Many-Body Effects in FeN4 Center Embedded in Graphene

 ,
,
Abstract
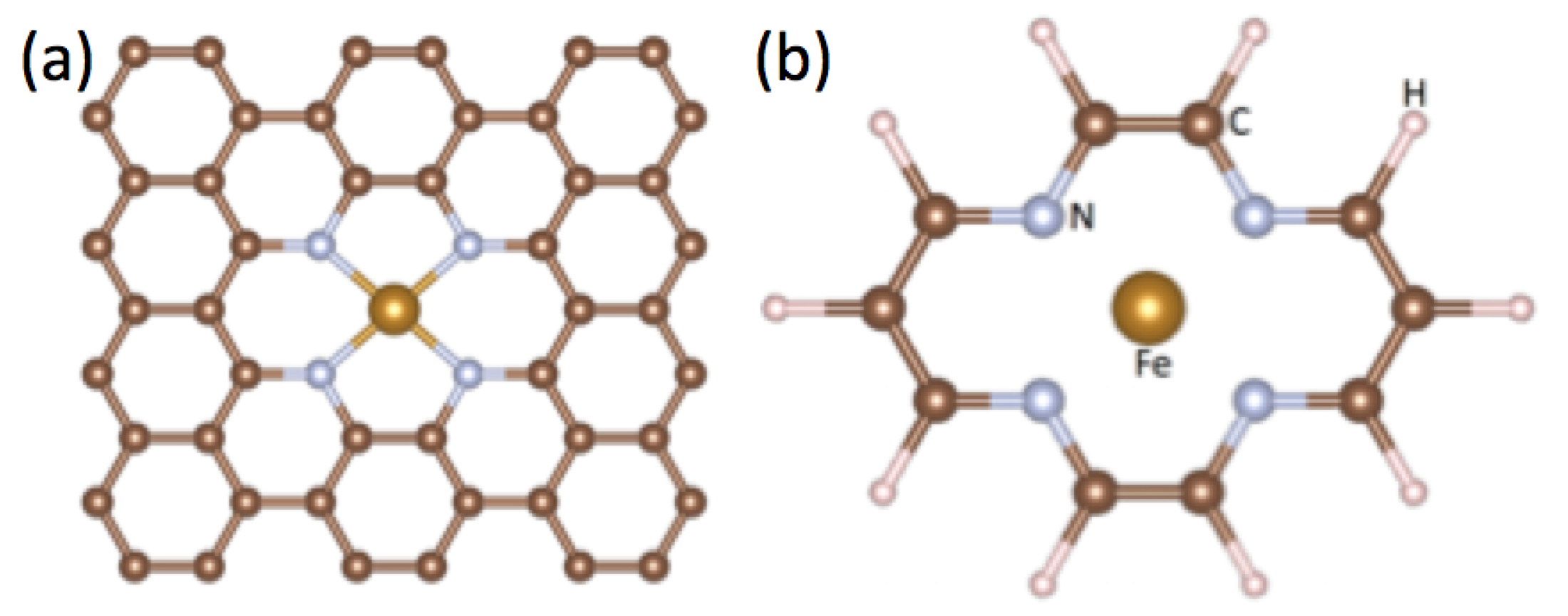
:1. Introduction
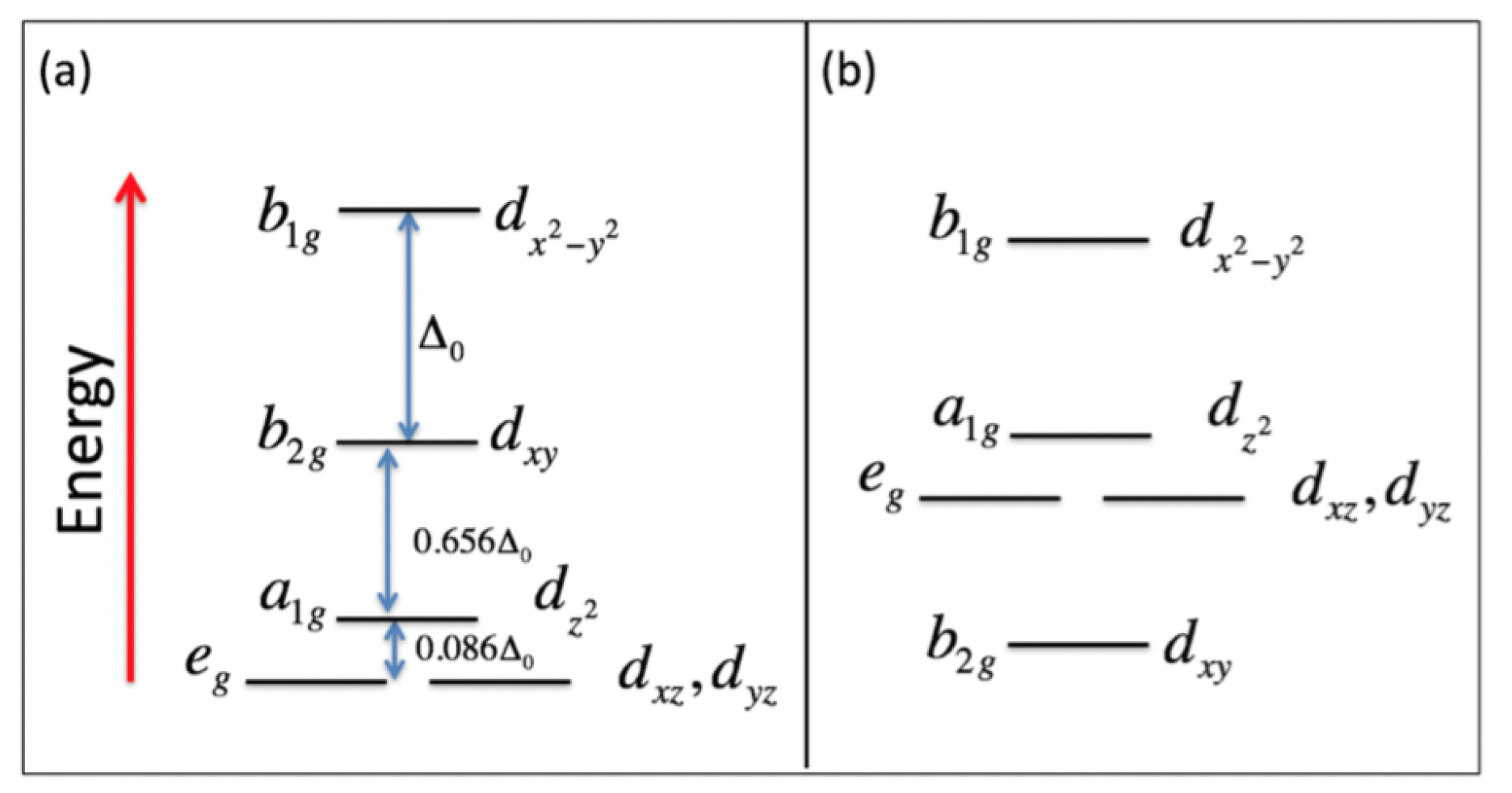
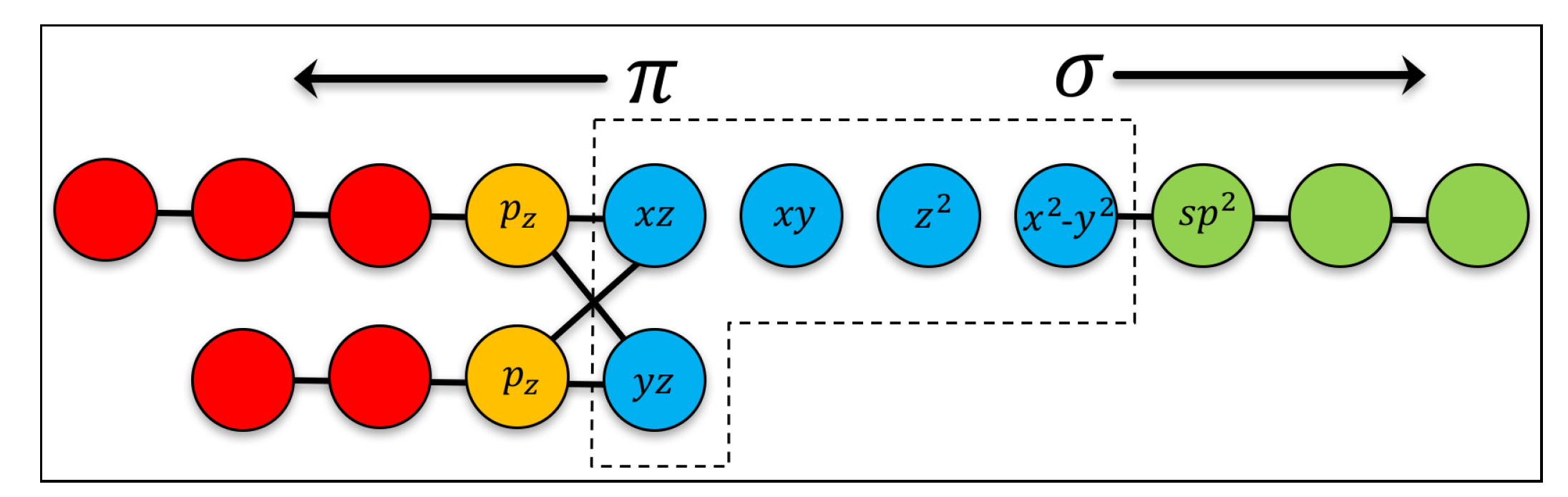
2. Model Hamiltonian
3. Method
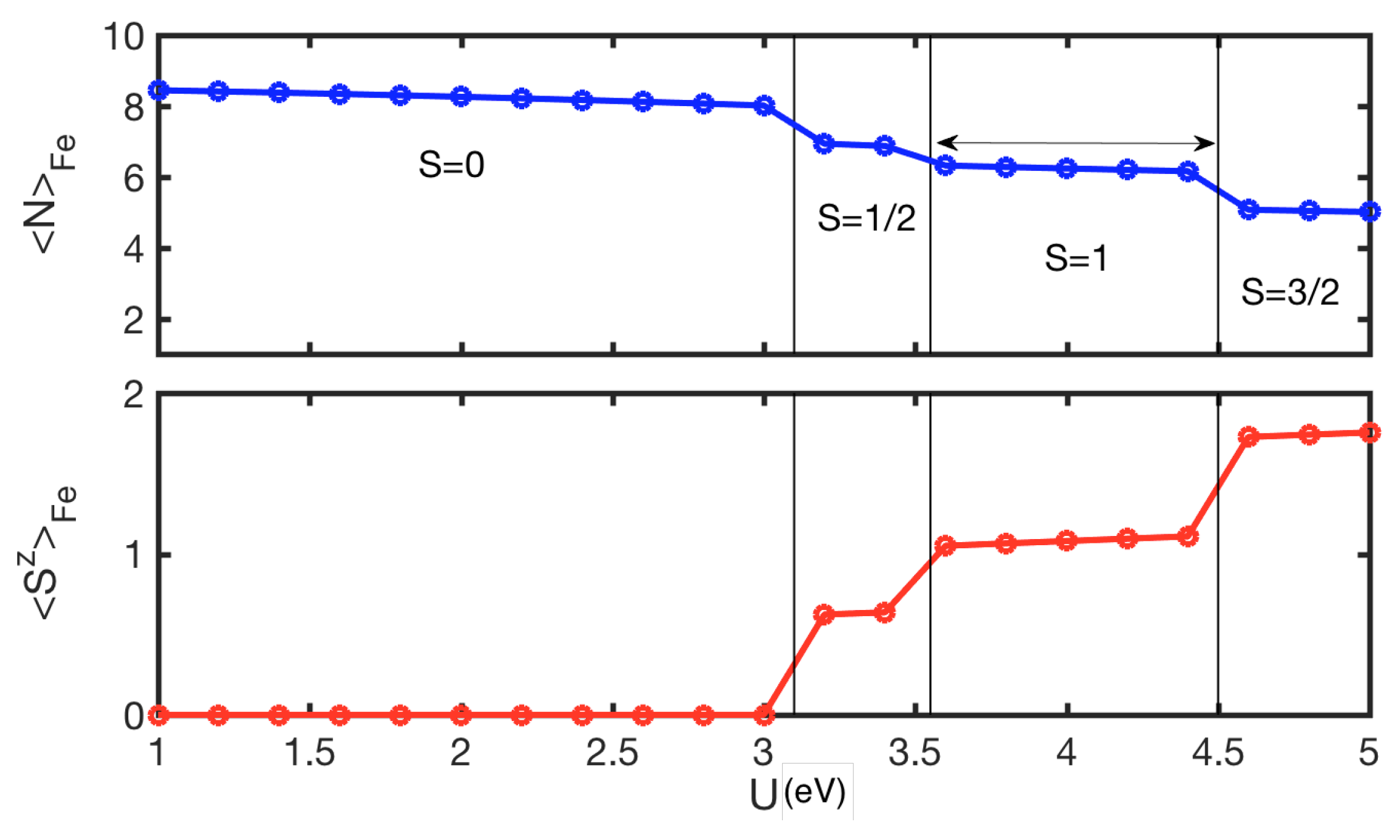
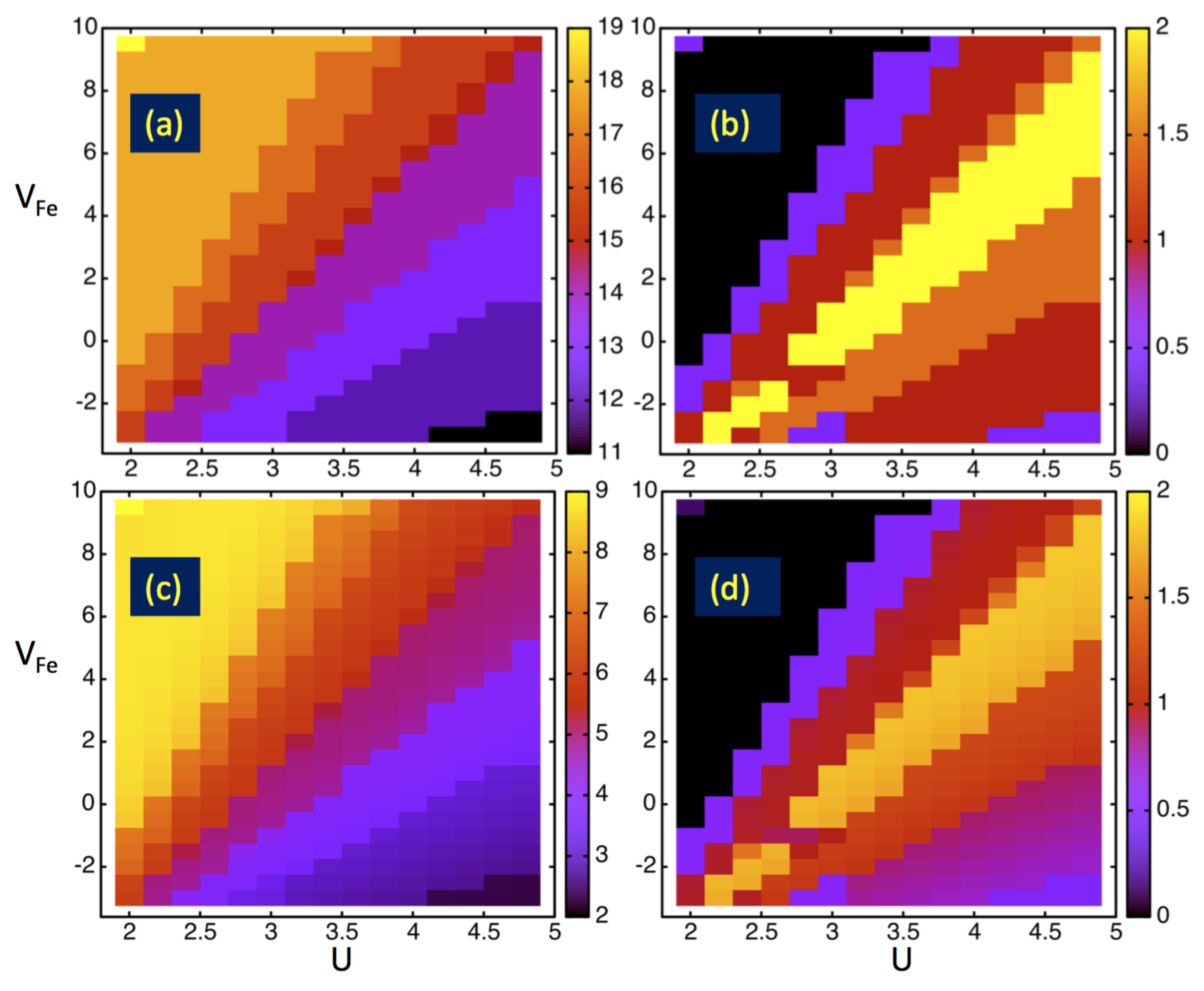
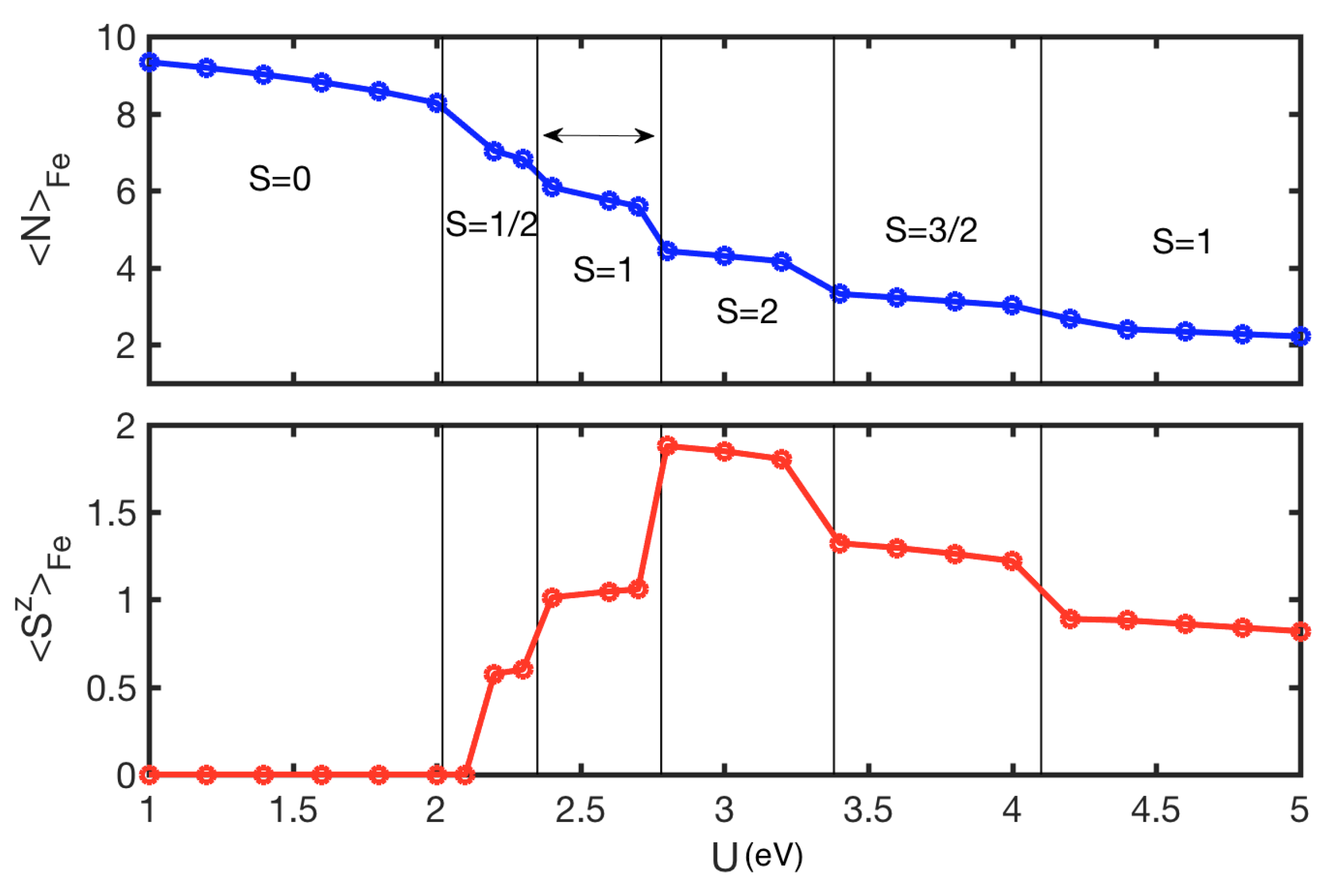
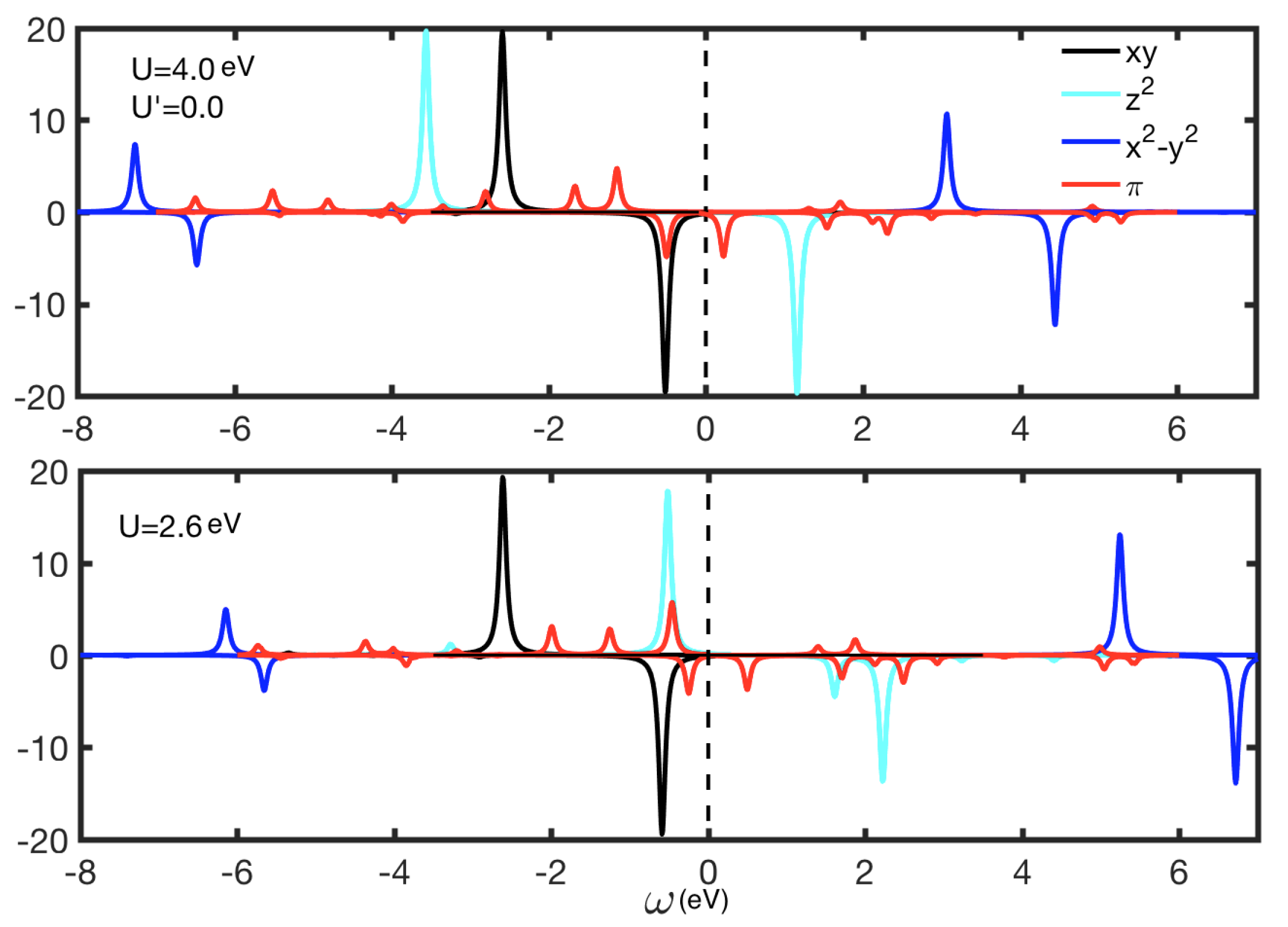
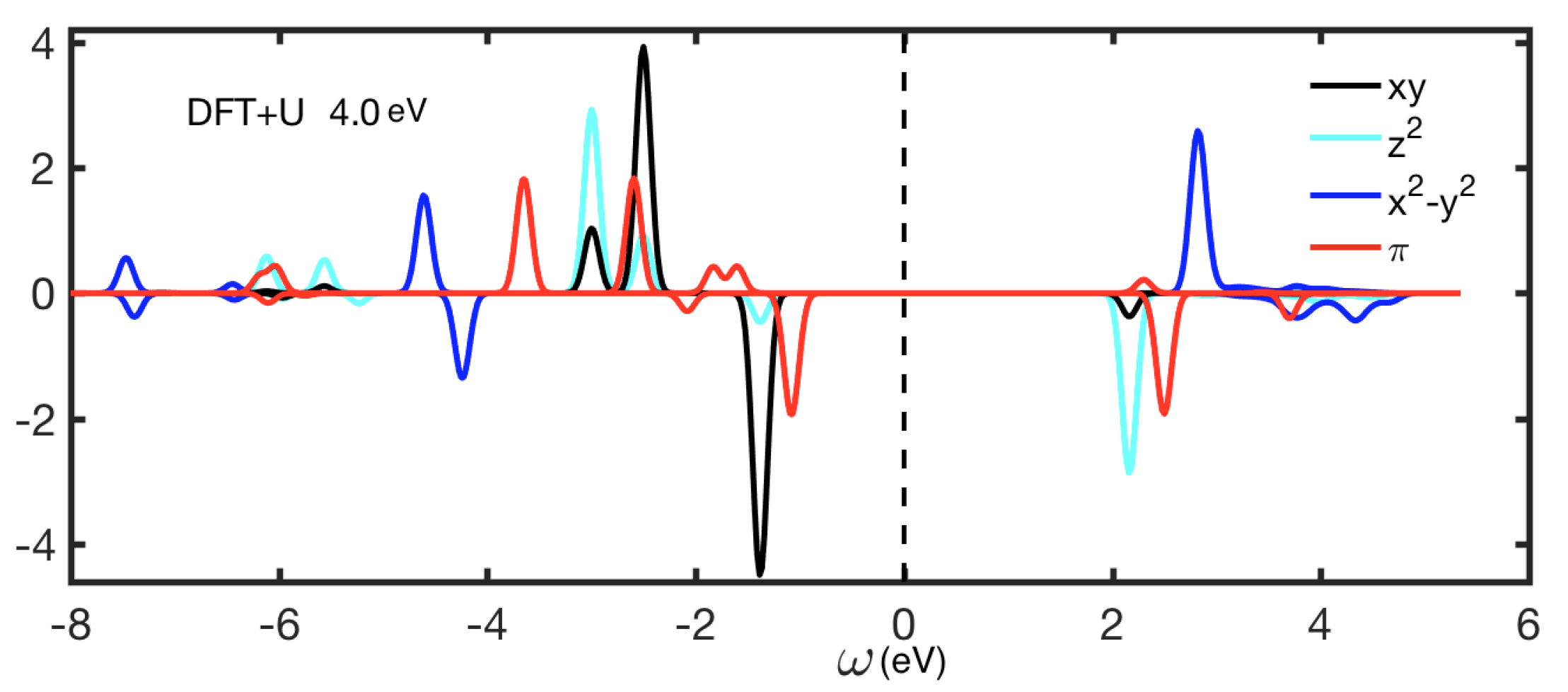
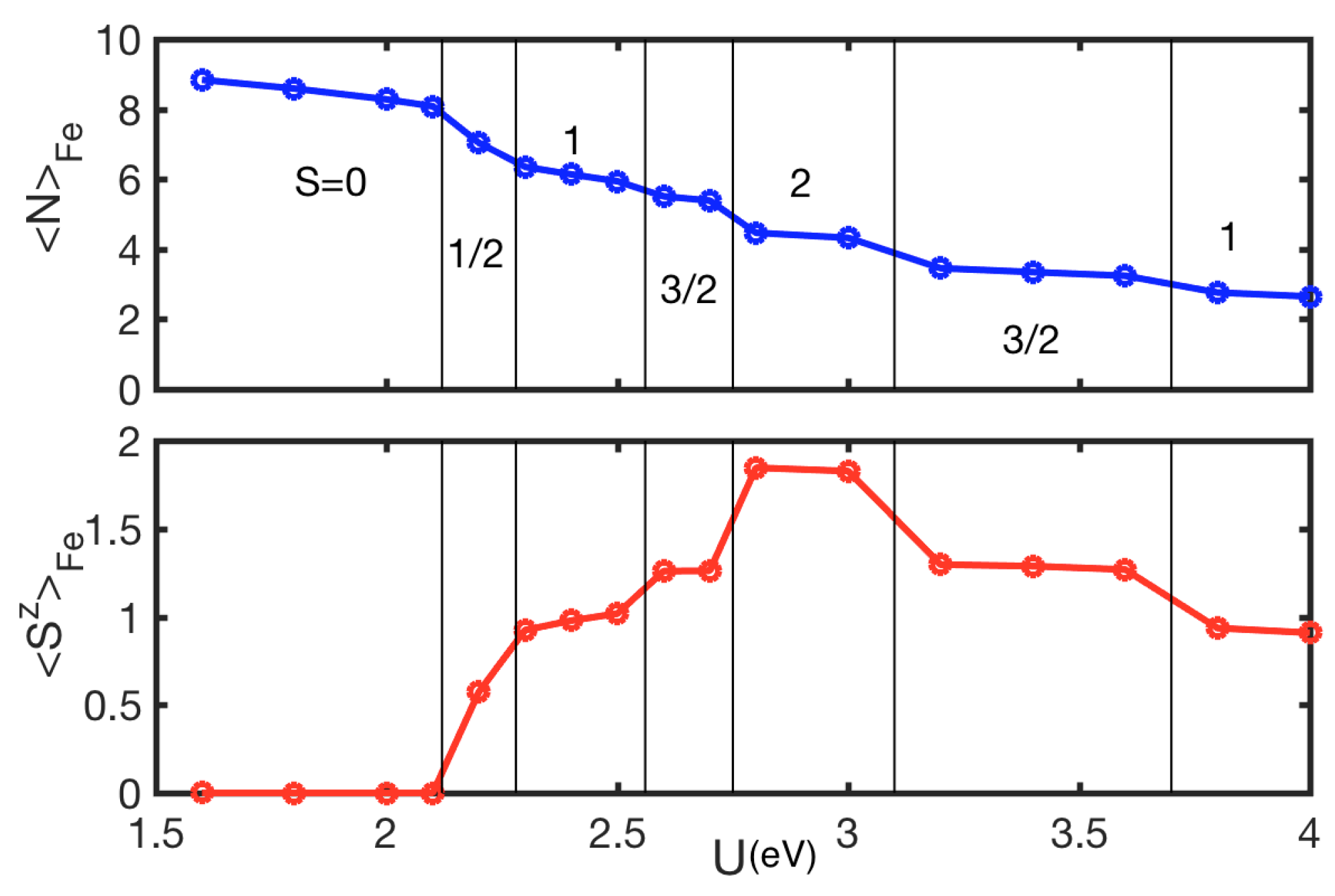
4. Results
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
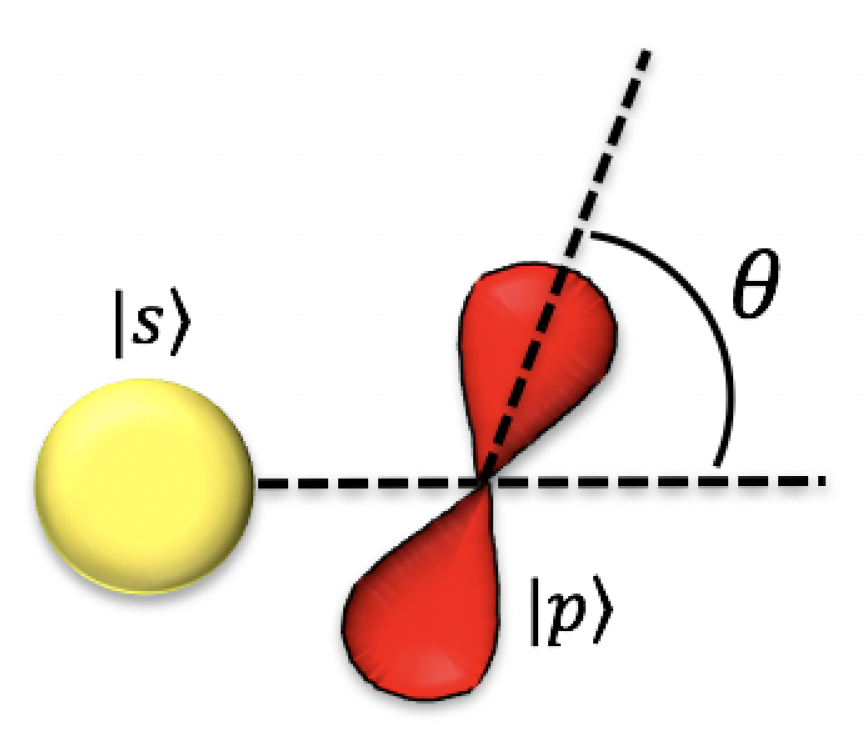
Appendix A. Tight-Binding Model for Graphene’s σ-Bands

References
- De Boer, R.W.I.; Stassen, A.F.; Craciun, M.F.; Mulder, C.L.; Molinari, A.; Rogge, S.; Morpurgo, A.F. Ambipolar Cu- and Fe-Phthalocyanine Single-Crystal Field-Effect Transistors. Appl. Phys. Lett. 2005, 86, 262109. [Google Scholar] [CrossRef] [Green Version]
- Zeis, R.; Siegrist, T.; Kloc, C. Single-Crystal Field- Effect Transistors Based on Copper Phthalocyanine. Appl. Phys. Lett. 2005, 86, 022103. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.X.; Li, H.X.; Liu, Y.L.; Hu, W.P. High-Performance Air-Stable n-Type Transistors with an Asymmetrical Device Configuration Based on Or- ganic Single-Crystalline Submicrometer/Nanometer Rib- 8 bons. J. Am. Chem. Soc. 2006, 128, 14634. [Google Scholar] [CrossRef] [PubMed]
- Asano, M.S.; Shibuki, M.; Otsuka, T. Prolonged Lifetime of Near-IR Emission due to a Reservoir State in a Phenylene-linked Copper(II) Porphyrin–Zinc(II) Porphyrin Dimer. Chem. Lett. 2016, 45, 1114–1116. [Google Scholar] [CrossRef]
- Wende, H.; Bernien, M.; Luo, J.; Sorg, C.; Ponpandian, N.; Kurde, J.; Miguel, J.; Piantek, M.; Xu, X.; Eckhold, P.; et al. Substrate-induced magnetic ordering and switching of iron porphyrin molecules. Nat. Mater. 2007, 6, 516–520. [Google Scholar] [CrossRef]
- Bogani, L.; Wernsdorfer, W. Molecular spintronics using single-molecule magnets. Nat. Mater. 2008, 7, 179–186. [Google Scholar] [CrossRef]
- Warner, B.; Oberg, J.; Gill, T.; El, H.F.; Hirjibehedin, C.; Serri, M.; Heutz, S.; Arrio, M.A.; Sainctavit, P.; Mannini, M.; et al. Temperature- and Light-Induced Spin Crossover Observed by X-ray Spectroscopy on Isolated Fe(II) Complexes on Gold. J. Phys. Chem. Lett. 2013, 4, 1546–1552. [Google Scholar] [CrossRef] [Green Version]
