Spotlight on the Transglutaminase 2-Heparan Sulfate Interaction



Abstract
:1. Extracellular Transglutaminase 2 in Human Pathology
2. The Diversity of Heparan Sulfate Proteoglycans Functions in the Cells
3. Involvement of Heparan Sulfate in Pathology
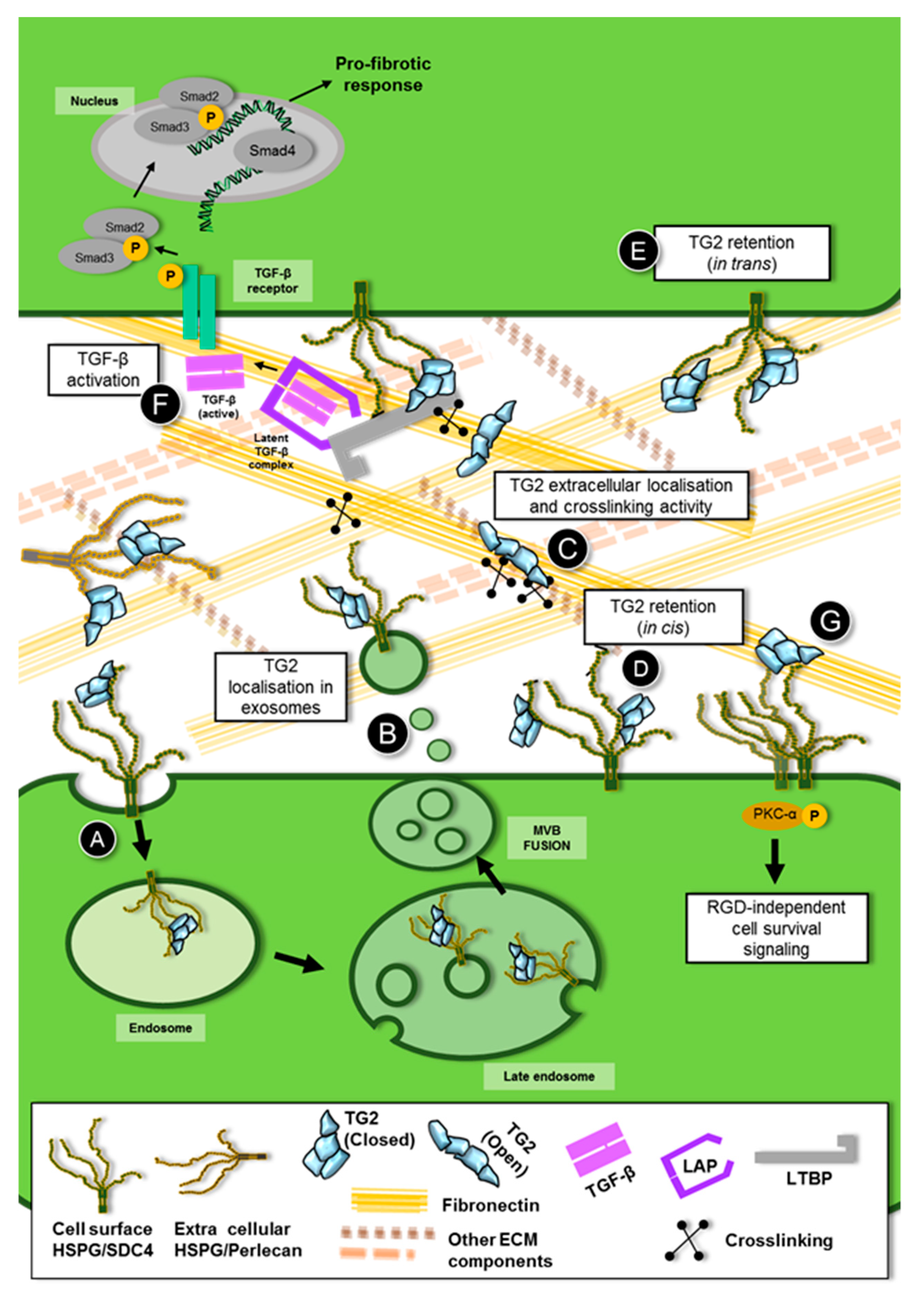
4. Role of Heparan Sulfate /Syndecan-4 in the Trafficking and Extracellular Function of TG2
5. Partnership of Transglutaminase-2 and Heparan Sulfate/Syndecan-4 in Disease
6. The Heparin Binding Site of Transglutaminase-2
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fesus, L.; Piacentini, M. Transglutaminase 2: An enigmatic enzyme with diverse functions. Trends Biochem. Sci. 2002, 27, 534–539. [Google Scholar] [CrossRef]
- Tatsukawa, H.; Furutani, Y.; Hitomi, K.; Kojima, S. Transglutaminase 2 has opposing roles in the regulation of cellular functions as well as cell growth and death. Cell Death Dis. 2016, 7, e2244. [Google Scholar] [CrossRef] [PubMed]
- Beninati, S.; Piacentini, M.; Bergamini, C.M. Transglutaminase 2, a double face enzyme. Amino Acids 2017, 49, 415–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, M.; Casadio, R.; Bergamini, C.M. Transglutaminases: Nature’s biological glues. Biochem. J. 2002, 368, 377–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savoca, M.P.; Tonoli, E.; Atobatele, A.G.; Verderio, E.A.M. Biocatalysis by Transglutaminases: A Review of Biotechnological Applications. Micromachines 2018, 9. [Google Scholar] [CrossRef]
- Csosz, E.; Mesko, B.; Fesus, L. Transdab wiki: The interactive transglutaminase substrate database on web 2.0 surface. Amino Acids 2009, 36, 615–617. [Google Scholar] [CrossRef]
- Tatsukawa, H.; Tani, Y.; Otsu, R.; Nakagawa, H.; Hitomi, K. Global identification and analysis of isozyme-specific possible substrates crosslinked by transglutaminases using substrate peptides in mouse liver fibrosis. Sci. Rep. 2017, 7, 45049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatsukawa, H.; Otsu, R.; Tani, Y.; Wakita, R.; Hitomi, K. Isozyme-specific comprehensive characterization of transglutaminase-crosslinked substrates in kidney fibrosis. Sci. Rep. 2018, 8, 7306. [Google Scholar] [CrossRef]
- Furini, G.; Schroeder, N.; Huang, L.; Boocock, D.; Scarpellini, A.; Coveney, C.; Tonoli, E.; Ramaswamy, R.; Ball, G.; Verderio, C.; et al. Proteomic Profiling Reveals the Transglutaminase-2 Externalization Pathway in Kidneys after Unilateral Ureteric Obstruction. J. Am. Soc. Nephrol. 2018, 29, 880–905. [Google Scholar] [CrossRef]
- Lortat-Jacob, H.; Burhan, I.; Scarpellini, A.; Thomas, A.; Imberty, A.; Vives, R.R.; Johnson, T.; Gutierrez, A.; Verderio, E.A. Transglutaminase-2 interaction with heparin: Identification of a heparin binding site that regulates cell adhesion to fibronectin-transglutaminase-2 matrix. J. Biol. Chem. 2012, 287, 18005–18017. [Google Scholar] [CrossRef]
- Nakaoka, H.; Perez, D.M.; Baek, K.J.; Das, T.; Husain, A.; Misono, K.; Im, M.J.; Graham, R.M. Gh: A GTP-binding protein with transglutaminase activity and receptor signaling function. Science 1994, 264, 1593–1596. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.S.; Slaughter, T.F.; Koropchak, C.M.; Haroon, Z.A.; Greenberg, C.S. C-terminal deletion of human tissue transglutaminase enhances magnesium-dependent GTP/ATPase activity. J. Biol. Chem. 1996, 271, 31191–31195. [Google Scholar] [CrossRef] [PubMed]
- Iismaa, S.E.; Chung, L.; Wu, M.J.; Teller, D.C.; Yee, V.C.; Graham, R.M. The core domain of the tissue transglutaminase Gh hydrolyzes GTP and ATP. Biochemistry 1997, 36, 11655–11664. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.; Grenard, P.; Aeschlimann, P.; Langley, M.; Blain, E.; Errington, R.; Kipling, D.; Thomas, D.; Aeschlimann, D. Crosslinking and G-protein functions of transglutaminase 2 contribute differentially to fibroblast wound healing responses. J. Cell Sci. 2004, 117, 3389–3403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, S.; Murphy, L.J. Tissue transglutaminase has intrinsic kinase activity: Identification of transglutaminase 2 as an insulin-like growth factor-binding protein-3 kinase. J. Biol. Chem. 2004, 279, 23863–23868. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Murphy, L.J. Phosphorylation of transglutaminase 2 by PKA at Ser216 creates 14-3-3 binding sites. Biochem. Biophys. Res. Commun. 2006, 347, 1166–1170. [Google Scholar] [CrossRef]
