google Scholar] [CrossRef] [Green Version]
- Boehnke, L.; Nilsson, F.; Aryasetiawan, F.; Werner, P. When strong correlations become weak: Consistent merging of GW and DMFT. Phys. Rev. B 2016, 94, 201106. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, F.; Boehnke, L.; Werner, P.; Aryasetiawan, F. Multitier self-consistent GW + EDMFT. Phys. Rev. Mater. 2017, 1, 043803. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Rinke, P.; Joas, C.; Scheffler, M. Random-phase approximation and its applications in computational chemistry and materials science. J. Mater. Sci. 2012, 47, 7447–7471. [Google Scholar] [CrossRef]
- Chen, G.P.; Voora, V.K.; Agee, M.M.; Balasubramani, S.G.; Furche, F. Random-Phase Approximation Methods. Annu. Rev. Phys. Chem. 2017, 68, 421–445. [Google Scholar] [CrossRef]
- Hanke, W.; Sham, L.J. Many-particle effects in the optical spectrum of a semiconductor. Phys. Rev. B 1980, 21, 4656–4673. [Google Scholar] [CrossRef]
- Rohlfing, M.; Louie, S.G. Excitonic Effects and the Optical Absorption Spectrum of Hydrogenated Si Clusters. Phys. Rev. Lett. 1998, 80, 3320–3323. [Google Scholar] [CrossRef]
- Benedict, L.X.; Shirley, E.L.; Bohn, R.B. Optical Absorption of Insulators and the Electron-Hole Interaction: An Ab Initio Calculation. Phys. Rev. Lett. 1998, 80, 4514–4517. [Google Scholar] [CrossRef] [Green Version]
- Strinati, G. Effects of dynamical screening on resonances at inner-shell thresholds in semiconductors. Phys. Rev. B 1984, 29, 5718–5726. [Google Scholar] [CrossRef]
- Rohlfing, M.; Louie, S.G. Electron-Hole Excitations in Semiconductors and Insulators. Phys. Rev. Lett. 1998, 81, 2312–2315. [Google Scholar] [CrossRef]
- Albrecht, S.; Reining, L.; Del Sole, R.; Onida, G. Ab Initio Calculation of Excitonic Effects in the Optical Spectra of Semiconductors. Phys. Rev. Lett. 1998, 80, 4510–4513. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G. Effects of the Electron-Hole Interaction on the Optical Properties of Materials: The Bethe-Salpeter Equation. Phys. Scr. 2004, T109, 141. [Google Scholar] [CrossRef]
- Blase, X.; Duchemin, I.; Jacquemin, D. The Bethe-Salpeter equation in chemistry: Relations with TD-DFT, applications and challenges. Chem. Soc. Rev. 2018, 47, 1022–1043. [Google Scholar] [CrossRef]
- Sharifzadeh, S. Many-body perturbation theory for understanding optical excitations in organic molecules and solids. J. Phys. Condens. Matter 2018, 30, 153002. [Google Scholar] [CrossRef] [Green Version]
- Rohlfing, M.; Louie, S.G. Electron-hole excitations and optical spectra from first principles. Phys. Rev. B 2000, 62, 4927–4944. [Google Scholar] [CrossRef]
- Tiago, M.L.; Chelikowsky, J.R. Optical excitations in organic molecules, clusters, and defects studied by first-principles Green’s function methods. Phys. Rev. B 2006, 73, 205334. [Google Scholar] [CrossRef] [Green Version]
