First-Principles Study of the Contact Resistance at 2D Metal/2D Semiconductor Heterojunctions


Abstract
:1. Introduction
2. Computational Methods
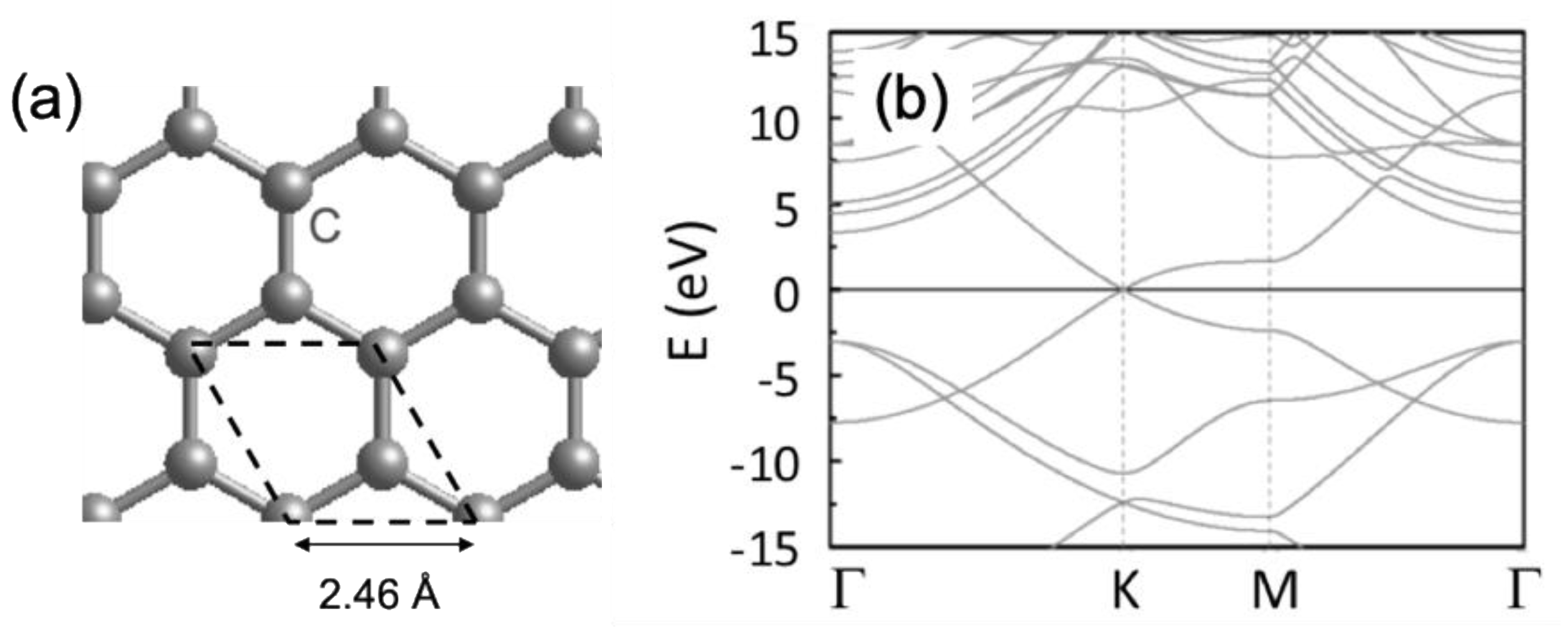
3. Structural and Electronic Properties of Graphene and MoS2
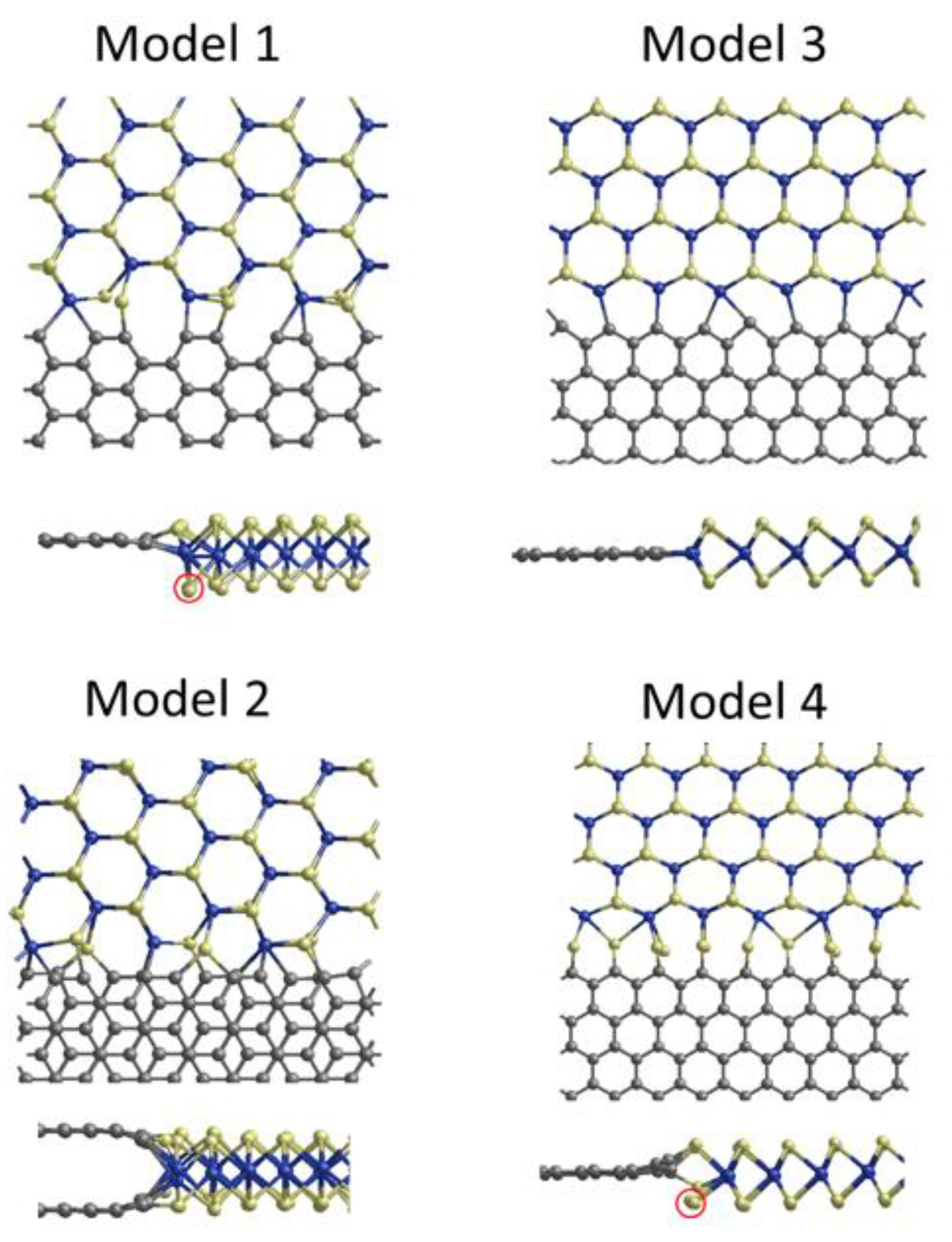
4. Graphene/2H-MoS2 Edge Contacts
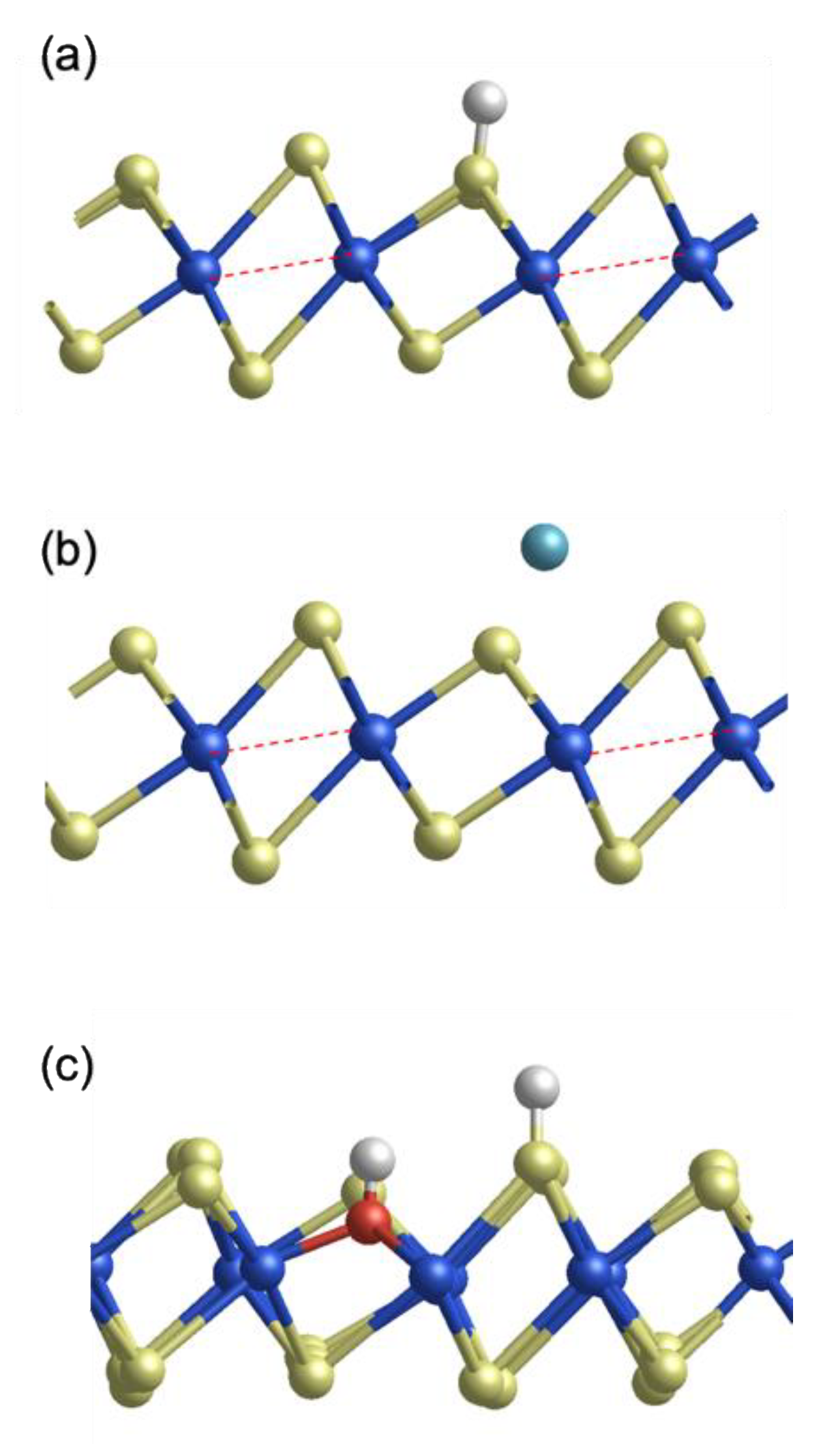
5. T/2H MoS2 Edge Contacts
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Butler, S.Z.; Hollen, S.M.; Cao, L.Y.; Cui, Y.; Gupta, J.A.; Gutiérrez, H.R.; Heinz, T.F.; Hong, S.S.; Huang, J.X.; Ismach, A.F.; et al. Progress, Challenges, and Opportunities in Two-Dimensional Materials Beyond Graphene. ACS Nano 2013, 7, 2898–2926. [Google Scholar] [CrossRef] [PubMed]
- Fiori, G.; Bonaccorso, F.; Iannaccone, G.; Palacios, T.; Neumaier, D.; Seabaugh, A.; Banerjee, S.K.; Colombo, L. Electronics Based on Two-Dimensional Materials. Nat. Nanotechnol. 2014, 9, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Miro, P.; Audiffred, M.; Heine, T. An Atlas of Two-Dimensional Materials. Chem. Soc. Rev. 2014, 43, 6537–6554. [Google Scholar] [CrossRef] [PubMed]
- Schwierz, F.; Pezoldt, J.; Granzner, R. Two-Dimensional Materials and their Prospects in Transistor Electronics. Nanoscale 2015, 7, 8261–8283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molle, A.; Goldberger, J.; Houssa, M.; Xu, Y.; Zhang, S.C.; Akinwande, D. Buckled Two-Dimensional Xene Sheets. Nat. Mater. 2017, 16, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tao, L.; Chen, Z.; Fang, H.; Li, X.; Wang, X.; Xu, J.B.; Zhu, H. Graphene and Related Two-dimensional Materials: Structure-Property Relationships for Electronics and Optoelectronics. Appl. Phys. Rev. 2017, 4, 021306. [Google Scholar] [CrossRef]
- Robinson, J.A. 2D for Beyond CMOS. APL Mater. 2018, 6, 058202. [Google Scholar] [CrossRef] [Green Version]
- Geim, A.K.; Grigorieva, I.V. Van der Waals Heterostructure. Nature 2013, 499, 419–425. [Google Scholar] [CrossRef]
- Huang, C.; Wu, S.; Sanchez, A.M.; Peters, J.J.P.; Beanland, R.; Ross, J.S.; Rivera, P.; Yao, W.; Cobden, D.H.; Xu, X. Lateral Heterojunctions with Monolayer MoSe2-WSe2 Semiconductors. Nat. Mater. 2014, 13, 1096–1101. [Google Scholar] [CrossRef]