- Bernien, M.; Miguel, J.; Weis, C.; Ali, M.E.; Kurde, J.; Krumme, B.; Panchmatia, P.M.; Sanyal, B.; Piantek, M.; Srivastava, P.; et al. Tailoring the Nature of Magnetic Coupling of Fe-Porphyrin Molecules to Ferromagnetic Substrates. Phys. Rev. Lett. 2009, 102, 047202. [Google Scholar] [CrossRef] [Green Version]
- Bhandary, S.; Ghosh, S.; Herper, H.; Wende, H.; Eriksson, O.; Sanyal, B. Graphene as a Reversible Spin Manipulator of Molecular Magnets. Phys. Rev. Lett. 2011, 107, 257202. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Chen, K.Q. Magnetic configuration dependence of magnetoresistance in a Fe-porphyrin-like carbon nanotube spintronic device. Appl. Phys. Lett. 2014, 104, 033104. [Google Scholar] [CrossRef]
- Mittra, K.; Sengupta, K.; Singha, A.; Bandyopadhyay, S.; Chatterjee, S.; Rana, A.; Samanta, S.; Dey, A. Second sphere control of spin state: Differential tuning of axial ligand bonds in ferric porphyrin complexes by hydrogen bonding. J. Inorg. Biochem. 2016, 155, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, D.; Quesne, M.G.; de Visser, S.P.; Rath, S.P. Hydrogen-Bonding Interactions Trigger a Spin-Flip in Iron(III) Porphyrin Complexes. Angew. Chem. Int. Ed. 2015, 54, 4796–4800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Merino-Díez, N.; Carbonell-Sanromà, E.; Vilas-Varela, M.; de Oteyza, D.G.; Peña, D.; Corso, M.; Pascual, J.I. Survival of spin state in magnetic porphyrins contacted by graphene nanoribbons. Sci. Adv. 2018, 4, eaaq0582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio-Verdú, C.; Sarasola, A.; Choi, D.J.; Majzik, Z.; Ebeling, R.; Calvo, M.R.; Ugeda, M.M.; Garcia-Lekue, A.; Sánchez-Portal, D.; Pascual, J.I. Orbital-selective spin excitation of a magnetic porphyrin. Commun. Phys. 2018, 1, 15. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, H.S.; Pate, B.D.; Choi, S.M. Magnetic alignment of discotic liquid crystals on substrates. Phys. B Condens. Matter 2006, 385–386, 798–800. [Google Scholar] [CrossRef]
- Sergeyev, S.; Pisula, W.; Geerts, Y.H. Discotic liquid crystals: A new generation of organic semiconductors. Chem. Soc. Rev. 2007, 36, 1902–1929. [Google Scholar] [CrossRef]
