Enamine Barbiturates and Thiobarbiturates as a New Class of Bacterial Urease Inhibitors


 , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. In Vitro Urease Inhibition Assay Protocol
2.2. DFT Calculation
2.3. Molecular Docking
3. Results and Discussion
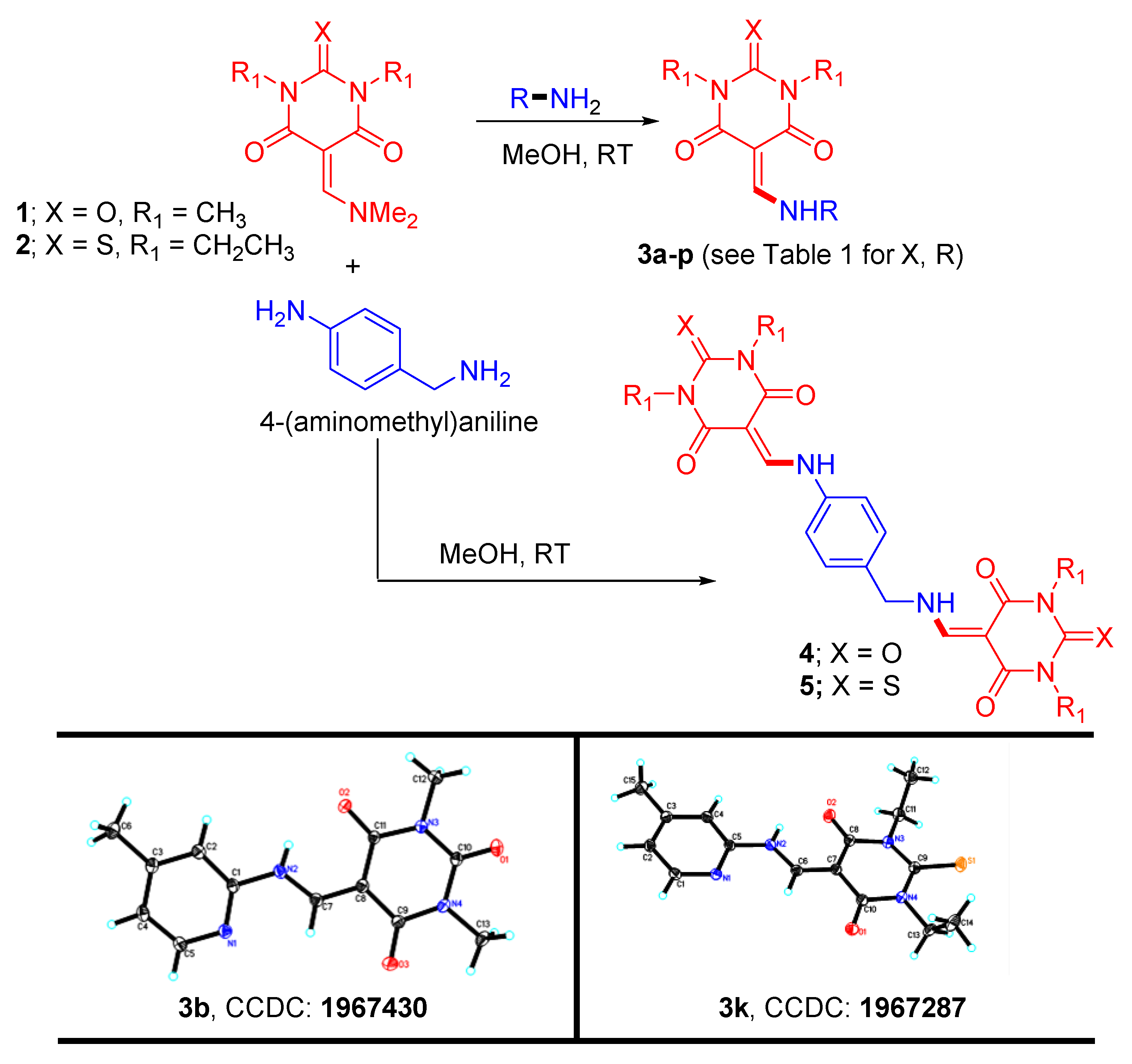
3.1. Synthesis
3.2. In Vitro Evaluation of Urease Inhibition
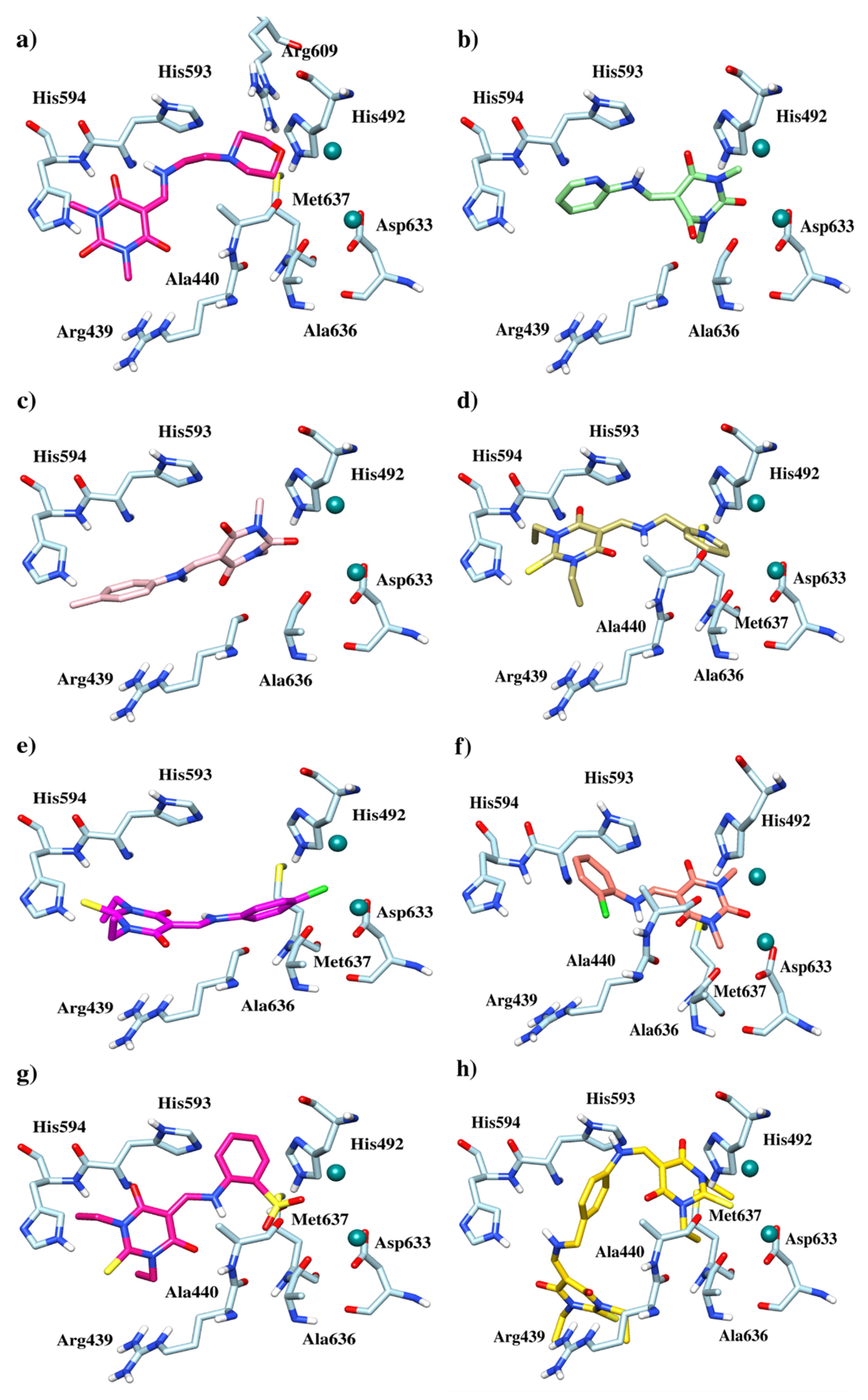
3.3. Docking Study
3.4. Density Functional Theory (DFT)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mobley, H.L.; Hausinger, R.P. Microbial ureases: Significance, regulation, and molecular characterization. Microbiol. Mol. Biol. Rev. 1989, 53, 85–108. [Google Scholar] [CrossRef] [Green Version]
- Zonia, L.E.; Stebbins, N.E.; Polacco, J.C. Essential role of urease in germination of nitrogen-limited Arabidopsis thaliana seeds. Plant Physiol. 1995, 107, 1097–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulvaney, R.L.; Bremner, J.M. Soil Biochemistry; Paul, E.A., Ladd, J.N., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 1981; pp. 153–196. [Google Scholar]
- Kataria, R.; Khatkar, A. In-silico Designing, ADMET Analysis, Synthesis and Biological Evaluation of Novel Derivatives of Diosmin Against Urease Protein and Helicobacter pylori Bacterium. Curr. Top. Med. Chem. 2019, 19, 2658–2675. [Google Scholar] [CrossRef] [PubMed]
- Li, W.Y.; Ni, W.W.; Ye, Y.X.; Fang, H.L.; Pan, X.M.; He, J.L.; Zhou, T.-L.; Yi, J.; Liu, S.-S.; Zhou, M.; et al. N-monoarylacetothioureas as potent urease inhibitors: Synthesis, SAR, and biological evaluation. J. Enzyme Inhib. Med. Chem. 2020, 35, 404–413. [Google Scholar] [CrossRef] [Green Version]
- Qamar, N.; Sultan, H.; Raheel, A.; Ashfaq, M.; Azmat, R.; Naz, R.; Raheela, L.M.; Khan, K.M.; Arshad, T. Heterochelates of metals as an effective anti-Urease agents couple with their docking studies. Pak. J. Pharm. Sci. 2019, 32, 1179–1183. [Google Scholar]
- Shah, S.R.; Shah, Z.; Khiat, M.; Khan, A.; Hill, L.R.; Khan, S.; Hussain, J.; Csuk, R.; Anwar, M.U.; Al-Harrasi, A. Complexes of N-and O-Donor Ligands as Potential Urease Inhibitors. ACS Omega 2020, 17, 10200–10206. [Google Scholar] [CrossRef]
