Peak Fitting Applied to Fourier Transform Infrared and Raman Spectroscopic Analysis of Proteins



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Sample Preparation
2.3. Attenuated Total Reflectance (ATR) FTIR Spectroscopy
2.4. FT-Raman Measurement
2.5. Peak Fitting
3. Results
3.1. Secondary Structure Estimation for Standard Proteins
3.2. Secondary Structure Estimation for Gluten and Zein
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Susi, H. [22] Infrared spectroscopy—Conformation. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1972; Volume 26, pp. 455–472. [Google Scholar]
- Susi, H.; Byler, D.M. [13] Resolution-enhanced fourier transform infrared spectroscopy of enzymes. In Bioluminescence and Chemiluminescence; Elsevier: Amsterdam, The Netherlands, 1986; Volume 130, pp. 290–311. [Google Scholar]
- Elliott, A.; Ambrose, E.J. Evidence of chain folding in polypeptides and proteins. Discuss. Faraday Soc. 1950, 9, 246–251. [Google Scholar] [CrossRef]
- Surewicz, W.K.; Mantsch, H.H. New insight into protein secondary structure from resolution-enhanced infrared spectra. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. Enzym. 1988, 952, 115–130. [Google Scholar] [CrossRef]
- Dong, A.; Huang, P.; Caughey, W.S. Protein secondary structures in water from second-derivative amide I infrared spectra. Biochemistry 1990, 29, 3303–3308. [Google Scholar] [CrossRef]
- Kong, J.; Yu, S. Fourier transform infrared spectroscopic analysis of protein secondary structures. Acta Biochim. Biophys. Sin. 2007, 39, 549–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.T.; Wu, C.-S.C.; Yang, J.T. Circular dichroic analysis of protein conformation: Inclusion of the β-turns. Anal. Biochem. 1978, 91, 13–31. [Google Scholar] [CrossRef]
- Provencher, S.W.; Gloeckner, J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry 1981, 20, 33–37. [Google Scholar] [CrossRef]
- Hennessey, J.P.; Johnson, W.C. Information content in the circular dichroism of proteins. Biochemistry 1981, 20, 1085–1094. [Google Scholar] [CrossRef]
- Sadat, A.; Corradini, M.G.; Joye, I. Molecular spectroscopy to assess protein structures within cereal systems. Curr. Opin. Food Sci. 2019, 25, 42–51. [Google Scholar] [CrossRef]
- Wellner, N.; Mills, E.N.C.; Brownsey, G.; Wilson, R.H.; Brown, N.A.; Freeman, J.; Halford, N.G.; Shewry, P.R.; Belton, P.S. Changes in Protein Secondary Structure during Gluten Deformation Studied by Dynamic Fourier Transform Infrared Spectroscopy. Biomacromolecules 2005, 6, 255–261. [Google Scholar] [CrossRef]
- Li, W.; Dobraszczyk, B.J.; Dias, A.; Gil, A.M. Polymer Conformation Structure of Wheat Proteins and Gluten Subfractions Revealed by ATR-FTIR. Cereal Chem. J. 2006, 83, 407–410. [Google Scholar] [CrossRef]
- Robertson, G.H.; Gregorski, K.S.; Cao, T.K. Changes in Secondary Protein Structures During Mixing Development of High Absorption (90%) Flour and Water Mixtures. Cereal Chem. J. 2006, 83, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Jazaeri, S.; Bock, J.E.; Bagagli, M.P.; Iametti, S.; Bonomi, F.; Seetharaman, K. Structural Modifications of Gluten Proteins in Strong and Weak Wheat Dough During Mixing. Cereal Chem. J. 2015, 92, 105–113. [Google Scholar] [CrossRef]
- Bagagli, M.P.; Jazaeri, S.; Bock, J.E.; Seetharaman, K.; Sato, H.H. Effect of Transglutaminase, Citrate Buffer, and Temperature on a Soft Wheat Flour Dough System. Cereal Chem. J. 2014, 91, 460–465. [Google Scholar] [CrossRef]