- Yella, A.; Lee, H.W.; Tsao, H.N.; Yi, C.; Chandiran, A.K.; Nazeeruddin, M.K.; Diau, E.W.G.; Yeh, C.Y.; Zakeeruddin, S.M.; Grätzel, M. Porphyrin-sensitized solar cells with cobalt (II/III)-based redox electrolyte exceed 12 percent efficiency. Science 2011, 334, 629–634. [Google Scholar] [CrossRef]
- Walter, M.G.; Rudine, A.B.; Wamser, C.C. Por- phyrins and Phthalocyanines in Solar Photovoltaic Cells. J. Porphyr. Phthalocyanines 2010, 14, 759. [Google Scholar] [CrossRef]
- Siebbeles, L.D.A.; Huijser, A.; Savenije, T.J. Effects of Molecular Organization on Exciton Diffusion in Thin Films of Bioinspired Light-Harvesting Molecules. J. Mater. Chem. 2009, 19, 6067–6072. [Google Scholar] [CrossRef]
- Najafov, H.; Lee, B.; Zhou, Q.; Feldman, L.C.; Podzorov, V. Observation of Long-Range Exciton Diffusion in Highly Ordered Organic Semiconductors. Nat. Mater. 2010, 9, 938–943. [Google Scholar] [CrossRef]
- Hains, A.W.; Liang, Z.; Woodhouse, M.A.; Gregg, B.A. Molecular Semiconductors in Organic Photo-voltaic Cells. Chem. Rev. 2010, 110, 6689–6735. [Google Scholar] [CrossRef] [PubMed]
- Higashino, T.; Kawamoto, K.; Sugiura, K.; Fujimori, Y.; Tsuji, Y.; Kurotobi, K.; Ito, S.; Imahori, H. Effects of Bulky Substituents of Push-Pull Porphyrins on Photovoltaic Properties of Dye-Sensitized Solar Cells. ACS Appl. Mater. Interfaces 2016, 8, 15379–15390. [Google Scholar] [CrossRef]
- Ishikawa, N. Functional Phthalocyanine Molecular Materials; Springer: Berlin/Heidelberg, Germany, 2010; Volume 135. [Google Scholar]
- Bartolomé, J.; Luis, F.; Fernández, J.F. (Eds.) Molecular Magnets; Springer: Berlin, Germany, 2013. [Google Scholar]
- Shimizu, S.; Shin, J.Y.; Furuta, H.; Ismael, R.; Osuka, A. Perfluorinated meso-Aryl-Substituted Expanded Porphyrins. Angew. Chem. Int. Ed. 2003, 115, 1521–3757. [Google Scholar] [CrossRef]
- Asano, M.S.; Yamashita, K.; Kitabayashi, M.; Kusama, K.; Kagotaa, D.; Sugiura, K. Superexchange mediated energy transfer in zinc(II) porphyrin–free base porphyrin dimers: Comparison of m- and p-bis(phenylethynyl) phenylene linked dimers. J. Chem. Chem. Phys. 2011, 13, 12712–12715. [Google Scholar] [CrossRef] [PubMed]