- Hanif, M.; Saleem, M.; Hussain, M.T.; Rama, N.H.; Zaib, S.; Aslam MA, M.; Iqbal, J. Synthesis, urease inhibition, antioxidant and antibacterial studies of some 4-amino-5-aryl-3H-1,2,4-triazole-3-thiones and their 3,6-disubstituted 1,2,4-triazolo[3,4-b]1,3,4-thiadiazole derivatives. J. Braz. Chem. Soc. 2012, 23, 854–860. [Google Scholar] [CrossRef] [Green Version]
- Kafarski, P.; Talma, M. Recent advances in design of new urease inhibitors: A review. J. Adv. Res. 2018, 13, 101–112. [Google Scholar] [CrossRef]
- Shamim, S.; Khan, K.M.; Salar, U.; Ali, F.; Lodhi, M.A.; Taha, M.; Perveen, S. 5-Acetyl-6-methyl-4-aryl-3,4-dihydropyrimidin-2(1H)-ones: As potent urease inhibitors; synthesis, in vitro screening, and molecular modeling study. Bioorg. Chem. 2018, 76, 37–52. [Google Scholar] [CrossRef]
- Perveen, S.; Khan, K.M.; Lodhi, M.A.; Choudhary, M.I.; Voelter, W. Urease and α-chymotrypsin inhibitory effects of selected urea derivatives. Lett. Drug Des. Discov. 2008, 5, 401–405. [Google Scholar] [CrossRef]
- Pervez, H.; Chohan, Z.H.; Ramzan, M.; Nasim FU, H.; Khan, K.M. Synthesis and biological evaluation of some new N(4)-substituted isatin-3-thiosemicarbazones. J. Enzyme Inhib. Med. Chem. 2009, 24, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Aslam MA, S.; Mahmood, S.U.; Shahid, M.; Saeed, A.; Iqbal, J. Synthesis, biological assay in vitro and molecular docking studies of new Schiff base derivatives as potential urease inhibitors. Eur. J. Med. Chem. 2011, 46, 5473–5479. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Issar, U.; Kakkar, R. In silico study of the active site of Helicobacter pylori urease and its inhibition by hydroxamic acids. J. Mol. Graph. Model. 2018, 83, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.; Wadood, A. Synthesis and molecular docking study of piperazine derivatives as potent urease inhibitors. Bioorg. Chem. 2018, 78, 411–417. [Google Scholar]
- Khan, K.M.; Iqbal, S.; Lodhi, M.A.; Maharvi, G.M.; Choudhary, M.I.; Perveen, S. Biscoumarin: New class of urease inhibitors; economical synthesis and activity. Bioorg. Med. Chem. 2004, 12, 1963–1968. [Google Scholar] [CrossRef]
- Menteşe, E.; Bektaş, H.; Sokmen, B.B.; Emirik, M.; Çakır, D.; Kahveci, B. Synthesis and molecular docking study of some 5,6-dichloro-2-cyclopropyl-1H-benzimidazole derivatives bearing triazole, oxadiazole, and imine functionalities as potent inhibitors of urease. Bioorg. Med. Chem. Lett. 2017, 27, 3014–3018. [Google Scholar]