- Georget, D.M.R.; Belton, P.S. Effects of Temperature and Water Content on the Secondary Structure of Wheat Gluten Studied by FTIR Spectroscopy. Biomacromolecules 2006, 7, 469–475. [Google Scholar] [CrossRef]
- Belton, P.; Colquhoun, I.; Grant, A.; Wellner, N.; Field, J.; Shewry, P.; Tatham, A. FTIR and NMR studies on the hydration of a high-Mr subunit of glutenin. Int. J. Boil. Macromol. 1995, 17, 74–80. [Google Scholar] [CrossRef]
- Bock, J.E.; Damodaran, S. Bran-induced changes in water structure and gluten conformation in model gluten dough studied by Fourier transform infrared spectroscopy. Food Hydrocoll. 2013, 31, 146–155. [Google Scholar] [CrossRef]
- Krimm, S.; Bandekar, J. Vibrational Spectroscopy and Conformation of Peptides, Polypeptides, and Proteins. In Cytokines; Elsevier: Amsterdam, The Netherlands, 1986; Volume 38, pp. 181–364. [Google Scholar]
- Venyaminov, S.Y.; Kalnin, N.N. Quantitative IR Spectrophotometry of Peptide Compounds in Water (H2O) Solutions. II. Amide Absorption Bands of Polypeptides and Fibrous Proteins in A-, B-, and Random Coil Conformations. Biopolym. Orig. Res. Biomol. 1990, 30, 1259–1271. [Google Scholar] [CrossRef]
- Venyaminov, S.Y.; Kalnin, N.N. Quantitative IR spectrophotometry of peptide compounds in water (H2O) solutions. I. Spectral parameters of amino acid residue absorption bands. Biopolym. Orig. Res. Biomol. 1990, 30, 1243–1257. [Google Scholar] [CrossRef]
- Kalnin, N.N.; Baikalov, I.A.; Venyaminov, S.Y. Quantitative IR Spectrophotometry of Peptide Compounds in Water (H2O) Solutions. III. Estimation of the Protein Secondary Structure. Biopolym. Orig. Res. Biomol. 1990, 30, 1273–1280. [Google Scholar] [CrossRef]
- Byler, D.M.; Susi, H. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolym. Orig. Res. Biomol. 1986, 25, 469–487. [Google Scholar] [CrossRef]
- Susi, H.; Byler, D.M. Protein structure by Fourier transform infrared spectroscopy: Second derivative spectra. Biochem. Biophys. Res. Commun. 1983, 115, 391–397. [Google Scholar] [CrossRef]
- Cioni, P.; Strambini, G.B. Effect of Heavy Water on Protein Flexibility. Biophys. J. 2002, 82, 3246–3253. [Google Scholar] [CrossRef] [Green Version]
- Efimova, Y.M.; Haemers, S.; Wierczinski, B.; Norde, W.; Van Well, A. Stability of globular proteins in H2O and D2O. Biopolym. Orig. Res. Biomol. 2007, 85, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.C. Fundamentals of Fourier Transform Infrared Spectroscopy; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Ma, C.-Y.; Phillips, D.L. FT-Raman Spectroscopy and Its Applications in Cereal Science. Cereal Chem. J. 2002, 79, 171–177. [Google Scholar] [CrossRef]
- Thygesen, L.G.; Løkke, M.M.; Micklander, E.; Engelsen, S.B. Vibrational microspectroscopy of food. Raman vs. FT-IR. Trends Food Sci. Technol. 2003, 14, 50–57. [Google Scholar] [CrossRef]
- Lórenz-Fonfría, V.; Padrós, E.; Lórenz-Fonfría, V.A.; Padrós, E. Maximum Entropy Deconvolution of Infrared Spectra: Use of a Novel Entropy Expression without Sign Restriction. Appl. Spectrosc. 2005, 59, 474–486. [Google Scholar] [CrossRef]
- DeAragao, B.J.G.; Messaddeq, Y. Peak separation by derivative spectroscopy applied to ftir analysis of hydrolized silica. J. Braz. Chem. Soc. 2008, 19, 1582–1594. [Google Scholar] [CrossRef]
- Dong, A.; Malecki, J.M.; Lee, L.; Carpenter, J.F.; Lee, J.C. Ligand-Induced Conformational and Structural Dynamics Changes inEscherichia coliCyclic AMP Receptor Protein†. Biochemistry 2002, 41, 6660–6667. [Google Scholar] [CrossRef]