- Mishra, S.; Melino, G.; Murphy, L.J. Transglutaminase 2 kinase activity facilitates protein kinase A-induced phosphorylation of retinoblastoma protein. J. Biol. Chem. 2007, 282, 18108–18115. [Google Scholar] [CrossRef]
- Piacentini, M.; Farrace, M.G.; Piredda, L.; Matarrese, P.; Ciccosanti, F.; Falasca, L.; Rodolfo, C.; Giammarioli, A.M.; Verderio, E.; Griffin, M.; et al. Transglutaminase overexpression sensitizes neuronal cell lines to apoptosis by increasing mitochondrial membrane potential and cellular oxidative stress. J. Neurochem. 2002, 81, 1061–1072. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, G.; Suwa, M.; Ichikawa, Y.; Ohtsuka, T.; Kumagai, S.; Kikuchi, M.; Sato, Y.; Saito, Y. A novel function of tissue-type transglutaminase: Protein disulphide isomerase. Biochem. J. 2003, 373, 793–803. [Google Scholar] [CrossRef]
- Mastroberardino, P.G.; Farrace, M.G.; Viti, I.; Pavone, F.; Fimia, G.M.; Melino, G.; Rodolfo, C.; Piacentini, M. “Tissue” transglutaminase contributes to the formation of disulphide bridges in proteins of mitochondrial respiratory complexes. Biochim. Biophys. Acta 2006, 1757, 1357–1365. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, G.; Farrace, M.G.; Mastroberardino, P.G.; Viti, I.; Fimia, G.M.; Van Beeumen, J.; Devreese, B.; Melino, G.; Molinaro, G.; Busceti, C.L.; et al. Transglutaminase 2 ablation leads to defective function of mitochondrial respiratory complex I affecting neuronal vulnerability in experimental models of extrapyramidal disorders. J. Neurochem. 2007, 100, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, R.P.; Degano, P.; Godkin, A.J.; Jewell, D.P.; Hill, A.V. In vivo antigen challenge in celiac disease identifies a single transglutaminase-modified peptide as the dominant A-gliadin T-cell epitope. Nat. Med. 2000, 6, 337–342. [Google Scholar] [CrossRef]
- Arentz-Hansen, H.; Korner, R.; Molberg, O.; Quarsten, H.; Vader, W.; Kooy, Y.M.; Lundin, K.E.; Koning, F.; Roepstorff, P.; Sollid, L.M.; et al. The intestinal T cell response to alpha-gliadin in adult celiac disease is focused on a single deamidated glutamine targeted by tissue transglutaminase. J. Exp. Med. 2000, 191, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Dorum, S.; Qiao, S.W.; Sollid, L.M.; Fleckenstein, B. A quantitative analysis of transglutaminase 2-mediated deamidation of gluten peptides: Implications for the T-cell response in celiac disease. J. Proteome Res. 2009, 8, 1748–1755. [Google Scholar] [CrossRef] [PubMed]
- Sollid, L.M.; Jabri, B. Celiac disease and transglutaminase 2: A model for posttranslational modification of antigens and HLA association in the pathogenesis of autoimmune disorders. Curr. Opin. Immunol. 2011, 23, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Klöck, C.; DiRaimondo, T.R.; Khosla, C. Role of transglutaminase 2 in celiac disease pathogenesis. Semin. Immunopathol. 2012, 34, 513–522. [Google Scholar] [CrossRef]
- Barsigian, C.; Stern, A.M.; Martinez, J. Tissue (type II) transglutaminase covalently incorporates itself, fibrinogen, or fibronectin into high molecular weight complexes on the extracellular surface of isolated hepatocytes. Use of 2-[(2-oxopropyl)thio] imidazolium derivatives as cellular transglutaminase inactivators. J. Biol. Chem. 1991, 266, 22501–22509. [Google Scholar] [PubMed]
- Upchurch, H.F.; Conway, E.; Patterson, M.K., Jr.; Maxwell, M.D. Localization of cellular transglutaminase on the extracellular matrix after wounding: Characteristics of the matrix bound enzyme. J. Cell. Physiol. 1991, 149, 375–382. [Google Scholar] [CrossRef]