- Saeed, A.; Mahmood, S.U.; Rafiq, M.; Ashraf, Z.; Jabeen, F.; Seo, S.Y. Iminothiazoline-sulfonamide hybrids as Jack bean urease inhibitors; synthesis, kinetic mechanism and computational molecular modeling. Chem. Biol. Drug. Des. 2016, 87, 434–443. [Google Scholar] [CrossRef]
- Mabkhot, Y.N.; Barakat, A.; Yousuf, S. Substituted thieno [2, 3-b] thiophenes and related congeners: Synthesis, β-glucuronidase inhibition activity, crystal structure, and POM analyses. Bioorg. Med. Chem. 2014, 22, 6715–6725. [Google Scholar] [CrossRef]
- Mabkhot, Y.N.; Al-Majid, A.M.; Barakat, A.; Alshahrani, S.; Siddiqui, Y. 1, 1′-(3-Methyl-4-phenylthieno [2–b] thiophene-2, 5-diyl) diethanone as a building block in heterocyclic synthesis. novel synthesis of some pyrazole and pyrimidine derivatives. Molecules 2011, 16, 6502–6511. [Google Scholar] [CrossRef] [Green Version]
- Fırıncı, R.; Fırıncı, E.; Başbülbül, G.; Dabanca, M.B.; Celepci, D.B.; Günay, M.E. Enamines of 1, 3-dimethylbarbiturates and their symmetrical palladium (II) complexes: Synthesis, characterization and biological activity. Trans. Met. Chem. 2019, 44, 391–397. [Google Scholar]
- Cox, D.S.; Scott, K.R.; Gao, H.; Eddington, N.D. Effect of P-glycoprotein on the pharmacokinetics and tissue distribution of enaminone anticonvulsants: Analysis by population and physiological approaches. J. Pharmacol. Exp. Ther. 2002, 302, 1096–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidwai, M.; Thakur, R.; Mohan, R. Ecofriendly synthesis of novel antifungal (thio)- barbituric acid derivatives. Acta Chim. Solv. 2005, 52, 88–92. [Google Scholar]
- Figueiredo, J.; Serrano, J.L.; Cavalheiro, E.; Keurulainen, L.; Yli-Kauhaluoma, J.; Moreira, V.M.; Almeida, P. Trisubstituted barbiturates and thiobarbiturates: Synthesis and biological evaluation as xanthine oxidase inhibitors, antioxidants, antibacterial and anti-proliferative agents. Eur. J. Med. Chem. 2018, 143, 829–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabholkar, V.V.; Ravi, T.D. Synthesis of Biginelli products of thiobarbituric acids and their antimicrobial activity. J. Serb. Chem. Soc. 2010, 75, 1033–1040. [Google Scholar] [CrossRef]