- Kauppinen, J.K.; Moffatt, D.J.; Mantsch, H.H.; Cameron, D.G. Fourier Self-Deconvolution: A Method for Resolving Intrinsically Overlapped Bands. Appl. Spectrosc. 1981, 35, 271–276. [Google Scholar] [CrossRef]
- Rodionova, O.Y.; Brereton, R.G. Chemometrics: Data Analysis for the Laboratory and Chemical Plant, Chichester: Wiley, 2003, 489 pp. J. Anal. Chem. 2005, 60, 994–996. [Google Scholar] [CrossRef]
- Gautam, R.; Vanga, S.; Ariese, F.; Umapathy, S. Review of multidimensional data processing approaches for Raman and infrared spectroscopy. EPJ Tech. Instrum. 2015, 2, 1–38. [Google Scholar] [CrossRef] [Green Version]
- Di Rocco, H.O.; Iriarte, D.I.; Pomarico, J. General Expression for the Voigt Function That is of Special Interest for Applied Spectroscopy. Appl. Spectrosc. 2001, 55, 822–826. [Google Scholar] [CrossRef]
- Arrondo, J.L.; Young, N.; Mantsch, H. The solution structure of concanavalin A probed by FT-IR spectroscopy. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. Enzym. 1988, 952, 261–268. [Google Scholar] [CrossRef]
- Shewry, P.R.; Lookhart, G.L. Wheat Gluten Protein Analysis; American Association of Cereal Chemists: Saint Paul, MN, USA, 2003. [Google Scholar]
- Ngarize, S.; Herman, H.; Adams, A.; Howell, N. Comparison of Changes in the Secondary Structure of Unheated, Heated, and High-Pressure-Treated β-Lactoglobulin and Ovalbumin Proteins Using Fourier Transform Raman Spectroscopy and Self-Deconvolution. J. Agric. Food Chem. 2004, 52, 6470–6477. [Google Scholar] [CrossRef] [PubMed]
- Levitt, M.; Chothia, C. Structural patterns in globular proteins. Nature 1976, 261, 552–558. [Google Scholar] [CrossRef]
- Richardson, J.S. The Anatomy and Taxonomy of Protein Structure. In Advances in Protein Chemistry; Elsevier: Amsterdam, The Netherlands, 1981; Volume 34, pp. 167–339. [Google Scholar]
- Levitt, M.; Greer, J. Automatic identification of secondary structure in globular proteins. J. Mol. Boil. 1977, 114, 181–239. [Google Scholar] [CrossRef]
- Wellner, N.; Belton, P.S.; Tatham, A.S. Fourier transform IR spectroscopic study of hydration-induced structure changes in the solid state of ω-gliadins. Biochem. J. 1996, 319, 741–747. [Google Scholar] [CrossRef]
- Pézolet, M.; Bonenfant, S.; Dousseau, F.; Popineau, Y. Conformation of wheat gluten proteins Comparison between functional and solution states as determined by infrared spectroscopy. FEBS Lett. 1992, 299, 247–250. [Google Scholar] [CrossRef] [Green Version]
- Aboul-Enein, Y.; Bunaciu, A.A.; Fleschin, S. Evaluation of the Protein Secondary Structures Using Fourier Transform Infrared Spectroscopy. Gazi Univ. J. Sci. 2014, 27, 637–644. [Google Scholar]
- Susi, H.; Byler, D.M. Fourier Deconvolution of the Amide I Raman Band of Proteins as Related to Conformation. Appl. Spectrosc. 1988, 42, 819–826. [Google Scholar] [CrossRef]
- Lippert, J.L.; Tyminski, D.; Desmeules, P.J. Determination of the secondary structure of proteins by laser Raman spectroscopy. J. Am. Chem. Soc. 1976, 98, 7075–7080. [Google Scholar] [CrossRef] [PubMed]
- Edelman, G.M.; Cunningham, B.A.; Reeke, G.N.; Becker, J.W.; Waxdal, M.J.; Wang, J.L. The Covalent and Three-Dimensional Structure of Concanavalin A. Proc. Natl. Acad. Sci. USA 1972, 69, 2580–2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccirilli, F.; Schirò, G.; Vetri, V.; Lupi, S.; Perucchi, A.; Militello, V. Decoding vibrational states of Concanavalin A amyloid fibrils. Biophys. Chem. 2015, 199, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.-T.; Liu, C.; O’Shea, D. Laser Raman spectroscopy and the conformation of insulin and proinsulin. J. Mol. Boil. 1972, 70, 117–132. [Google Scholar] [CrossRef]