- Körbel, S.; Boulanger, P.; Duchemin, I.; Blase, X.; Marques, M.A.L.; Botti, S. Benchmark Many-Body GW and Bethe-Salpeter Calculations for Small Transition Metal Molecules. J. Chem. Theory Comput. 2014, 10, 3934–3943. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Hummer, K.; Franchini, C. Stacking effects on the electronic and optical properties of bilayer transition metal dichalcogenides MoS2, MoSe2, WS2, and WSe2. Phys. Rev. B 2014, 89, 075409. [Google Scholar] [CrossRef]
- Cunningham, B.; Grüning, M.; Azarhoosh, P.; Pashov, D.; van Schilfgaarde, M. Effect of ladder diagrams on optical absorption spectra in a quasiparticle self-consistent GW framework. Phys. Rev. Mater. 2018, 2, 034603. [Google Scholar] [CrossRef] [Green Version]
- Franchini, C.; Sanna, A.; Marsman, M.; Kresse, G. Structural, vibrational, and quasiparticle properties of the Peierls semiconductor BaBiO3: A hybrid functional and self-consistent GW + vertex-corrections study. Phys. Rev. B 2010, 81, 085213. [Google Scholar] [CrossRef]
- Sponza, L.; Véniard, V.; Sottile, F.; Giorgetti, C.; Reining, L. Role of localized electrons in electron-hole interaction: The case of SrTiO3. Phys. Rev. B 2013, 87, 235102. [Google Scholar] [CrossRef]
- Gatti, M.; Sottile, F.; Reining, L. Electron-hole interactions in correlated electron materials: Optical properties of vanadium dioxide from first principles. Phys. Rev. B 2015, 91, 195137. [Google Scholar] [CrossRef]
- Sander, T.; Maggio, E.; Kresse, G. Beyond the Tamm-Dancoff approximation for extended systems using exact diagonalization. Phys. Rev. B 2015, 92, 045209. [Google Scholar] [CrossRef]
- Gross, E.K.U.; Kohn, W. Local density-functional theory of frequency-dependent linear response. Phys. Rev. Lett. 1985, 55, 2850–2852. [Google Scholar] [CrossRef]
- Runge, E.; Gross, E.K.U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Botti, S.; Schindlmayr, A.; Sole, R.D.; Reining, L. Time-dependent density-functional theory for extended systems. Rep. Prog. Phys. 2007, 70, 357–407. [Google Scholar] [CrossRef]
- Marini, A.; Del Sole, R.; Rubio, A. Bound Excitons in Time-Dependent Density-Functional Theory: Optical and Energy-Loss Spectra. Phys. Rev. Lett. 2003, 91, 256402. [Google Scholar] [CrossRef] [Green Version]
- Reining, L.; Olevano, V.; Rubio, A.; Onida, G. Excitonic Effects in Solids Described by Time-Dependent Density-Functional Theory. Phys. Rev. Lett. 2002, 88, 066404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sottile, F.; Olevano, V.; Reining, L. Parameter-Free Calculation of Response Functions in Time-Dependent Density-Functional Theory. Phys. Rev. Lett. 2003, 91, 056402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adragna, G.; Del Sole, R.; Marini, A. Ab initio calculation of the exchange-correlation kernel in extended systems. Phys. Rev. B 2003, 68, 165108. [Google Scholar] [CrossRef]
- Sharma, S.; Dewhurst, J.K.; Sanna, A.; Gross, E.K.U. Bootstrap Approximation for the Exchange-Correlation Kernel of Time-Dependent Density-Functional Theory. Phys. Rev. Lett. 2011, 107, 186401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigamonti, S.; Botti, S.; Veniard, V.; Draxl, C.; Reining, L.; Sottile, F. Estimating Excitonic Effects in the Absorption Spectra of Solids: Problems and Insight from a Guided Iteration Scheme. Phys. Rev. Lett. 2015, 114, 146402. [Google Scholar] [CrossRef]