- Gong, Y.; Lin, J.; Wang, X.; Shi, G.; Lei, S.; Lin, Z.; Zou, X.; Ye, G.; Vajtai, R.; Yakobson, B.I.; et al. Vertical and In-plane Heterostructures from WS2/MoS2 Monolayers. Nat. Mater. 2014, 13, 1135–1142. [Google Scholar] [CrossRef] [Green Version]
- Vu, Q.A.; Lee, J.H.; Nguyen, V.L.; Shin, Y.S.; Lim, S.C.; Lee, K.; Heo, J.; Park, S.; Kim, K.; Lee, Y.H. Tuning Carrier Tunneling in Van der Waals Heterostructures for Ultrahigh Detectivity. Nano Lett. 2017, 17, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, M.Y.; Tersoff, J.; Han, Y.; Su, Y.; Li, L.J.; Muller, D.A.; Shih, C.K. Strain Distributions and their Influence in Electronic Structures of WSe2-MoS2 Laterally Strained Heterojunctions. Nat. Nanotech. 2018, 13, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Unuchek, D.; Ciarrocchi, A.; Avsar, A.; Watanabe, K.; Taniguchi, T.; Kis, A. Room-Temperature Electrical Control of Exciton Flux in a Van der Waals Heterostructure. Nature 2018, 560, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Allain, A.; Kang, J.; Banerjee, K.; Kis, A. Electrical Contacts to Two-Dimensional Semiconductors. Nat. Mater. 2015, 14, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Schulman, D.S.; Arnold, A.J.; Das, S. Contact Engineering for 2D Materials and Devices. Chem. Soc. Rev. 2018, 47, 3037–3058. [Google Scholar] [CrossRef]
- Kappera, R.; Voiry, D.; Yalcin, S.E.; Branch, B.; Gupta, G.; Mohite, A.D.; Chhowalla, M. Phase-Engineered Low-Resistance Contacts for Ultrathin MoS2 Transistors. Nat. Mater. 2014, 13, 1128–1134. [Google Scholar] [CrossRef]
- Lin, Y.C.; Dumcenco, D.O.; Hung, Y.S.; Suenaga, K. Atomic Mechanism of the Semiconducting-to-Metallic Phase Transition in Single Layered MoS2. Nat. Nanotech. 2014, 9, 391–396. [Google Scholar] [CrossRef]
- Meng, J.; Song, H.D.; Li, C.Z.; Jin, Y.; Tang, L.; Liu, D.; Liao, Z.M.; Xiu, F.; Yu, D.P. Lateral Graphene P-N Junctions Formed by the Graphene/MoS2 Hybrid Interface. Nanoscale 2015, 7, 11611–11619. [Google Scholar] [CrossRef]