- Belitz, I.H.-D.; Grosch, I.W. Food Chemistry; Springer Science & Business Media: Berlin, Germany, 2013. [Google Scholar]
- Delcour, J.A.; Hoseney, R.C. Principles of Cereal Science and Technology. In Principles of Cereal Science and Technology; AACC International Inc.: St. Paul, MN, USA, 2010; pp. 229–235. [Google Scholar]
- Delcour, J.A.; Joye, I.J.; Pareyt, B.; Wilderjans, E.; Brijs, K.; Lagrain, B. Wheat Gluten Functionality as a Quality Determinant in Cereal-Based Food Products. Annu. Rev. Food Sci. Technol. 2012, 3, 469–492. [Google Scholar] [CrossRef]
- Tatham, A.S.; Miflin, B.J.; Shewry, P.R. The Beta-Turn Conformation in Wheat Gluten Proteins: Relationship to Gluten Elasticity. Cereal Chem. 1985, 62, 405–412. [Google Scholar]
- Belton, P. Mini Review: On the Elasticity of Wheat Gluten. J. Cereal Sci. 1999, 29, 103–107. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhao, D.; Foster, T.J.; Liu, Y.; Wang, Y.; Nirasawa, S.; Tatsumi, E.; Cheng, Y. Konjac glucomannan-induced changes in thiol/disulphide exchange and gluten conformation upon dough mixing. Food Chem. 2014, 143, 163–169. [Google Scholar] [CrossRef]
- Mejia, C.D.; Mauer, L.J.; Hamaker, B.R. Similarities and differences in secondary structure of viscoelastic polymers of maize α-zein and wheat gluten proteins. J. Cereal Sci. 2007, 45, 353–359. [Google Scholar] [CrossRef]
- Nawrocka, A.; Szymańska-Chargot, M.; Miś, A.; Kowalski, R.J.; Gruszecki, W.I. Raman studies of gluten proteins aggregation induced by dietary fibres. Food Chem. 2016, 194, 86–94. [Google Scholar] [CrossRef]
- Van Velzen, E.J.J.; Van Duynhoven, J.; Pudney, P.; Weegels, P.L.; Van Der Maas, J.H. Factors Associated with Dough Stickiness as Sensed by Attenuated Total Reflectance Infrared Spectroscopy. Cereal Chem. J. 2003, 80, 378–382. [Google Scholar] [CrossRef]
- Lawton, J.W. Viscoelasticity of Zein-Starch Doughs. Cereal Chem. 1992, 69, 351–355. [Google Scholar]
- Argos, P.; Pedersen, K.; Marks, M.D.; Larkins, B.A. A structural model for maize zein proteins. J. Boil. Chem. 1982, 257, 9984–9990. [Google Scholar]
- Momany, F.; Sessa, D.J.; Lawton, J.W.; Selling, G.W.; Hamaker, S.A.H.; Willett, J.L. Structural Characterization of α-Zein. J. Agric. Food Chem. 2006, 54, 543–547. [Google Scholar] [CrossRef]
- Shewry, P.R.; Tatham, A.S. The prolamin storage proteins of cereal seeds: Structure and evolution. Biochem. J. 1990, 267, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Athamneh, A.I.; Griffin, M.B.; Whaley, M.; Barone, J.R. Conformational Changes and Molecular Mobility in Plasticized Proteins. Biomacromolecules 2008, 9, 3181–3187. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, R.; Liu, J. Effects of different concentrations of ethanol and isopropanol on physicochemical properties of zein-based films. Ind. Crop. Prod. 2014, 53, 140–147. [Google Scholar] [CrossRef]


 ), (
), (  ), (
), (  ), and (
), and (  ), respectively.
), ( ), ( ), and ( ), respectively.
), respectively.
), ( ), ( ), and ( ), respectively.

 ), (
), (  ), (
), (  ), and (
), and (  ), respectively.
), ( ), ( ), and ( ), respectively.
), respectively.
), ( ), ( ), and ( ), respectively.
 ), (
), (  ), (
), (  ), respectively.
), ( ), ( ), respectively.
), respectively.
), ( ), ( ), respectively.
 ), (
), (  ), (
), (  ), respectively.
), ( ), ( ), respectively.
), respectively.
), ( ), ( ), respectively.