- Saha-Dasgupta, T.; Oppeneer, P.M. Computational design of magnetic metal-organic complexes and coordination polymers with spin-switchable functionalities. MRS Bull. 2014, 39, 614–620. [Google Scholar] [CrossRef]
- Rawat, N.; Pan, Z.; Manning, L.W.; Lamarche, C.J.; Cour, I.; Headrick, R.L.; Waterman, R.; Woll, A.R.; Furis, M.I. Macroscopic Molecular Ordering and Exciton Delocalization in Crystalline Phthalocyanine Thin Films. J. Phys. Chem. Lett. 2015, 6, 1834–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepper, M.; Schmitt, T.; Gurrath, M.; Raschmann, M.; Zhang, L.; Stark, M.; Hölzel, H.; Jux, N.; Meyer, B.; Schneider, M.A.; et al. Adsorption Behavior of a Cyano-Functionalized Porphyrin on Cu(111) and Ag(111): From Molecular Wires to Ordered Supramolecular Two-Dimensional Aggregates. J. Phys. Chem. C 2017, 121, 26361–26371. [Google Scholar] [CrossRef] [Green Version]
- Urtizberea, A.; Natividad, E.; Alonso, P.J.; Andrés, M.A.; Gascón, I.; Goldmann, M.; Roubeau, O. A Porphyrin Spin Qubit and Its 2D Framework Nanosheets. Adv. Funct. Mater. 2018, 28, 1801695. [Google Scholar] [CrossRef]
- Iancu, V.; Deshpande, A.; Hla, S.W. Manipulating Kondo Temperature via Single Molecule Switching. Nano Lett. 2006, 6, 820–823. [Google Scholar] [CrossRef] [Green Version]
- Dias da Silva, L.G.G.V.; Tiago, M.L.; Ulloa, S.E.; Reboredo, F.A.; Dagotto, E. Many-body electronic structure and Kondo properties of cobalt-porphyrin molecules. Phys. Rev. B 2009, 80, 155443. [Google Scholar] [CrossRef] [Green Version]
- Minamitani, E.; Tsukahara, N.; Matsunaka, D.; Kim, Y.; Takagi, N.; Kawai, M. Symmetry-Driven Novel Kondo Effect in a Molecule. Phys. Rev. Lett. 2012, 109, 086602. [Google Scholar] [CrossRef] [PubMed]
- Lobos, A.M.; Romero, M.; Aligia, A.A. Spectral evolution of the SU(4) Kondo effect from the single impurity to the two-dimensional limit. Phys. Rev. B 2014, 89, 121406. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Wehling, T.O.; Werner, P. Electronic excitation spectra of the five-orbital Anderson impurity model: From the atomic limit to itinerant atomic magnetism. Phys. Rev. B 2014, 89, 245104. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Pang, R.; Kuang, G.; Shi, X.; Shang, X.; Liu, P.N.; Lin, N. Intramolecularly resolved Kondo resonance of high-spin Fe(II)-porphyrin adsorbed on Au(111). Phys. Rev. B 2015, 91, 045440. [Google Scholar] [CrossRef]