- Guimaraes, M.H.D.; Gao, H.; Han, Y.; Kang, K.; Xie, S.; Kim, C.J.; Muller, D.A.; Ralph, D.C.; Park, J. Atomically Thin Ohmic Edge Contacts Between Two-Dimensional Materials. ACS Nano 2016, 10, 6392–6399. [Google Scholar] [CrossRef] [Green Version]
- Nourbakhsh, A.; Zubair, A.; Sajjad, R.N.; Tavakkoli, A.; Chen, W.; Fang, S.; Ling, X.; Kong, J.; Dresselhaus, M.S.; Kaxiras, E.; et al. MoS2 Field-Effect Transistor with Sub-10 nm Channel Length. Nano Lett. 2016, 16, 7798–7806. [Google Scholar] [CrossRef]
- Zhao, M.; Ye, Y.; Han, Y.; Xia, Y.; Zhu, H.; Wang, S.; Wang, Y.; Muller, D.A.; Zhang, X. Large-Scale Chemical Assembly of Atomically Thin Transistors and Circuits. Nat. Nanotech. 2016, 11, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Mahapatra, S. Atomistic Modeling of The Metallic-To-Semiconducting Phase Boundaries in Monolayer MoS2. Appl. Phys. Lett. 2016, 108, 25316. [Google Scholar] [CrossRef] [Green Version]
- Saha, D.; Mahapatra, S. Asymmetric Junctions in Metallic-Semiconducting-Metallic Heterophase MoS2. IEEE Trans. Electron Dev. 2017, 64, 2457–2460. [Google Scholar] [CrossRef]
- Chen, W.; Yang, Y.; Zhang, Z.; Kaxiras, E. Properties of In-Plane Graphene/MoS2 Heterojunctions. 2D Mater. 2017, 4, 045001. [Google Scholar] [CrossRef]
- Liu, X.; Gao, J.; Zhang, G.; Zhang, Y.W. MoS2-Graphene In-Plane Contact for High Interfacial Thermal Conduction. Nano Res. 2017, 10, 2944–2953. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.Q.; Jiang, X.W.; Luo, J.W.; Jiao, L.Y.; Huang, R.; Li, S.S.; Wang, L.W. In-Plane Schottky-Barrier Field-Effect Transistors Based on 1T/2H Heterojunctions of Transition Metal Dichalcogenides. Phys. Rev. B 2017, 96, 165402. [Google Scholar] [CrossRef] [Green Version]
- Bai, H.F.; Xu, L.C.; Di, M.Y.; Hao, L.Y.; Yang, Z.; Liu, R.P.; Li, X.Y. The Intrinsic Interface Properties of the Top and Edge 1T/2H MoS2 Contact: A First-Principles Study. J. Appl. Phys. 2018, 123, 095301. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; Garcia, A.; Junquera, J.; Ordejon, P.; Sanchez-Portal, D. The Siesta Method for Ab-Initio Order-N Materials Simulations. J. Phys. Condens. Matter 2002, 14, 2745–2779. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Trouiller, N.; Martins, J.L. Efficient Pseudopotentials for Plane-Wave Calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef]