- Ma, L.; Li, S.; Zheng, H. Synthesis and biological activity of novel barbituric and thiobarbituric acid derivatives against non-alcoholic fatty liver disease. Eur. J. Med. Chem. 2011, 46, 2003. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.M.; Ali, M.; Farooqui, T.A.; Khan, M.; Taha, M.; Perveen, S. An improved method for the synthesis of 5- arylidene barbiturates using BiCl3. J. Chem. Soc. Pak. 2009, 31, 823–828. [Google Scholar]
- Archana, S.V.K.; Kumar, A. Synthesis of some newer derivatives of substitute quinazolinonyl-2-oxo/thiobarbituric acid as potent anticonvulsant agents. Bioorg. Med. Chem. 2004, 12, 1257–1264. [Google Scholar] [CrossRef]
- Hassan, M.; Khan Faidallah, K.A. Synthesis and biological evaluation of new barbituric and thiobarbituric acid fluoro analogs of benzenesulfonamides as antidiabetic and antibacterial agents. J. Fluorine Chem. 2012, 142, 96–104. [Google Scholar]
- Ali, M.; Barakat, A.; El-Faham, A.; Al-Rasheed, H.H.; Dahlous, K.; Al-Majid, A.M.; Choudhary, M.I. Synthesis and characterisation of thiobarbituric acid enamine derivatives, and evaluation of their α-glucosidase inhibitory and anti-glycation activity. Enzyme Inhib. Med. Chem. 2020, 35, 692–701. [Google Scholar] [CrossRef]
- Puerta, D.T.; Cohen, S.M.A. A bioinorganic perspective on matrix metalloproteinase inhibition. Curr. Topic. Med. Chem. 2004, 4, 1551–1573. [Google Scholar] [CrossRef]
- Hameed, A.; Al-Rashida, M.; Uroos, M. A patent update on therapeutic applications of urease inhibitors (2012–2018). Expert Opin. Rapeutic Pat. 2019, 29, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Soliman, S.M.; Ali, M.; Elmarghany, A.; Al-Majid, A.M.; Yousuf, S.; El-Faham, A. Synthesis, crystal structure, evaluation of urease inhibition potential and the docking studies of cobalt(III) complex based on barbituric acid Schiff base ligand. Inorg. Chim. Acta. 2020, 503, 119405. [Google Scholar] [CrossRef]
- Barakat, A.; Ali, M.; Al-Majid, A.M.; Yousuf, S.; Choudhary, M.I.; Khalil, R.; Ul-Haq, Z. Synthesis of thiobarbituric acid derivatives: In vitro α-glucosidase inhibition and molecular docking studies. Bioorg. Chem. 2017, 75, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Al-Majid, A.M.; Al-Najjar, H.J.; Mabkhot, Y.N.; Javaid, S.; Yousuf, S.; Choudhary, M.I. Zwitterionic pyrimidinium adducts as antioxidants with therapeutic potential as nitric oxide scavenger. Eur. J. Med. Chem. 2014, 84, 146. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Islam, M.S.; Al-Majid, A.M.; Ghabbour, H.A.; Fun, H.K.; Javed, K.; Wadood, A. Synthesis, in vitro biological activities and in silico study of dihydropyrimidines derivatives. Bioorg.Med. Chem. 2015, 23, 6740. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Al-Majid, A.M.; Soliman, S.M.; Lotfy, G.; Ghabbour, H.A.; Fun, H.K.; Sloop, J.C. New diethyl ammonium salt of thiobarbituric acid derivative: Synthesis, molecular structure investigations and docking studies. Molecules 2015, 20, 20642–20658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barakat, A.; Al-Majid, A.M.; Lotfy, G.; Arshad, F.; Yousuf, S.; Choudhary, M.I.; Ul-Haq, Z. Synthesis and Dynamics Studies of Barbituric Acid Derivatives as Urease Inhibitors. Chem. Cent. J. 2015, 9, 63. [Google Scholar] [CrossRef] [Green Version]
- Badria, F.A.; Atef, S.; Al-Majid, A.M.; Ali, M.; Elshaier, Y.A.; Ghabbour, H.A.; Barakat, A. Synthesis and inhibitory effect of some indole-pyrimidine-based hybrid heterocycles on α-glucosidase and α-amylase as potential hypoglycemic agents. ChemistryOpen 2019, 8, 1288–1297. [Google Scholar] [CrossRef]
- Barakat, A.; Islam, M.S.; Al-Majid, A.M.; Ghabbour, H.A.; Yousuf, S.; Ashraf, M.; Ul-Haq, Z. Synthesis of pyrimidine-2,4,6-trione derivatives: Anti-oxidant, Anti-cancer, α-glucosidase, ß-glucuronidase inhibiton and their molecular docking studies. Bioorg. Chem. 2016, 86, 72–79. [Google Scholar] [CrossRef]
- Barakat, A.; Soliman, S.M.; Elshaier, Y.A.M.M. Molecular structure investigation, hypoglycemic/anticancer and docking studies of 5-((4-fluorophenyl)(2-hydroxy-6-oxocyclohex-1-en-1-yl)methyl)-6-hydroxy-1,3-dimethylpyrimidine-2,4(1H,3H)-dione. J. Mol. Struc. 2017, 1134, 99–111. [Google Scholar] [CrossRef]
- Ul-Haq, Z.; Ashraf, S.; Al-Majid, A.M.; Barakat, A. 3D-QSAR Studies on Barbituric Acid Derivatives as Urease Inhibitors and the Effect of Charges on the Quality of a Model. Int. J. Mol. Sci. 2016, 17, 657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, K.M.; Rahim, F.; Khan, A. Synthesis and structure–activity relationship of thiobarbituric acid derivatives as potent inhibitors of urease. Bioorg. Med. Chem. 2014, 22, 4119. [Google Scholar] [CrossRef] [PubMed]