- Zemskov, E.A.; Janiak, A.; Hang, J.; Waghray, A.; Belkin, A.M. The role of tissue transglutaminase in cell-matrix interactions. Front. Biosci. 2006, 11, 1057–1076. [Google Scholar] [CrossRef]
- Belkin, A.M. Extracellular TG2: Emerging functions and regulation. FEBS J. 2011, 278, 4704–4716. [Google Scholar] [CrossRef]
- Akimov, S.S.; Krylov, D.; Fleischman, L.F.; Belkin, A.M. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J. Cell Biol. 2000, 148, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Verderio, E.A.; Telci, D.; Okoye, A.; Melino, G.; Griffin, M. A novel RGD-independent cel adhesion pathway mediated by fibronectin-bound tissue transglutaminase rescues cells from anoikis. J. Biol. Chem. 2003, 278, 42604–42614. [Google Scholar] [CrossRef] [PubMed]
- Telci, D.; Wang, Z.; Li, X.; Verderio, E.A.; Humphries, M.J.; Baccarini, M.; Basaga, H.; Griffin, M. Fibronectin-tissue transglutaminase matrix rescues RGD-impaired cell adhesion through syndecan-4 and beta1 integrin co-signaling. J. Biol. Chem. 2008, 283, 20937–20947. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Collighan, R.J.; Gross, S.R.; Danen, E.H.; Orend, G.; Telci, D.; Griffin, M. RGD-independent cell adhesion via a tissue transglutaminase-fibronectin matrix promotes fibronectin fibril deposition and requires syndecan-4/2 α5β1 integrin co-signaling. J. Biol. Chem. 2010, 285, 40212–40229. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Telci, D.; Griffin, M. Importance of syndecan-4 and syndecan-2 in osteoblast cell adhesion and survival mediated by a tissue transglutaminase-fibronectin complex. Exp. Cell Res. 2011, 317, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Kanchan, K.; Fuxreiter, M.; Fesus, L. Physiological, pathological, and structural implications of non-enzymatic protein-protein interactions of the multifunctional human transglutaminase 2. Cell. Mol. Life Sci. 2015, 72, 3009–3035. [Google Scholar] [CrossRef] [PubMed]
- Verderio, E.A.; Furini, G.; Burhan, I.W.; Johnson, T.S. Transglutaminases: Expression in kidney and relation to kidney fibrosis. In Transglutaminases: Multiple Functional Modifiers and Targets for New Drug Discovery; Hitomi, K., Kojima, S., Fesus, L., Eds.; Springer: Tokyo, Japan, 2015; pp. 229–262. [Google Scholar]
- Aeschlimann, D.; Paulsson, M. Cross-linking of laminin-nidogen complexes by tissue transglutaminase. A novel mechanism for basement membrane stabilization. J. Biol. Chem. 1991, 266, 15308–15317. [Google Scholar] [PubMed]
- Jones, R.A.; Nicholas, B.; Mian, S.; Davies, P.J.; Griffin, M. Reduced expression of tissue transglutaminase in a human endothelial cell line leads to changes in cell spreading, cell adhesion and reduced polymerisation of fibronectin. J. Cell Sci. 1997, 110 Pt 19, 2461–2472. [Google Scholar]
- Verderio, E.; Nicholas, B.; Gross, S.; Griffin, M. Regulated expression of tissue transglutaminase in Swiss 3T3 fibroblasts: Effects on the processing of fibronectin, cell attachment, and cell death. Exp. Cell Res. 1998, 239, 119–138. [Google Scholar] [CrossRef]
- Johnson, T.S.; Skill, N.J.; El Nahas, A.M.; Oldroyd, S.D.; Thomas, G.L.; Douthwaite, J.A.; Haylor, J.L.; Griffin, M. Transglutaminase transcription and antigen translocation in experimental renal scarring. J. Am. Soc. Nephrol. 1999, 10, 2146–2157. [Google Scholar]
- Aeschlimann, D.; Thomazy, V. Protein crosslinking in assembly and remodelling of extracellular matrices: The role of transglutaminases. Connect. Tissue Res. 2000, 41, 1–27. [Google Scholar] [CrossRef] [PubMed]
- De Laurenzi, V.; Melino, G. Gene disruption of tissue transglutaminase. Mol. Cell. Biol. 2001, 21, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Nanda, N.; Iismaa, S.E.; Owens, W.A.; Husain, A.; Mackay, F.; Graham, R.M. Targeted inactivation of Gh/tissue transglutaminase II. J. Biol. Chem. 2001, 276, 20673–20678. [Google Scholar] [CrossRef]