- Fernández, J.; Aligia, A.A.; Lobos, A.M. Valence fluctuations in a lattice of magnetic molecules: Application to iron(II) phthalocyanine molecules on Au(111). EPL Europhys. Lett. 2015, 109, 37011. [Google Scholar] [CrossRef] [Green Version]
- Fernández, J.; Roura-Bas, P.; Camjayi, A.; Aligia, A.A. Two-stage three-channel Kondo physics for an FePc molecule on the Au(111) surface. J. Phys. Condens. Matter 2018, 30, 374003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Shaikh, A.U.; Tsui, E.Y.; Swager, T.M. Cobalt Porphyrin Functionalized Carbon Nanotubes for Oxygen Reduction. Chem. Mater. 2009, 21, 3234–3241. [Google Scholar] [CrossRef]
- Lee, D.H.; Lee, W.J.; Lee, W.J.; Kim, S.O.; Kim, Y.H. Theory, Synthesis, and Oxygen Reduction Catalysis of Fe-Porphyrin-Like Carbon Nanotube. Phys. Rev. Lett. 2011, 106, 175502. [Google Scholar] [CrossRef]
- Chung, H.T.; Won, J.H.; Zelenay, P. Active and stable carbon nanotube/nanoparticle composite electrocatalyst for oxygen reduction. Nat. Commun. 2013, 4, 1922. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Dong, S. Recent progress in graphene-based nanomaterials as advanced electrocatalysts towards oxygen reduction reaction. Nanoscale 2013, 5, 1753–1767. [Google Scholar] [CrossRef]
- Orellana, W. Catalytic Properties of Transition Metal-N4 Moieties in Graphene for the Oxygen Reduction Reaction: Evidence of Spin-Dependent Mechanisms. J. Phys. Chem. C 2013, 117, 9812–9818. [Google Scholar] [CrossRef]
- Jia, Q.; Ramaswamy, N.; Hafiz, H.; Tylus, U.; Strickland, K.; Wu, G. Experimental Observation of Redox-Induced Fe–N Switching Behavior as a Determinant Role for Oxygen Reduction Activity. ACS Nano 2015, 9, 12496–12505. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Ramaswamy, N.; Tylus, U.; Strickland, K.; Li, J.; Serov, A.; Artyushkova, K.; Atanassov, P.; Anibal, J.; Gumeci, C.; et al. Spectroscopic insights into the nature of active sites in iron–nitrogen–carbon electrocatalysts for oxygen reduction in acid. Nano Energy 2016, 29, 65–82. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Hu, R.; Bai, F. DFT Study of the Oxygen Reduction Reaction Activity on Fe-N4-Patched Carbon Nanotubes: The Influence of the Diameter and Length. Materials 2017, 10, 549. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, S.; Kaiwa, J.; Chantngarm, P.; Tanibayashi, S.; Saito, H.; Hasegawa, M.; Nishidate, K. Oxygen reduction reaction of FeN4 center embedded in graphene and carbon nanotube: Density functional calculations. AIP Adv. 2018, 8, 115113. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Zhu, G.; Yang, D.; Jia, D.; Jin, F.; Wang, W. Systematic exploration of N, C configurational effects on the ORR performance of Fe–N doped graphene catalysts based on DFT calculations. RSC Adv. 2019, 9, 22656–22667. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.T.; Kang, J.; Wei, S.H.; Chang, K.J.; Kim, Y.H. Carrier-mediated long-range ferromagnetism in electron-doped Fe-C4 and Fe-N4 incorporated graphene. Phys. Rev. B 2012, 86, 165403. [Google Scholar] [CrossRef] [Green Version]
- Groot, M.J.D.; Havenith, R.W.A.; Vinkers, H.M.; Zwaans, R.; Vermeulen, N.P.E.; Lenthe, J.H.V. Ab initio calculations on iron-porphyrin model systems for intermediates in the oxidative cycle of cytochrome P450s. J. Comput. Aided Mol. Des. 1998, 12, 183–193. [Google Scholar] [CrossRef]
- Johansson, M.P.; Sundholm, D. Spin and charge distribution in iron porphyrin models: A coupled cluster and density- functional study. J. Chem. Phys. 2004, 120, 3229. [Google Scholar] [CrossRef]
- Scherlis, D.A.; Cococcioni, M.; Sit, P.; Marzari, N. Simulation of Heme Using DFT + U: A Step toward Accurate Spin-State Energetics. J. Phys. Chem. B 2007, 111, 7384–7391. [Google Scholar] [CrossRef] [Green Version]
- Kramm, U.I.; Herranz, J.; Larouche, N.; Arruda, T.M.; Lefèvre, M.; Jaouen, F.; Bogdanoff, P.; Fiechter, S.; Abs-Wurmbach, I.; Mukerjee, S.; et al. Structure of the catalytic sites in Fe/N/C-catalysts for O2-reduction in PEM fuel cells. Phys. Chem. Chem. Phys. 2012, 14, 11673–11688. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Harrison, N.M.; Fisher, A.J. Electronic structure and exchange interactions in cobalt-phthalocyanine chains. Phys. Rev. B 2013, 88, 024426. [Google Scholar] [CrossRef]