- Altowyan, M.S.; Barakat, A.; Soliman, S.M.; Al-Majid, A.M.; Ali, M.; Elshaier, Y.A.; Ghabbour, H.A. A New Barbituric Acid Derivatives as Reactive Oxygen Scavenger: Experimental and Theoretical Investigations. J. Mol. Struc. 2019, 1175, 524–535. [Google Scholar] [CrossRef]
- Weatherburn, M.W. Phenol-hypochlorite reaction for determination of ammonia. Anal. Chem. 1967, 39, 971. [Google Scholar] [CrossRef]
- Mabkhot, Y.N.; Aldawsari, F.D.; Al-Showiman, S.S.; Barakat, A.; Soliman, S.M.; Choudhary, M.I.; Hadda, T.B. Novel enaminone derived from thieno [2–b] thiene: Synthesis, x-ray crystal structure, HOMO, LUMO, NBO analyses and biological activity. Chem. Cent. J. 2015, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.K.; Saify, Z.S.; Arif Lodhi, M.; Butt, N.; Perveen, S.; Murtaza Maharvi, G.; Atta-Ur-Rahman. Piperidines: A new class of urease inhibitors. Nat. Prod. Res. 2006, 20, 523. [Google Scholar] [CrossRef]
- Begum, A.; Banumathi, S.; Choudhary, M.I.; Betzel, C. Crystallographic structure analysis of urease from Jack bean (Canavalia ensiformis) at 1.49 A Resolution. Macromolecules 2012. [Google Scholar] [CrossRef]
- Kulinich, A.V.; Derevyanko, N.A.; Ishchenko, A.A. Synthesis and spectral properties of cyanine dyes-derivatives of 10,10-dimethyl-7,8,9,10-tetrahydro-6H-pyrido[1–a]indolium. J. Photochem. Photobiol. 2008, 198, 119–125. [Google Scholar] [CrossRef]
- Gorobets, N.Y.; Yousefi, B.H.; Belaj, F.; Kappe, C.O. Rapid microwave-assisted solution phase synthesis of substituted 2-pyridone libraries. Tetrahedron 2004, 60, 8633–8644. [Google Scholar] [CrossRef]
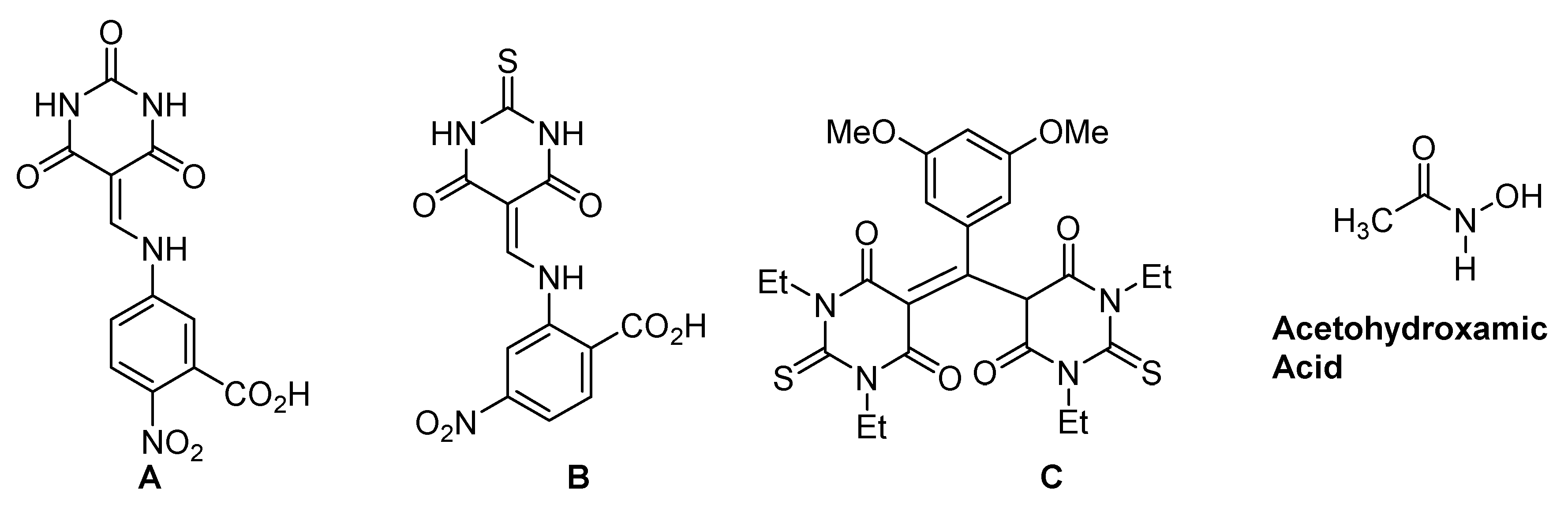
- Kumar, S.; Kayastha, A.M. Acetohydroxamic Acid—A Competitive Inhibitor of Urease from Soybean “Glycine max”. J. Proteins Proteom. 2013, 1, 3–8. [Google Scholar]
- Krajewska, B.; Zaborska, W. Jack bean urease: The effect of active-site binding inhibitors on the reactivity of enzyme thiol groups. Bioorg. Chem. 2007, 35, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group. Molecular Operating Environment (MOE) 2016.0810; Chemical Computing Group Inc.: Montreal, QC, Canada, 2016; Available online: https://www.chemcomp.com/Products.htm (accessed on 19 May 2020).