- Bengtsson, L. Dipole Correction for Surface Supercell Calculations. Phys. Rev. B 1999, 59, 12301–12304. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical, GGA-Type Density Functional Constructed with Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Brandbyge, M.; Mozos, J.L.; Ordejon, P.; Taylor, J.; Stokbro, K. Density-Functional Method for Nonequilibrium Electron Transport. Phys. Rev. B 2002, 65, 165401. [Google Scholar] [CrossRef] [Green Version]
- Topsakal, M.; Bagci, V.M.K.; Ciraci, S. Current-Voltage (I-V) Characteristics of Armchair Graphene Nanoribbons Under Uniaxial Strain. Phys. Rev. B 2010, 81, 205437. [Google Scholar] [CrossRef] [Green Version]
- Van den Broek, B.; Houssa, M.; Iordanidou, K.; Pourtois, G.; Afanas’ev, V.V.; Stesmans, A. Functional silicene and stanene nanoribbons compared to graphene: Electronic structure and transport. 2D Mater. 2016, 3, 015001. [Google Scholar] [CrossRef]
- Iordanidou, K.; Houssa, M.; van den Broek, B.; Pourtois, G.; Afanas’ev, V.V.; Stesmans, A. Impact of point defects on the electronic and transport properties of silicene nanoribbons. J. Phys. Condens. Matter 2016, 28, 035302. [Google Scholar] [CrossRef] [PubMed]
- Büttiker, M.; Imry, Y.; Landauer, R.; Pinhas, S. Generalized Many-Channel Conductance Formula with Application to Small Rings. Phys. Rev. B 1985, 31, 6207–6215. [Google Scholar] [CrossRef] [Green Version]
- Datta, S. Electronic Transport in Mesoscopic Systems; Cambridge University Press: Cambridge, UK, 1995. [Google Scholar]
- Markussen, T.; Rurali, R.; Jauho, A.P.; Brandbyge, M. Scaling Theory Put Into Practice: First-Principles Modeling of Transport in Doped Silicon Nanowires. Phys. Rev. Lett. 2007, 99, 076803. [Google Scholar] [CrossRef] [Green Version]