- Kattel, S.; Atanassov, P.; Kiefer, B. Stability, Electronic and Magnetic Properties of In-Plane Defects in Graphene: A First-Principles Study. J. Phys. Chem. C 2012, 116, 8161–8166. [Google Scholar] [CrossRef]
- Kattel, S.; Wang, G. A density functional theory study of oxygen reduction reaction on Me–N4 (Me = Fe, Co, or Ni) clusters between graphitic pores. J. Mater. Chem. A 2013, 1, 10790–10797. [Google Scholar] [CrossRef]
- Berryman, V.E.J.; Boyd, R.J.; Johnson, E.R. Balancing Exchange Mixing in Density-Functional Approximations for Iron Porphyrin. J. Chem. Theory Comput. 2015, 11, 3022–3028. [Google Scholar] [CrossRef]
- Weber, C.; Cole, D.J.; O’Regan, D.D.; Payne, M.C. Renormalization of myoglobin–ligand binding energetics by quantum many-body effects. Proc. Natl. Acad. Sci. USA 2014, 111, 5790–5795. [Google Scholar] [CrossRef] [Green Version]
- Weber, C.; O’Regan, D.D.; Hine, N.D.M.; Littlewood, P.B.; Kotliar, G.; Payne, M.C. Importance of Many-Body Effects in the Kernel of Hemoglobin for Ligand Binding. Phys. Rev. Lett. 2013, 110, 106402. [Google Scholar] [CrossRef] [Green Version]
- Koseki, J.; Maezono, R.; Tachikawa, M.; Towler, M.D.; Needs, R.J. Quantum Monte Carlo study of porphyrin transition metal complexes. J. Chem. Phys. 2008, 129, 085103. [Google Scholar] [CrossRef]
- Aspuru-guzik, A.; Akramine, O.E.; Grossman, J.C.; Lester, W.A., Jr. Quantum Monte Carlo for electronic excitations of free-base porphyrin. J. Chem. Phys. 2004, 120, 3049. [Google Scholar] [CrossRef]
- Rovira, C.; Parrinello, M. First-principles molecular dynamics simulations of models for the myoglobin active center. Int. J. Quantum Chem. 2000, 80, 1172–1180. [Google Scholar] [CrossRef]
- LaBute, M.X.; Kulkarni, R.V.; Endres, R.G.; Cox, D.L. Strong electron correlations in cobalt valence tautomers. J. Chem. Phys. 2002, 116, 3681–3689. [Google Scholar] [CrossRef] [Green Version]
- White, S.R. Density matrix formulation for quantum renormalization groups. Phys. Rev. Lett. 1992, 69, 2863–2866. [Google Scholar] [CrossRef] [PubMed]
- White, S.R. Density-matrix algorithms for quantum renormalization groups. Phys. Rev. B 1993, 48, 10345–10356. [Google Scholar] [CrossRef] [PubMed]
- Peschel, I.; Wang, X.; Kaulke, M.; Hallberg, K. (Eds.) Density-Matrix Renormalization—A New Numerical Method in Physics, Springer: Berlin, Germany, 1999.