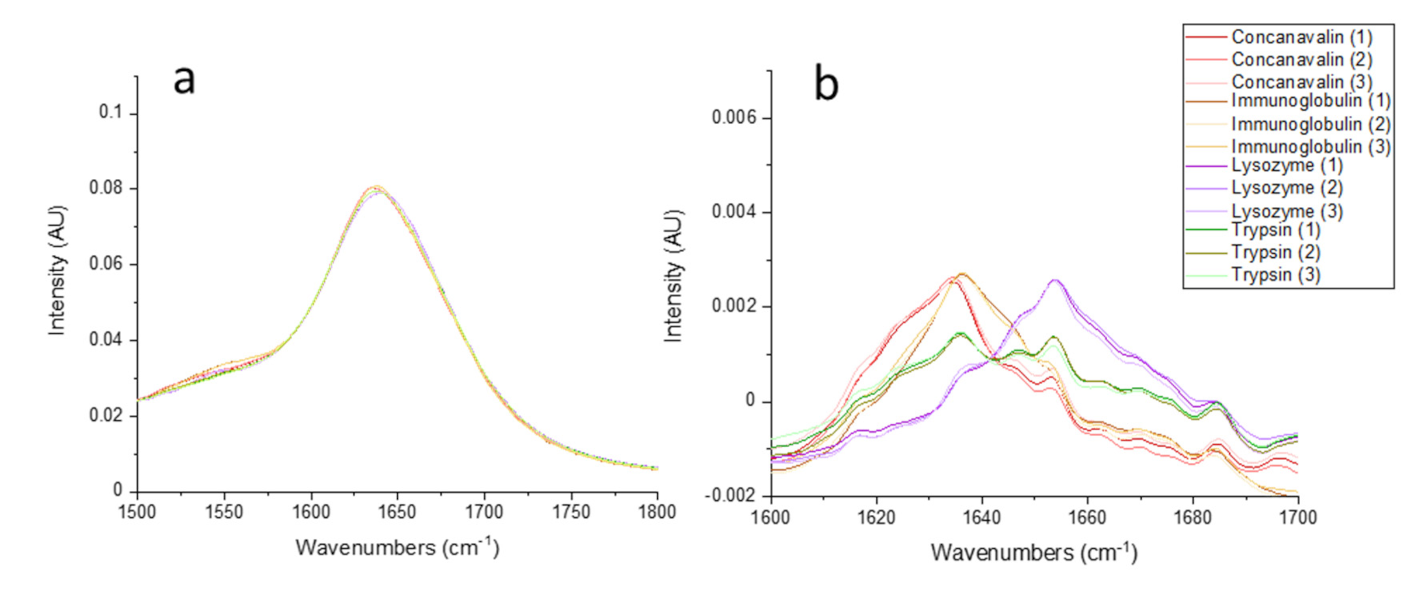{kind=link}
{kind=link}
{kind=link}
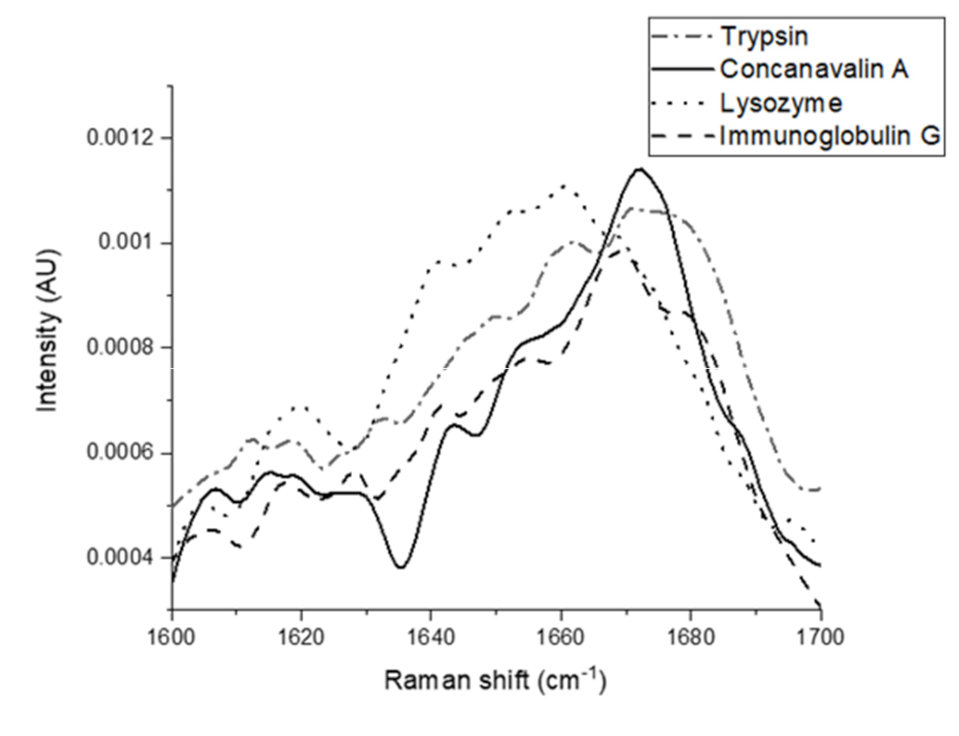{kind=link}
{kind=link}
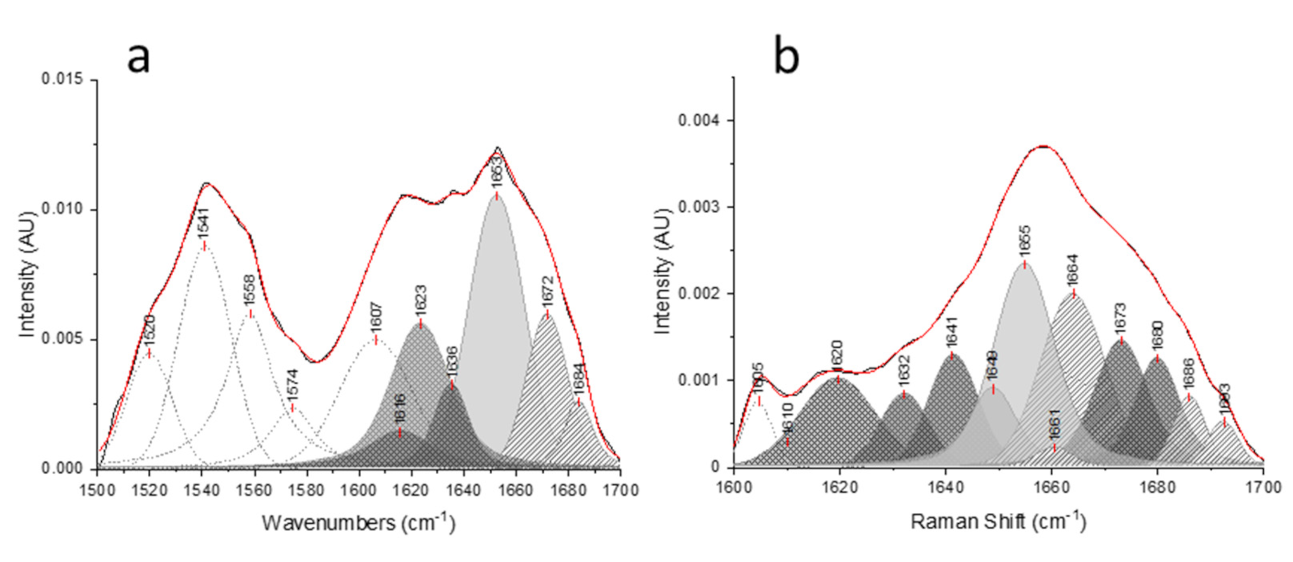{kind=link}
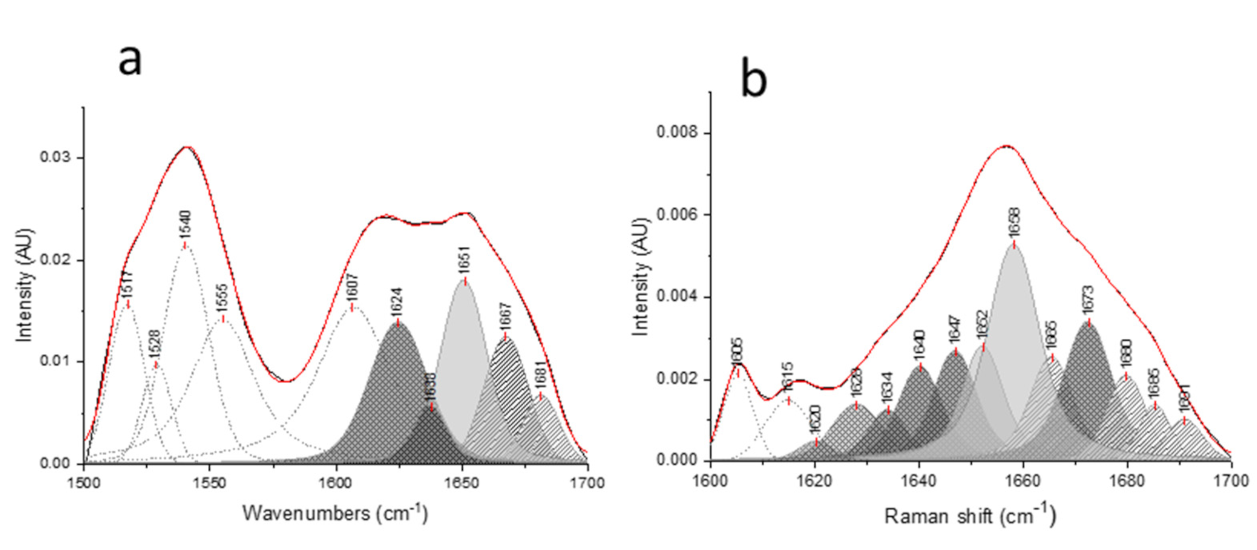{kind=link}
| Amide I Position (cm−1) | Assignment | |
|---|---|---|
| FTIR | 1610–1627 | Intermolecular β-sheet |
| 1628–1642 | β-sheet | |
| 1643–1650 | Random coil | |
| 1650–1659 | α-helix and Gln sidechain | |
| 1660–1699 | β-turn | |
| Raman | 1620–1648 | β-sheet |
| 1649–1660 | α-helix | |
| 1660–1665 | Random coil | |
| 1665–1680 | β-sheet | |
| 1658–1680 | β-turn | |
| 1680–1699 | β-turn |
| Protein | Secondary Structure (%) | Method | |||
|---|---|---|---|---|---|
| α-Helix | β-Sheet | β-Turn | Random Coil | ||
| Immunoglobulin G | 7 ± 1 | 67 ± 2 | 19 ± 3 | 7 ± 1 | FTIR |