- Verderio, E.A.; Johnson, T.; Griffin, M. Tissue transglutaminase in normal and abnormal wound healing: Review article. Amino Acids 2004, 26, 387–404. [Google Scholar] [CrossRef]
- Chau, D.Y.; Collighan, R.J.; Verderio, E.A.; Addy, V.L.; Griffin, M. The cellular response to transglutaminase-cross-linked collagen. Biomaterials 2005, 26, 6518–6529. [Google Scholar] [CrossRef] [PubMed]
- Quan, G.; Choi, J.Y.; Lee, D.S.; Lee, S.C. TGF-β1 up-regulates transglutaminase two and fibronectin in dermal fibroblasts: A possible mechanism for the stabilization of tissue inflammation. Arch. Dermatol. Res. 2005, 297, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.S.; Griffin, M.; Thomas, G.L.; Skill, J.; Cox, A.; Yang, B.; Nicholas, B.; Birckbichler, P.J.; Muchaneta-Kubara, C.; Meguid El Nahas, A. The role of transglutaminase in the rat subtotal nephrectomy model of renal fibrosis. J. Clin. Investig. 1997, 99, 2950–2960. [Google Scholar] [CrossRef]
- Johnson, T.S.; El-Koraie, A.F.; Skill, N.J.; Baddour, N.M.; El Nahas, A.M.; Njloma, M.; Adam, A.G.; Griffin, M. Tissue transglutaminase and the progression of human renal scarring. J. Am. Soc. Nephrol. 2003, 14, 2052–2062. [Google Scholar] [CrossRef]
- Johnson, T.S.; Abo-Zenah, H.; Skill, J.N.; Bex, S.; Wild, G.; Brown, C.B.; Griffin, M.; El Nahas, A.M. Tissue transglutaminase: A mediator and predictor of chronic allograft nephropathy? Transplantation 2004, 77, 1667–1675. [Google Scholar] [CrossRef]
- El Nahas, A.M.; Abo-Zenah, H.; Skill, N.J.; Bex, S.; Wild, G.; Griffin, M.; Johnson, T.S. Elevated ε-(γ-glutamyl)lysine in human diabetic nephropathy results from increased expression and cellular release of tissue transglutaminase. Nephron Clin. Pract. 2004, 97, c108–c117. [Google Scholar] [CrossRef]
- Elli, L.; Bergamini, C.M.; Bardella, M.T.; Schuppan, D. Transglutaminases in inflammation and fibrosis of the gastrointestinal tract and the liver. Dig. Liver Dis. 2009, 41, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Grenard, P.; Bresson-Hadni, S.; El Alaoui, S.; Chevallier, M.; Vuitton, D.A.; Ricard-Blum, S. Transglutaminase-mediated cross-linking is involved in the stabilization of extracellular matrix in human liver fibrosis. J. Hepatol. 2001, 35, 367–375. [Google Scholar] [CrossRef]
- Wang, Z.; Stuckey, D.J.; Murdoch, C.E.; Camelliti, P.; Lip, G.Y.H.; Griffin, M. Cardiac fibrosis can be attenuated by blocking the activity of transglutaminase 2 using a selective small-molecule inhibitor. Cell Death Dis. 2018, 9, 613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, M.; Smith, L.L.; Wynne, J. Changes in transglutaminase activity in an experimental model of pulmonary fibrosis induced by paraquat. Br. J. Exp. Pathol. 1979, 60, 653–661. [Google Scholar] [PubMed]
- Olsen, K.C.; Sapinoro, R.E.; Kottmann, R.M.; Kulkarni, A.A.; Iismaa, S.E.; Johnson, G.V.; Thatcher, T.H.; Phipps, R.P.; Sime, P.J. Transglutaminase 2 and its role in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Kojima, S.; Nara, K.; Rifkin, D.B. Requirement for transglutaminase in the activation of latent transforming growth factor-β in bovine endothelial cells. J. Cell Biol. 1993, 121, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Nunes, I.; Gleizes, P.E.; Metz, C.N.; Rifkin, D.B. Latent transforming growth factor-β binding protein domains involved in activation and transglutaminase-dependent cross-linking of latent transforming growth factor-β. J. Cell Biol. 1997, 136, 1151–1163. [Google Scholar] [CrossRef]
- Verderio, E.; Gaudry, C.; Gross, S.; Smith, C.; Downes, S.; Griffin, M. Regulation of cell surface tissue transglutaminase: Effects on matrix storage of latent transforming growth factor-β binding protein-1. J. Histochem. Cytochem. 1999, 47, 1417–1432. [Google Scholar] [CrossRef]