- Schollwöck, U. The density-matrix renormalization group. Rev. Mod. Phys. 2005, 77, 259–315. [Google Scholar] [CrossRef] [Green Version]
- Feiguin, A.E. The density matrix renormalization group. In Strongly Correlated Systems: Numerical Methods; Avella, A., Mancini, F., Eds.; Springer: Berlin, Germany, 2013; Chapter 2; pp. 31–65. [Google Scholar]
- Jones, R.O.; Gunnarsson, O. The density functional formalism, its applications and prospects. Rev. Mod. Phys. 1989, 61, 689–746. [Google Scholar] [CrossRef]
- Mackintosh, A.; Andersen, O. The electronic structure of transition metals. In Electrons at the Fermi Surface; Springford, M., Ed.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 1980; Volume Chapter 5, pp. 149–224. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Barbiellini, B.; Moroni, E.G.; Jarlborg, T. Effects of gradient corrections on electronic structure in metals. J. Phys. Condens. Matter 1990, 2, 7597–7611. [Google Scholar] [CrossRef]
- Panchmatia, P.M.; Sanyal, B.; Oppeneer, P.M. GGA+U modeling of structural, electronic, and magnetic properties of iron porphyrin-type molecules. Chem. Phys. 2008, 343, 47–60. [Google Scholar] [CrossRef]
- Kumar, M.; Pati, Y.A.; Ramasesha, S. A density matrix renormalization group method study of optical properties of porphines and metalloporphines. J. Chem. Phys. 2012, 136, 014112. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.; Pati, Y.A.; Ramasesha, S. Linear and Nonlinear Optical Properties of Expanded Porphyrins: A DMRG Study. J. Phys. Chem. A 2013, 117, 7804–7809. [Google Scholar] [CrossRef]
- LaBute, M.X.; Endres, R.G.; Cox, D. An Anderson impurity model for efficient sampling of adiabatic potential energy surfaces of transition metal complexes. J. Chem. Phys. 2004, 121, 8221–8230. [Google Scholar] [CrossRef] [Green Version]
- Jahn, H.A.; Teller, E. Stability of Polyatomic Molecules in Degenerate Electronic States. I. Orbital Degeneracy. Proc. R. Soc. Lond. A Math. Phys. Eng. Sci. 1937, 161, 220–235. [Google Scholar] [CrossRef]
- Dagotto, E.; Hotta, T.; Moreo, A. Colossal magnetoresistant materials: The key role of phase separation. Phys. Rep. 2001, 344, 1–153. [Google Scholar] [CrossRef] [Green Version]
- De’ Medici, L.; Capone, M. Modeling Many-Body Physics with Slave-Spin Mean-Field: Mott and Hund’s Physics in Fe-Superconductors. In The Iron Pnictide Superconductors: An Introduction and Overview; Mancini, F., Citro, R., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 115–185. [Google Scholar] [CrossRef] [Green Version]
- Kanamori, J. Electron Correlation and Ferromagnetism of Transition Metals. Prog. Theor. Phys. 1963, 30, 275–289. [Google Scholar] [CrossRef] [Green Version]
- Frésard, R.; Kotliar, G. Interplay of Mott transition and ferromagnetism in the orbitally degenerate Hubbard model. Phys. Rev. B 1997, 56, 12909–12915. [Google Scholar] [CrossRef] [Green Version]
- Weissbluth, M. Hemoglobin (Cooperativity and Electronic Properties); Springer: Berlin/Heidelberg, Germany, 1974. [Google Scholar]
- Eder, R. Multiplets in Transition Metal Ions. In Correlated Electrons: From Models to Materials. Modeling and Simulation; Pavarini, E., Koch, E., Anders, F., Jarrell, M., Eds.; Forschungszentrum Jülich: Jülich, Germany, 2012; Chapter 8; Volume 2. [Google Scholar]
- Day, R.P.; Levy, G.; Michiardi, M.; Zwartsenberg, B.; Zonno, M.; Ji, F.; Razzoli, E.; Boschini, F.; Chi, S.; Liang, R.; et al. Influence of Spin-Orbit Coupling in Iron-Based Superconductors. Phys. Rev. Lett. 2018, 121, 076401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poteryaev, A.I.; Ferrero, M.; Georges, A.; Parcollet, O. Effect of crystal-field splitting and interband hybridization on the metal-insulator transitions of strongly correlated systems. Phys. Rev. B 2008, 78, 045115. [Google Scholar] [CrossRef] [Green Version]
- Werner, P.; Gull, E.; Millis, A.J. Metal-insulator phase diagram and orbital selectivity in three-orbital models with rotationally invariant Hund coupling. Phys. Rev. B 2009, 79, 115119. [Google Scholar] [CrossRef] [Green Version]
- Mazza, G.; Amaricci, A.; Capone, M.; Fabrizio, M. Field-Driven Mott Gap Collapse and Resistive Switch in Correlated Insulators. Phys. Rev. Lett. 2016, 117, 176401. [Google Scholar] [CrossRef] [Green Version]
- Büsser, C.A.; Martins, G.B.; Feiguin, A.E. Lanczos transformation for quantum impurity problems in d-dimensional lattices: Application to graphene nanoribbons. Phys. Rev. B 2013, 88, 245113. [Google Scholar] [CrossRef] [Green Version]
- Allerdt, A.; Büsser, C.A.; Martins, G.B.; Feiguin, A.E. Kondo versus indirect exchange: Role of lattice and actual range of RKKY interactions in real materials. Phys. Rev. B 2015, 91, 085101. [Google Scholar] [CrossRef] [Green Version]
- Allerdt, A.; Feiguin, A.E. A Numerically Exact Approach to Quantum Impurity Problems in Realistic Lattice Geometries. Front. Phys. 2019, 7, 67. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Bhandary, S.; Schüler, M.; Thunström, P.; di Marco, I.; Brena, B.; Eriksson, O.; Wehling, T.; Sanyal, B. Correlated electron behavior of metal-organic molecules: Insights from density functional theory combined with many-body effects using exact diagonalization. Phys. Rev. B 2016, 93, 155158. [Google Scholar] [CrossRef] [Green Version]
- Kühner, T.D.; White, S.R. Dynamical correlation functions using the density matrix renormalization group. Phys. Rev. B 1999, 60, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Jeckelmann, E. Dynamical density-matrix renormalization-group method. Phys. Rev. B 2002, 66, 045114. [Google Scholar] [CrossRef] [Green Version]
- Chan, G.K.L.; Sharma, S. The Density Matrix Renormalization Group in Quantum Chemistry. Annu. Rev. Phys. Chem. 2011, 62, 465–481. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Chan, G.K.L. Spin-adapted density matrix renormalization group algorithms for quantum chemistry. J. Chem. Phys. 2012, 136, 124121. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Sivalingam, K.; Neese, F.; Chan, G.K.L. Low-energy spectrum of iron-sulfur clusters directly from many-particle quantum mechanics. Nat. Chem. 2014, 6, 927–933. [Google Scholar] [CrossRef] [Green Version]
- Olivares-Amaya, R.; Hu, W.; Nakatani, N.; Sharma, S.; Yang, J.; Chan, G.K.L. The ab-initio density matrix renormalization group in practice. J. Chem. Phys. 2015, 142, 034102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, K.; Malmqvist, P.A.; Roos, B.O. Second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218–1226. [Google Scholar] [CrossRef]
- Yanai, T.; Saitow, M.; Xiong, X.G.; Chalupský, J.; Kurashige, Y.; Guo, S.; Sharma, S. Multistate Complete-Active-Space Second-Order Perturbation Theory Based on Density Matrix Renormalization Group Reference States. J. Chem. Theory Comput. 2017, 13, 4829–4840. [Google Scholar] [CrossRef] [PubMed]
- Hübsch, A.; Lin, J.C.; Pan, J.; Cox, D.L. Correlated Hybridization in Transition-Metal Complexes. Phys. Rev. Lett. 2006, 96, 196401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z. (Ed.) Graphene: Properties, Synthesis, and Applications; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2011. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| m | m | J | |
|---|---|---|---|
| xy, xz, yz | xy, xz, yz | A − 2B + C | 3B + C |
| xz, yz | z | A + 2B + C | B + C |
| xz, yz | x − y | A − 2B + C | 3B + C |
| xy | z | A − 4B + C | 4B + C |
| xy | x − y | A + 4B + C | C |
| x − y, z | x − y, z | A − 4B + C | 4B + C |
| Ion | B | C |
|---|---|---|
| Fe | 0.114 | 0.501 |
| Fe | 0.126 | 0.595 |
| DFT+U | Full H | |||||
|---|---|---|---|---|---|---|
| Orbital | ||||||
| 1.79 | 0.05 | 2.00 | 0.00 | 2.00 | 0.00 | |
| 1.07 | 0.40 | 1.00 | 0.50 | 1.0 | 0.50 | |
| 0.83 | 0.04 | 0.77 | 0.04 | 0.53 | 0.03 | |
| 1.35 | 0.26 | 1.26 | 0.27 | 1.11 | 0.26 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allerdt, A.; Hafiz, H.; Barbiellini, B.; Bansil, A.; Feiguin, A.E. Many-Body Effects in FeN4 Center Embedded in Graphene. Appl. Sci. 2020, 10, 2542. https://doi.org/10.3390/app10072542
Allerdt A, Hafiz H, Barbiellini B, Bansil A, Feiguin AE. Many-Body Effects in FeN4 Center Embedded in Graphene. Applied Sciences. 2020; 10(7):2542. https://doi.org/10.3390/app10072542
Chicago/Turabian StyleAllerdt, Andrew, Hasnain Hafiz, Bernardo Barbiellini, Arun Bansil, and Adrian E. Feiguin. 2020. "Many-Body Effects in FeN4 Center Embedded in Graphene" Applied Sciences 10, no. 7: 2542. https://doi.org/10.3390/app10072542