- Yang, Z.H.; Sottile, F.; Ullrich, C.A. Simple screened exact-exchange approach for excitonic properties in solids. Phys. Rev. B 2015, 92, 035202. [Google Scholar] [CrossRef] [Green Version]
- Refaely-Abramson, S.; Jain, M.; Sharifzadeh, S.; Neaton, J.B.; Kronik, L. Solid-state optical absorption from optimally tuned time-dependent range-separated hybrid density functional theory. Phys. Rev. B 2015, 92, 081204. [Google Scholar] [CrossRef] [Green Version]
- Wing, D.; Haber, J.B.; Noff, R.; Barker, B.; Egger, D.A.; Ramasubramaniam, A.; Louie, S.G.; Neaton, J.B.; Kronik, L. Comparing time-dependent density functional theory with many-body perturbation theory for semiconductors: Screened range-separated hybrids and the GW plus Bethe-Salpeter approach. Phys. Rev. Mater. 2019, 3, 064603. [Google Scholar] [CrossRef]
- Sun, J.; Yang, J.; Ullrich, C.A. Low-cost alternatives to the Bethe-Salpeter equation: Towards simple hybrid functionals for excitonic effects in solids. Phys. Rev. Res. 2020, 2, 013091. [Google Scholar] [CrossRef] [Green Version]
- Bokdam, M.; Sander, T.; Stroppa, A.; Picozzi, S.; Sarma, D.D.; Franchini, C.; Kresse, G. Role of Polar Phonons in the Photo Excited State of Metal Halide Perovskites. Sci. Rep. 2016, 6, 28618. [Google Scholar] [CrossRef] [Green Version]
- Tal, A.; Liu, P.; Kresse, G.; Pasquarello, A. Accurate optical spectra through time-dependent density functional theory based on screening-dependent hybrid functionals. Phys. Rev. Res. 2020, 2, 032019. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Reticcioli, M.; Kim, B.; Continenza, A.; Kresse, G.; Sarma, D.D.; Chen, X.Q.; Franchini, C. Electron and hole doping in the relativistic Mott insulator Sr2IrO4: A first-principles study using band unfolding technique. Phys. Rev. B 2016, 94, 195145. [Google Scholar] [CrossRef] [Green Version]
- Beznosikov, B.V.; Aleksandrov, K.S. Perovskite-like crystals of the Ruddlesden-Popper series. Crystallogr. Rep. 2000, 45, 792–798. [Google Scholar] [CrossRef]
- Crawford, M.K.; Subramanian, M.A.; Harlow, R.L.; Fernandez-Baca, J.A.; Wang, Z.R.; Johnston, D.C. Structural and magnetic studies of Sr2IrO4. Phys. Rev. B 1994, 49, 9198–9201. [Google Scholar] [CrossRef]
- Subramanian, M.; Crawford, M.; Harlow, R. Single crystal structure determination of double layered strontium iridium oxide [Sr3Ir2O7]. Mater. Res. Bull. 1994, 29, 645–650. [Google Scholar] [CrossRef]
- Longo, J.; Kafalas, J.; Arnott, R. Structure and properties of the high and low pressure forms of SrIrO3. J. Solid State Chem. 1971, 3, 174–179. [Google Scholar] [CrossRef]
- Zhao, J.G.; Yang, L.X.; Yu, Y.; Li, F.Y.; Yu, R.C.; Fang, Z.; Chen, L.C.; Jin, C.Q. High-pressure synthesis of orthorhombic SrIrO3 perovskite and its positive magnetoresistance. J. Appl. Phys. 2008, 103, 103706. [Google Scholar] [CrossRef]
- Zeb, M.A.; Kee, H.Y. Interplay between spin-orbit coupling and Hubbard interaction in SrIrO3 and related Pbnm perovskite oxides. Phys. Rev. B 2012, 86, 085149. [Google Scholar] [CrossRef] [Green Version]