- Telci, D.; Collighan, R.J.; Basaga, H.; Griffin, M. Increased TG2 expression can result in induction of transforming growth factor β1, causing increased synthesis and deposition of matrix proteins, which can be regulated by nitric oxide. J. Biol. Chem. 2009, 284, 29547–29558. [Google Scholar] [CrossRef]
- Shweke, N.; Boulos, N.; Jouanneau, C.; Vandermeersch, S.; Melino, G.; Dussaule, J.C.; Chatziantoniou, C.; Ronco, P.; Boffa, J.J. Tissue transglutaminase contributes to interstitial renal fibrosis by favoring accumulation of fibrillar collagen through TGF-β activation and cell infiltration. Am. J. Pathol. 2008, 173, 631–642. [Google Scholar] [CrossRef]
- Huang, L.; Haylor, J.L.; Fisher, M.; Hau, Z.; El Nahas, A.M.; Griffin, M.; Johnson, T.S. Do changes in transglutaminase activity alter latent transforming growth factor β activation in experimental diabetic nephropathy? Nephrol. Dial. Transplant. 2010, 25, 3897–3910. [Google Scholar] [CrossRef] [PubMed]
- Burhan, I.; Furini, G.; Lortat-Jacob, H.; Atobatele, A.G.; Scarpellini, A.; Schroeder, N.; Atkinson, J.; Maamra, M.; Nutter, F.H.; Watson, P.; et al. Interplay between transglutaminases and heparan sulphate in progressive renal scarring. Sci. Rep. 2016, 6, 31343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, T.S.; Fisher, M.; Haylor, J.L.; Hau, Z.; Skill, N.J.; Jones, R.; Saint, R.; Coutts, I.; Vickers, M.E.; El Nahas, A.M.; et al. Transglutaminase inhibition reduces fibrosis and preserves function in experimental chronic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 3078–3088. [Google Scholar] [CrossRef] [PubMed]
- Iismaa, S.E.; Mearns, B.M.; Lorand, L.; Graham, R.M. Transglutaminases and disease: Lessons from genetically engineered mouse models and inherited disorders. Physiol. Rev. 2009, 89, 991–1023. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, D.; Campisi, A.; Curro, M.; Li Volti, G.; Vanella, A.; Ientile, R. Excitotoxic and post-ischemic neurodegeneration: Involvement of transglutaminases. Amino Acids 2004, 27, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Junn, E.; Ronchetti, R.D.; Quezado, M.M.; Kim, S.Y.; Mouradian, M.M. Tissue transglutaminase-induced aggregation of α-synuclein: Implications for Lewy body formation in Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 2003, 100, 2047–2052. [Google Scholar] [CrossRef] [PubMed]
- Mastroberardino, P.G.; Iannicola, C.; Nardacci, R.; Bernassola, F.; De Laurenzi, V.; Melino, G.; Moreno, S.; Pavone, F.; Oliverio, S.; Fesus, L.; et al. ‘Tissue’ transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington’s disease. Cell Death Differ. 2002, 9, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmus, M.M.; Grunberg, S.C.; Bol, J.G.; van Dam, A.M.; Hoozemans, J.J.; Rozemuller, A.J.; Drukarch, B. Transglutaminases and transglutaminase-catalyzed cross-links colocalize with the pathological lesions in Alzheimer’s disease brain. Brain Pathol. 2009, 19, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Mangala, L.S.; Mehta, K. Tissue transglutaminase (TG2) in cancer biology. Prog. Exp. Tumor Res. 2005, 38, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Shao, M.; Schilder, J.; Guise, T.; Mohammad, K.S.; Matei, D. Tissue transglutaminase links TGF-β, epithelial to mesenchymal transition and a stem cell phenotype in ovarian cancer. Oncogene 2012, 31, 2521–2534. [Google Scholar] [CrossRef]
- Kumar, S.; Mehta, K. Tissue transglutaminase constitutively activates HIF-1α promoter and nuclear factor-κB via a non-canonical pathway. PLoS ONE 2012, 7, e49321. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.L.; Kaartinen, M.T.; Nurminskaya, M.; Belkin, A.M.; Colak, G.; Johnson, G.V.; Mehta, K. Transglutaminase regulation of cell function. Physiol. Rev. 2014, 94, 383–417. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.L.; Fisher, M.L.; Grun, D.; Adhikary, G.; Xu, W.; Kerr, C. Transglutaminase is a tumor cell and cancer stem cell survival factor. Mol. Carcinog. 