- Datta, S. Lessons From Nanoelectronics: A New Perspective on Transport; World Scientific: Singapore, 2012. [Google Scholar]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [Green Version]
- Geim, A.K.; Novoselov, K.S. The Rise of Graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef]
- Katsnelson, M.I. Graphene—Carbon in Two Dimensions; Cambridge University Press: Cambridge, UK, 2012. [Google Scholar]
- Foa Torres, L.E.; Roche, S.; Charlier, J.C. Introduction to Graphene-Based Nanomaterials; Cambridge University Press: Cambridge, UK, 2014. [Google Scholar]
- Chen, J.H.; Jang, C.; Xiao, S.D.; Ishigami, M.; Fuhrer, M.S. Intrinsic and Extrinsic Performance Limits of Graphene Devices on SiO2. Nat. Nanotech. 2008, 3, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Mayorov, A.S.; Gorbachev, R.V.; Morozov, S.V.; Britnell, L.; Jalil, R.; Ponomarenko, L.A.; Blake, P.; Novoselov, K.S.; Watanabe, K.; Taniguchi, T.; et al. Micrometer Scale Ballistic Transport in Encapsulated Graphene at Room Temperature. Nano Lett. 2011, 11, 2396–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baringhaus, J.; Ruan, M.; Edler, F.; Tejeda, A.; Sicot, M.; Taleb-Ibrahimi, A.; Li, A.P.; Jiang, Z.; Conrad, E.H.; Berger, C.; et al. Exceptional Ballistic Transport in Epitaxial Graphene Nanoribbons. Nature 2014, 506, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Single Layer MoS2 Transistors. Nat. Nanotech. 2011, 6, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Radisavljevic, B.; Whitwick, M.B.; Kis, A. Integrated Circuits and Logic Operations Based on Single-Layer MoS2. ACS Nano 2011, 5, 9934–9938. [Google Scholar] [CrossRef]
- Lopez-Sanchez, O.; Lembke, D.; Kayci, M.; Radenovic, A.; Kis, A. Ultrasensitive Photodectors based on Monolayer MoS2. Nat. Nanotech. 2013, 8, 497–501. [Google Scholar] [CrossRef]
- Molina-Mendoza, A.J.; Vaquero-Garzon, L.; Leret, S.; de Juan-Fernandez, L.; Perez, E.M.; Castellanos-Gomez, A. Engineering the Optoelectronic Properties of MoS2 Photodetectors through reversible noncovalent functionalization. Chem. Commun. 2016, 52, 14365. [Google Scholar] [CrossRef] [Green Version]