| 9 | 64 | 21 | 6 | FT-Raman | |
| 3 | 67 | 18 | 12 | X-ray [42] | |
| Concanavalin A | 9 ± 1 | 57 ± 1 | 22 ± 1 | 12 ± 0 | FTIR |
| 8 | 59 | 23 | 10 | FT-Raman | |
| 3 | 60 | 22 | 15 | X-ray [42] | |
| 8–20 | 41–67 | 8–15 | 6–36 | CD [7,8] | |
| Trypsin | 16 ± 2 | 46 ± 3 | 29 ± 2 | 9 ± 3 | FTIR |
| 14 | 44 | 29 | 12 | FT-Raman | |
| 9 | 56 | 24 | 11 | X-ray [42] | |
| Lysozyme | 46 ± 3 | 21 ± 1 | 26 ± 1 | 8 ± 0 | FTIR |
| 42 | 27 | 27 | 4 | FT-Raman | |
| 45 | 19 | 23 | 13 | X-ray [42] | |
| 45 | 21 | 26 | 8 | CD [8] | |
| Amino acid | Absorbance Band Assignment | IR Frequency Position (cm−1) |
|---|---|---|
| Asparagine | C=O (stretching) | 1678 ± 3 |
| NH bending | 1622 ± 2 | |
| Glutamine | C=O stretching | 1670 ± 4 |
| N-H bending | 1610 ± 4 | |
| Arginine | C=N asymmetric | 1673 ± 3 |
| C=N symmetric stretching | 1633 ± 3 | |
| Lysine | NH+ asymmetric bending | 1629 ± 1 |
| Tyrosine | Ring vibration | 1602 ± 2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadat, A.; Joye, I.J. Peak Fitting Applied to Fourier Transform Infrared and Raman Spectroscopic Analysis of Proteins. Appl. Sci. 2020, 10, 5918. https://doi.org/10.3390/app10175918
Sadat A, Joye IJ. Peak Fitting Applied to Fourier Transform Infrared and Raman Spectroscopic Analysis of Proteins. Applied Sciences. 2020; 10(17):5918. https://doi.org/10.3390/app10175918
Chicago/Turabian StyleSadat, Azin, and Iris J. Joye. 2020. "Peak Fitting Applied to Fourier Transform Infrared and Raman Spectroscopic Analysis of Proteins" Applied Sciences 10, no. 17: 5918. https://doi.org/10.3390/app10175918