2015, 54, 947–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, M.L.; Adhikary, G.; Xu, W.; Kerr, C.; Keillor, J.W.; Eckert, R.L. Type II transglutaminase stimulates epidermal cancer stem cell epithelial-mesenchymal transition. Oncotarget 2015, 6, 20525–20539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Condello, S.; Yakubov, B.; Emerson, R.; Caperell-Grant, A.; Hitomi, K.; Xie, J.; Matei, D. Tissue Transglutaminase Mediated Tumor-Stroma Interaction Promotes Pancreatic Cancer Progression. Clin. Cancer Res. 2015, 21, 4482–4493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condello, S.; Sima, L.; Ivan, C.; Cardenas, H.; Schiltz, G.; Mishra, R.K.; Matei, D. Tissue Tranglutaminase Regulates Interactions between Ovarian Cancer Stem Cells and the Tumor Niche. Cancer Res. 2018, 78, 2990–3001. [Google Scholar] [CrossRef]
- Ikura, K.; Yokota, H.; Sasaki, R.; Chiba, H. Determination of amino- and carboxyl-terminal sequences of guinea pig liver transglutaminase: Evidence for amino-terminal processing. Biochemistry 1989, 28, 2344–2348. [Google Scholar] [CrossRef]
- Ichinose, A.; Bottenus, R.E.; Davie, E.W. Structure of transglutaminases. J. Biol. Chem. 1990, 265, 13411–13414. [Google Scholar]
- Chou, C.Y.; Streets, A.J.; Watson, P.F.; Huang, L.; Verderio, E.A.; Johnson, T.S. A crucial sequence for transglutaminase type 2 extracellular trafficking in renal tubular epithelial cells lies in its N-terminal β-sandwich domain. J. Biol. Chem. 2011, 286, 27825–27835. [Google Scholar] [CrossRef]
- Esko, J.D.; Kimata, K.; Lindahl, U. Proteoglycans and Sulfated Glycosaminoglycans. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor: New York, NY, USA, 2009. [Google Scholar]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef]
- Raats, C.J.; Van Den Born, J.; Berden, J.H. Glomerular heparan sulfate alterations: Mechanisms and relevance for proteinuria. Kidney Int. 2000, 57, 385–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iozzo, R.V. Basement membrane proteoglycans: From cellar to ceiling. Nat. Rev. Mol. Cell Biol. 2005, 6, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Bernfield, M.; Gotte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef] [PubMed]
- Tkachenko, E.; Rhodes, J.M.; Simons, M. Syndecans: New kids on the signaling block. Circ. Res. 2005, 96, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Dews, I.C.; Mackenzie, K.R. Transmembrane domains of the syndecan family of growth factor coreceptors display a hierarchy of homotypic and heterotypic interactions. Proc. Natl. Acad. Sci. USA 2007, 104, 20782–20787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afratis, N.A.; Nikitovic, D.; Multhaupt, H.A.; Theocharis, A.D.; Couchman, J.R.; Karamanos, N.K. Syndecans—Key regulators of cell signaling and biological functions. FEBS J. 2017, 284, 27–41. [Google Scholar] [CrossRef]
- Elfenbein, A.; Simons, M. Syndecan-4 signaling at a glance. J. Cell Sci. 2013, 126, 3799–3804. [Google Scholar] [CrossRef] [Green Version]
- Whitelock, J.M.; Iozzo, R.V. Heparan sulfate: A complex polymer charged with biological activity. Chem. Rev. 2005, 105, 2745–2764. [Google Scholar] [CrossRef]
- Bishop, J.R.; Schuksz, M.; Esko, J.D. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 2007, 446, 1030–1037. [Google Scholar] [CrossRef]
- Sugahara, K.; Kitagawa, H. Heparin and heparan sulfate biosynthesis. IUBMB Life 2002, 54, 163–175. [Google Scholar] [CrossRef]
- Dreyfuss, J.L.; Regatieri, C.V.; Jarrouge, T.R.; Cavalheiro, R.P.; Sampaio, L.O.; Nader, H.B. Heparan sulfate proteoglycans: Structure, protein interactions and cell signaling. An. Acad. Bras. Cienc. 2009, 81, 409–429. [Google Scholar] [CrossRef] [PubMed]
- Spivak-Kroizman, T.; Lemmon, M.A.; Dikic, I.; Ladbury, J.E.; Pinchasi, D.; Huang, J.; Jaye, M.; Crumley, G.; Schlessinger, J.; Lax, I. Heparin-induced oligomerization of FGF molecules is responsible for FGF receptor dimerization, activation, and cell proliferation. Cell 1994, 79, 1015–1024. [Google Scholar] [CrossRef]