- Kuc, A. Low-Dimensional Transition Metal Dichalcogenides. Chem. Modelling 2014, 11, 1–29. [Google Scholar]
- Mak, K.F.; Lee, C.; Hone, J.; Shan, J.; Heinz, T.F. Atomically Thin MoS2: A New Direct Gap Semiconductor. Phys. Rev. Lett. 2010, 105, 136805. [Google Scholar] [CrossRef] [Green Version]
- Scalise, E.; Houssa, M.; Pourtois, G.; Afanas’ev, V.V.; Stesmans, A. Strain-Induced Semiconductor to Metal Transition in the Two-Dimensional Honeycomb Structure of MoS2. Nano Res. 2012, 5, 43–48. [Google Scholar] [CrossRef]
- Voiry, D.; Mohite, A.; Chhowalla, M. Phase Engineering of Transition Metal Dichalcogenides. Chem. Soc. Rev. 2015, 44, 2702–2712. [Google Scholar] [CrossRef] [PubMed]
- Enyashin, A.; Yadgarov, L.; Houben, L.; Popov, I.; Weidenbach, M.; Tenne, R.; Bar-Sadan, M.; Seifert, G. New Route for Stabilization of 1T-WS2 and MoS2 Phases. J. Phys. Chem. C 2011, 115, 24586–24591. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Liu, J.; Fu, L.; Li, J. Quantum Spin Hall Effect in Two-Dimensional Transition Metal Dichalcogenides. Science 2014, 346, 1344–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, D.; Mahapatra, S. Anisotropic Transport in 1T’ Monolayer MoS2 and its Metal Interfaces. Phys. Chem. Chem. Phys. 2017, 19, 10453–10461. [Google Scholar] [CrossRef]
- Houssa, M.; Iordanidou, K.; Dabral, A.; Lu, A.; Meng, R.; Pourtois, G.; Afanas’ev, V.V.; Stesmans, A. Contact Resistance at Graphene/MoS2 Lateral Heterostructures. Appl. Phys. Lett. 2019, 114, 163101. [Google Scholar] [CrossRef]
- Bollinger, M.V.; Jacobsen, K.W.; Nørskov, J.K. Atomic and Electronic Structure of MoS2 Nanoparticles. Phys. Rev. B 2003, 67, 085410. [Google Scholar] [CrossRef] [Green Version]
- Song, S.M.; Kim, T.Y.; Sul, O.J.; Shin, W.C.; Cho, B.J. Improvement of Graphene-Metal Contact Resistance by Introducing Edge Contacts at Graphene Under Metal. Appl. Phys. Lett. 2014, 104, 183506. [Google Scholar] [CrossRef]
- Houssa, M.; Iordanidou, K.; Dabral, A.; Lu, A.; Pourtois, G.; Afanas’ev, V.V.; Stesmans, A. Contact Resistance at MoS2-Based 2D Metal/Semiconductor Lateral Heterojunctions. ACS Appl. Nano Mater. 2019, 2, 760. [Google Scholar] [CrossRef]