- Andlauer, B.; Schneider, J.; Tolksdorf, W. Optical Absorption, Fluorescence, and Electron Spin Resonance of Ir4+ on Octahedral Sites in Y3Ga5O12. Phys. Status Solidi B 1976, 73, 533–540. [Google Scholar] [CrossRef]
- Carter, J.M.; Shankar, V.V.; Zeb, M.A.; Kee, H.Y. Semimetal and Topological Insulator in Perovskite Iridates. Phys. Rev. B 2012, 85, 115105. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Masumoto, H.; Goto, T. Structural, Electrical and Optical Characterization of SrIrO3 Thin Films Prepared by Laser-Ablation. Mater. Trans. 2005, 46, 100. [Google Scholar] [CrossRef] [Green Version]
- Nie, Y.F.; King, P.D.C.; Kim, C.H.; Uchida, M.; Wei, H.I.; Faeth, B.D.; Ruf, J.P.; Ruff, J.P.C.; Xie, L.; Pan, X.; et al. Interplay of Spin-Orbit Interactions, Dimensionality, and Octahedral Rotations in Semimetallic SrIrO3. Phys. Rev. Lett. 2015, 114, 016401. [Google Scholar] [CrossRef] [Green Version]
- De la Torre, A.; McKeown Walker, S.; Bruno, F.Y.; Riccó, S.; Wang, Z.; Gutierrez Lezama, I.; Scheerer, G.; Giriat, G.; Jaccard, D.; Berthod, C.; et al. Collapse of the Mott Gap and Emergence of a Nodal Liquid in Lightly Doped Sr2IrO4. Phys. Rev. Lett. 2015, 115, 176402. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Haule, K.; Vanderbilt, D. Effective J = 1/2 Insulating State in Ruddlesden-Popper Iridates: An LDA+DMFT Study. Phys. Rev. Lett. 2013, 111, 246402. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Wang, Q.; Waugh, J.A.; Reber, T.J.; Li, H.; Zhou, X.; Parham, S.; Plumb, N.C.; Rotenberg, E.; Bostwick, A.; et al. Hallmarks of the Mott-metal crossover in the hole-doped pseudospin-1/2 Mott insulator Sr2IrO4. Nat. Commun. 2016, 7, 11367. [Google Scholar] [CrossRef]
- Brouet, V.; Mansart, J.; Perfetti, L.; Piovera, C.; Vobornik, I.; Le Fèvre, P.; Bertran, F.M.C.; Riggs, S.C.; Shapiro, M.C.; Giraldo-Gallo, P.; et al. Transfer of spectral weight across the gap of Sr2IrO4 induced by La doping. Phys. Rev. B 2015, 92, 081117. [Google Scholar] [CrossRef] [Green Version]
- Cao, G.; Bolivar, J.; McCall, S.; Crow, J.E.; Guertin, R.P. Weak ferromagnetism, metal-to-nonmetal transition, and negative differential resistivity in single-crystal Sr2IrO4. Phys. Rev. B 1998, 57, R11039–R11042. [Google Scholar] [CrossRef]
- Boseggia, S.; Walker, H.C.; Vale, J.; Springell, R.; Feng, Z.; Perry, R.S.; Sala, M.M.; Ronnow, H.M.; Collins, S.P.; McMorrow, D.F. Locking of iridium magnetic moments to the correlated rotation of oxygen octahedra in Sr2IrO4 revealed by x-ray resonant scattering. J. Phys. Condens. Matter 2013, 25, 422202. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.H.; Khaliullin, G.; Min, B.I. Magnetic Couplings, Optical Spectra, and Spin-Orbit Exciton in 5d Electron Mott Insulator Sr2IrO4. Phys. Rev. Lett. 2012, 109, 167205. [Google Scholar] [CrossRef] [Green Version]
- Haskel, D.; Fabbris, G.; Zhernenkov, M.; Kong, P.P.; Jin, C.Q.; Cao, G.; van Veenendaal, M. Pressure Tuning of the Spin-Orbit Coupled Ground State in Sr2IrO4. Phys. Rev. Lett. 2012, 109, 027204. [Google Scholar] [CrossRef] [Green Version]