- Steinfeld, R.; Van Den Berghe, H.; David, G. Stimulation of fibroblast growth factor receptor-1 occupancy and signaling by cell surface-associated syndecans and glypican. J. Cell Biol. 1996, 133, 405–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobsson, L.; Kreuger, J.; Holmborn, K.; Lundin, L.; Eriksson, I.; Kjellen, L.; Claesson-Welsh, L. Heparan sulfate in trans potentiates VEGFR-mediated angiogenesis. Dev. Cell 2006, 10, 625–634. [Google Scholar] [CrossRef]
- Kirkpatrick, C.A.; Selleck, S.B. Heparan sulfate proteoglycans at a glance. J. Cell Sci. 2007, 120, 1829–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iozzo, R.V.; Zoeller, J.J.; Nystrom, A. Basement membrane proteoglycans: Modulators Par Excellence of cancer growth and angiogenesis. Mol. Cells 2009, 27, 503–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambaerts, K.; Wilcox-Adelman, S.A.; Zimmermann, P. The signaling mechanisms of syndecan heparan sulfate proteoglycans. Curr. Opin. Cell Biol. 2009, 21, 662–669. [Google Scholar] [CrossRef] [Green Version]
- Woods, A. Syndecans: Transmembrane modulators of adhesion and matrix assembly. J. Clin. Investig. 2001, 107, 935–941. [Google Scholar] [CrossRef]
- Morgan, M.R.; Humphries, M.J.; Bass, M.D. Synergistic control of cell adhesion by integrins and syndecans. Nat. Rev. Mol. Cell Biol. 2007, 8, 957–969. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.J.; Jang, B.; Yi, J.Y.; Han, I.O.; Oh, E.S. Syndecans play dual roles as cell adhesion receptors and docking receptors. FEBS Lett. 2012, 586, 2207–2211. [Google Scholar] [CrossRef] [Green Version]
- Echtermeyer, F.; Baciu, P.C.; Saoncella, S.; Ge, Y.; Goetinck, P.F. Syndecan-4 core protein is sufficient for the assembly of focal adhesions and actin stress fibers. J. Cell Sci. 1999, 112 Pt 20, 3433–3441. [Google Scholar]
- Saoncella, S.; Echtermeyer, F.; Denhez, F.; Nowlen, J.K.; Mosher, D.F.; Robinson, S.D.; Hynes, R.O.; Goetinck, P.F. Syndecan-4 signals cooperatively with integrins in a Rhodependent manner in the assembly of focal adhesions and actin stress fibers. Proc. Natl. Acad. Sci. USA 1999, 96, 2805–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, A.; Longley, R.L.; Tumova, S.; Couchman, J.R. Syndecan-4 binding to the high affinity heparin-binding domain of fibronectin drives focal adhesion formation in fibroblasts. Arch. Biochem. Biophys. 2000, 374, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, A.N.; Multhaupt, H.A.; Couchman, J.R. Syndecans in wound healing, inflammation and vascular biology. Int. J. Biochem. Cell Biol. 2007, 39, 505–528. [Google Scholar] [CrossRef]
- Mahalingam, Y.; Gallagher, J.T.; Couchman, J.R. Cellular adhesion responses to the heparin-binding (HepII) domain of fibronectin require heparan sulfate with specific properties. J. Biol. Chem. 2007, 282, 3221–3230. [Google Scholar] [CrossRef] [PubMed]
- Gallo, R.; Kim, C.; Kokenyesi, R.; Adzick, N.S.; Bernfield, M. Syndecans-1 and -4 are induced during wound repair of neonatal but not fetal skin. J. Investig. Dermatol. 1996, 107, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Brown, L.F.; Laham, R.J.; Volk, R.; Simons, M. Macrophage-dependent regulation of syndecan gene expression. Circ. Res. 1997, 81, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Tanino, Y.; Chang, M.Y.; Wang, X.; Gill, S.E.; Skerrett, S.; McGuire, J.K.; Sato, S.; Nikaido, T.; Kojima, T.; Munakata, M.; et al. Syndecan-4 regulates early neutrophil migration and pulmonary inflammation in response to lipopolysaccharide. Am. J. Respir. Cell Mol. Biol. 2012, 47, 196–202. [Google Scholar] [CrossRef]
- Xie, J.; Wang, J.; Li, R.; Dai, Q.; Yong, Y.; Zong, B.; Xu, Y.; Li, E.; Ferro, A.; Xu, B. Syndecan-4 over-expression preserves cardiac function in a rat model of myocardial infarction. J. Mol. Cell. Cardiol. 2012, 53, 250–258. [Google Scholar] [CrossRef]