- Chou, S.S.; Sai, N.; Lu, P.; Coker, E.N.; Liu, S.; Artyushkova, K.; Luk, T.S.; Kaehr, B.; Brinker, C.J. Understanding Catalysis in a Multiphasic Two-Dimensional Transition Metal Dichalcogenide. Nat. Commun. 2015, 6, 8311. [Google Scholar] [CrossRef]
- Dolui, K.; Rungger, I.; Pemmaraju, C.D.; Sanvito, S. Possible Doping Strategies for MoS2 Monolayers: An Ab-Initio Study. Phys. Rev. B 2013, 88, 075420. [Google Scholar] [CrossRef] [Green Version]
- Voiry, D.; Goswani, A.; Kappera, R.; de Carvalho Castro e Silva, C.; Kaplan, D.; Fujita, T.; Chen, M.; Asefa, T.; Chhowalla, M. Covalent Functionalization of Monolayered Transition Metal Dichalcogenides by Phase Engineering. Nat. Chem. 2014, 7, 45–49. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interface Model | γ (eV/Å) Mo-Rich Limit | γ (eV/Å) S-Rich Limit | RcW (Ω.μm) |
|---|---|---|---|
| Model 1 | 2.4 | 2.4 | 8.1 × 103 |
| Model 2 | 1.5 | 1.5 | 5.6 × 102 |
| Model 3 | 1.2 | 1.8 | 2.2 × 102 |
| Model 4 | 2.6 | 1.9 | 2.3 × 104 |
| Interface Model | RcW (Ω∙μm) | Eads (eV) | Electron Transfer to 1T-MoS2 (e) |
|---|---|---|---|
| Pristine armchair contact | 3.8 × 104 | / | / |
| Pristine zigzag contact | 2.9 × 104 | / | / |
| H-adsorbed contact | 2.8 × 102 | −0.75 | 0.05 |
| Li-adsorbed contact | 1.9 × 102 | −1.46 | 0.18 |
| H2O-adsorbed contact | 4.7 × 102 | −2.43 | 0.08 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Houssa, M.; Meng, R.; Afanas’ev, V.; Stesmans, A. First-Principles Study of the Contact Resistance at 2D Metal/2D Semiconductor Heterojunctions. Appl. Sci. 2020, 10, 2731. https://doi.org/10.3390/app10082731
Houssa M, Meng R, Afanas’ev V, Stesmans A. First-Principles Study of the Contact Resistance at 2D Metal/2D Semiconductor Heterojunctions. Applied Sciences. 2020; 10(8):2731. https://doi.org/10.3390/app10082731
Chicago/Turabian StyleHoussa, Michel, Ruishen Meng, Valery Afanas’ev, and André Stesmans. 2020. "First-Principles Study of the Contact Resistance at 2D Metal/2D Semiconductor Heterojunctions" Applied Sciences 10, no. 8: 2731. https://doi.org/10.3390/app10082731