- Groenendijk, D.J.; Manca, N.; Mattoni, G.; Kootstra, L.; Gariglio, S.; Huang, Y.; van Heumen, E.; Caviglia, A.D. Epitaxial growth and thermodynamic stability of SrIrO3/SrTiO3 heterostructures. Appl. Phys. Lett. 2016, 109, 041906. [Google Scholar] [CrossRef] [Green Version]
- Nishio, K.; Hwang, H.Y.; Hikita, Y. Thermodynamic guiding principles in selective synthesis of strontium iridate Ruddlesden-Popper epitaxial films. APL Mater. 2016, 4, 036102. [Google Scholar] [CrossRef]
- Sung, N.H.; Gretarsson, H.; Proepper, D.; Porras, J.; Tacon, M.L.; Boris, A.V.; Keimer, B.; Kim, B.J. Crystal growth and intrinsic magnetic behaviour of Sr2IrO4. Philos. Mag. (Abingdon) 2016, 96, 413–426. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, C.H.; Sandilands, L.J.; Sohn, C.H.; Matsuno, J.; Takagi, H.; Kim, K.W.; Lee, Y.S.; Moon, S.J.; Noh, T.W. Manipulation of electronic structure via alteration of local orbital environment in [(SrIrO3)m,(SrTiO3)](m = 0.16em0ex1,0.16em0ex2,and0.16em0ex∞) superlattices. Phys. Rev. B 2016, 94, 245113. [Google Scholar] [CrossRef]
- Paris, E.; Tseng, Y.; Pärschke, E.M.; Zhang, W.; Upton, M.H.; Efimenko, A.; Rolfs, K.; McNally, D.E.; Maurel, L.; Naamneh, M.; et al. Strain engineering of the charge and spin-orbital interactions in Sr2IrO4. Proc. Natl. Acad. Sci. USA 2020, 117, 24764–24770. [Google Scholar] [CrossRef]
- Kim, B.; Liu, P.; Franchini, C. Magnetic properties of bilayer Sr3Ir2O7: Role of epitaxial strain and oxygen vacancies. Phys. Rev. B 2017, 95, 024406. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Liu, P.; Franchini, C. Dimensionality-strain phase diagram of strontium iridates. Phys. Rev. B 2017, 95, 115111. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SrIrO | ||||||
| 2.30 | 1.72 | 1.57 | – | 0.23 | 0.22 | |
| 1.72 | 2.30 | 1.57 | 0.23 | – | 0.22 | |
| 1.57 | 1.57 | 2.03 | 0.22 | 0.22 | – | |
| SrIrO | ||||||
| 2.16 | 1.58 | 1.43 | – | 0.23 | 0.22 | |
| 1.58 | 2.16 | 1.43 | 0.23 | – | 0.22 | |
| 1.43 | 1.43 | 1.85 | 0.22 | 0.22 | – | |
| SrIrO | ||||||
| 1.78 | 1.21 | 1.19 | – | 0.22 | 0.22 | |
| 1.21 | 1.73 | 1.22 | 0.22 | – | 0.22 | |
| 1.19 | 1.22 | 1.74 | 0.22 | 0.22 | – | |
| SrIrO | SrIrO | SrIrO | |
|---|---|---|---|
| Space group | (142) | (68) | (62) |
| U | 1.82 | 1.67 | 1.39 |
| J | 0.22 | 0.22 | 0.22 |
| 10 | 3.70 | 3.80 | 4.24 |
| 0.47 | 0.49 | 0.45 | |
| Magnetic ordering | -canted AFM | c-collinear AFM | NM |
| DFT+U+SOC gap | 0.23 | 0.14 | metal |
| +SOC gap | 0.25 | 0.16 | metal |
| Expt. gap | 0.30 [18] | 0.13 [29] | metal [146,147] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, P.; Franchini, C. Advanced First-Principle Modeling of Relativistic Ruddlesden—Popper Strontium Iridates. Appl. Sci. 2021, 11, 2527. https://doi.org/10.3390/app11062527
Liu P, Franchini C. Advanced First-Principle Modeling of Relativistic Ruddlesden—Popper Strontium Iridates. Applied Sciences. 2021; 11(6):2527. https://doi.org/10.3390/app11062527
Chicago/Turabian StyleLiu, Peitao, and Cesare Franchini. 2021. "Advanced First-Principle Modeling of Relativistic Ruddlesden—Popper Strontium Iridates" Applied Sciences 11, no. 6: 2527. https://doi.org/10.3390/app11062527