- Rauf, A.; Shahzad, S.; Bajda, M.; Yar, M.; Ahmed, F.; Hussain, N.; Jończyk, J. Design and synthesis of new barbituric-and thiobarbituric acid derivatives as potent urease inhibitors: Structure activity relationship and molecular modeling studies. Bioorg. Med. Chem. 2015, 23, 6049–6058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.N.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | Urease Inhibition IC50 ± SEM [µM] |
|---|---|---|
| 3a |  | 9 ± 2 |
| 3b |  | NA |
| 3c |  | 26 ± 1 |
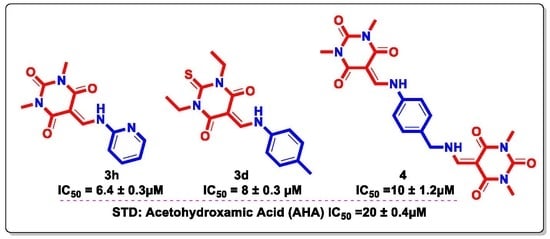
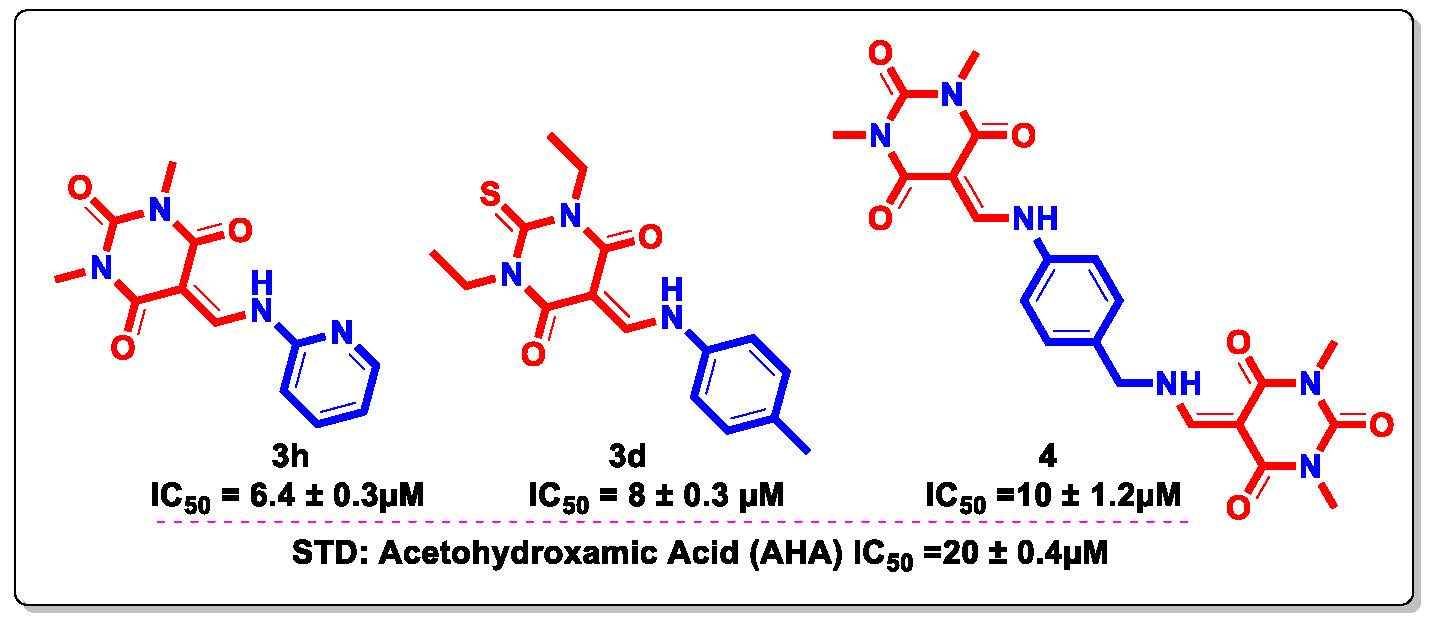
| 3d |  | 8 ± 0.3 |
| 3e |  | 11 ± 0.3 |
| 3f |  | 10 ± 0.6 |
| 3g |  | NA |
| 3h |  | 6.4 ± 0.3 |
| 3i |  | 66 ± 2.4 |
| 3j |  | 10 ± 0.9 |
| 3k |  | 11 ± 1.2 |
| 3l |  | 8 ± 0.4 |
| 3m |  | 15 ± 1.2 |
| 3n |  | 11 ± 1.1 |
| 3o |  | 9 ± 0.3 |
| 3p |  | 22 ± 0.8 |
| 4 |  | 10 ± 1.2 |
| 5 |  | 42 ± 2.3 |
| STD | Acetohydroxamic Acid (AHA) | 20 ± 0.4 |
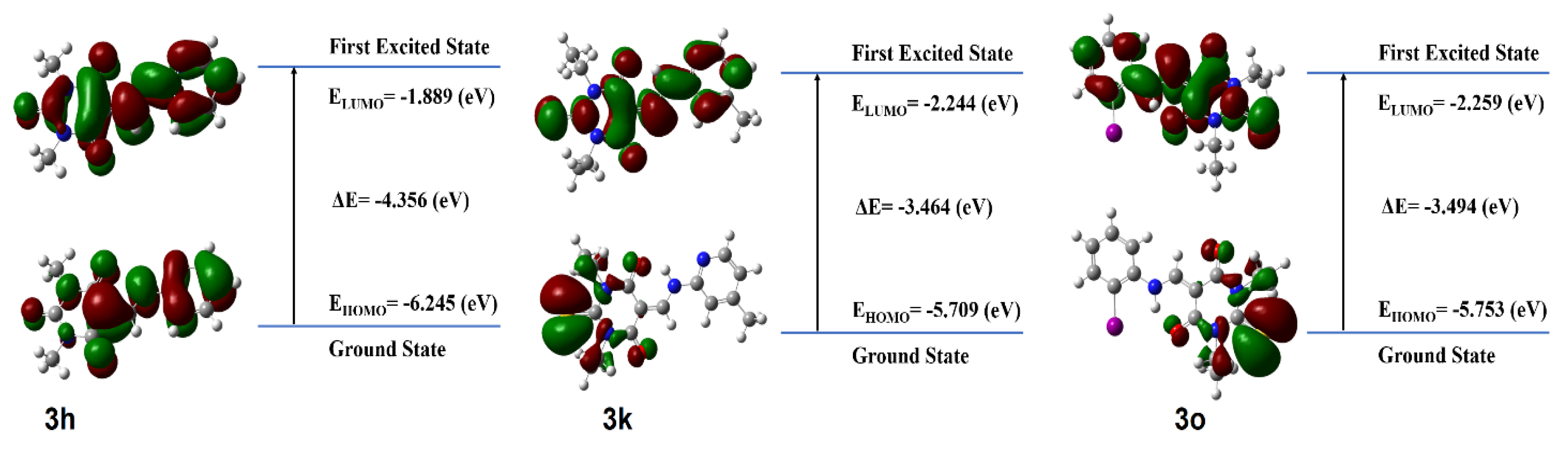
| Compound | IC50 | HOMO (eV) | LUMO (eV) | Egap (eV) |
|---|---|---|---|---|
| 3a | 9 ± 2 | −5.79330622969 | −1.21770997548 | −4.57559625421 |
| 3d | 8 ± 0.3 | −5.88881823 | −1.70370550982 | −4.76988562237 |
| 3f | 10 ± 0.6 | −6.2202530345 | −1.96248588673 | −4.25776714777 |
| 3h | 6 ± 0.3 | −6.24583174683 | −1.88955934518 | −4.35627240165 |
| 3k | 11 ± 1 | −5.70922301574 | −2.24439595033 | −3.46482706541 |
| 3o | 9 ± 0.3 | −5.75357759138 | −2.25963433214 | −3.49394325924 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, M.; Barakat, A.; El-Faham, A.; Al-Majid, A.M.; Yousuf, S.; Ashraf, S.; Ul-Haq, Z.; Choudhary, M.I.; de la Torre, B.G.; Albericio, F. Enamine Barbiturates and Thiobarbiturates as a New Class of Bacterial Urease Inhibitors. Appl. Sci. 2020, 10, 3523. https://doi.org/10.3390/app10103523
Ali M, Barakat A, El-Faham A, Al-Majid AM, Yousuf S, Ashraf S, Ul-Haq Z, Choudhary MI, de la Torre BG, Albericio F. Enamine Barbiturates and Thiobarbiturates as a New Class of Bacterial Urease Inhibitors. Applied Sciences. 2020; 10(10):3523. https://doi.org/10.3390/app10103523
Chicago/Turabian StyleAli, M., Assem Barakat, Ayman El-Faham, Abdullah Mohammed Al-Majid, Sammer Yousuf, Sajda Ashraf, Zaheer Ul-Haq, M. Iqbal Choudhary, Beatriz G. de la Torre, and Fernando Albericio. 2020. "Enamine Barbiturates and Thiobarbiturates as a New Class of Bacterial Urease Inhibitors" Applied Sciences 10, no. 10: 3523. https://doi.org/10.3390/app10103523