- Oh, E.S.; Woods, A.; Couchman, J.R. Multimerization of the cytoplasmic domain of syndecan-4 is required for its ability to activate protein kinase C. J. Biol. Chem. 1997, 272, 11805–11811. [Google Scholar] [CrossRef]
- Oh, E.S.; Woods, A.; Lim, S.T.; Theibert, A.W.; Couchman, J.R. Syndecan-4 proteoglycan cytoplasmic domain and phosphatidylinositol 4,5-bisphosphate coordinately regulate protein kinase C activity. J. Biol. Chem. 1998, 273, 10624–10629. [Google Scholar] [CrossRef] [PubMed]
- Bass, M.D.; Humphries, M.J. Cytoplasmic interactions of syndecan-4 orchestrate adhesion receptor and growth factor receptor signalling. Biochem. J. 2002, 368, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couchman, J.R.; Vogt, S.; Lim, S.T.; Lim, Y.; Oh, E.S.; Prestwich, G.D.; Theibert, A.; Lee, W.; Woods, A. Regulation of inositol phospholipid binding and signaling through syndecan-4. J. Biol. Chem. 2002, 277, 49296–49303. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.T.; Longley, R.L.; Couchman, J.R.; Woods, A. Direct binding of syndecan-4 cytoplasmic domain to the catalytic domain of protein kinase Cα (PKCα) increases focal adhesion localization of PKCα. J. Biol. Chem. 2003, 278, 13795–13802. [Google Scholar] [CrossRef] [PubMed]
- Dovas, A.; Yoneda, A.; Couchman, J.R. PKCβ-dependent activation of RhoA by syndecan-4 during focal adhesion formation. J. Cell Sci. 2006, 119, 2837–2846. [Google Scholar] [CrossRef] [PubMed]
- Bass, M.D.; Roach, K.A.; Morgan, M.R.; Mostafavi-Pour, Z.; Schoen, T.; Muramatsu, T.; Mayer, U.; Ballestrem, C.; Spatz, J.P.; Humphries, M.J. Syndecan-4-dependent Rac1 regulation determines directional migration in response to the extracellular matrix. J. Cell Biol. 2007, 177, 527–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, M.R.; Hamidi, H.; Bass, M.D.; Warwood, S.; Ballestrem, C.; Humphries, M.J. Syndecan-4 phosphorylation is a control point for integrin recycling. Dev. Cell 2013, 24, 472–485. [Google Scholar] [CrossRef]
- Tkachenko, E.; Simons, M. Clustering induces redistribution of syndecan-4 core protein into raft membrane domains. J. Biol. Chem. 2002, 277, 19946–19951. [Google Scholar] [CrossRef]
- Tkachenko, E.; Lutgens, E.; Stan, R.V.; Simons, M. Fibroblast growth factor 2 endocytosis in endothelial cells proceed via syndecan-4-dependent activation of Rac1 and a Cdc42-dependent macropinocytic pathway. J. Cell Sci. 2004, 117, 3189–3199. [Google Scholar] [CrossRef] [Green Version]
- Elfenbein, A.; Lanahan, A.; Zhou, T.X.; Yamasaki, A.; Tkachenko, E.; Matsuda, M.; Simons, M. Syndecan 4 regulates FGFR1 signaling in endothelial cells by directing macropinocytosis. Sci. Signal 2012, 5, ra36. [Google Scholar] [CrossRef]
- Christianson, H.C.; Belting, M. Heparan sulfate proteoglycan as a cell-surface endocytosis receptor. Matrix Biol. 2014, 35, 51–55. [Google Scholar] [CrossRef]
- Stanford, K.I.; Wang, L.; Castagnola, J.; Song, D.; Bishop, J.R.; Brown, J.R.; Lawrence, R.; Bai, X.; Habuchi, H.; Tanaka, M.; et al. Heparan sulfate 2-O-sulfotransferase is required for triglyceride-rich lipoprotein clearance. J. Biol. Chem. 2010, 285, 286–294. [Google Scholar] [CrossRef]
- Zimmermann, P.; Zhang, Z.; Degeest, G.; Mortier, E.; Leenaerts, I.; Coomans, C.; Schulz, J.; N’Kuli, F.; Courtoy, P.J.; David, G. Syndecan recycling is controlled by syntenin-PIP2 interaction and Arf6. Dev. Cell 2005, 9, 377–388. [Google Scholar] [CrossRef]
- Bass, M.D.; Williamson, R.C.; Nunan, R.D.; Humphries, J.D.; Byron, A.; Morgan, M.R.; Martin, P.; Humphries, M.J. A syndecan-4 hair trigger initiates wound healing through caveolin- and RhoG-regulated integrin endocytosis. Dev. Cell 2011, 21, 681–693. [Google Scholar] [CrossRef]
- Prieto-Sanchez, R.M.; Berenjeno, I.M.; Bustelo, X.R. Involvement of the Rho/Rac family member RhoG in caveolar endocytosis. Oncogene 2006, 25, 2961–2973. [Google Scholar] [CrossRef] [Green Version]
- Elfenbein, A.; Rhodes, J.M.; Meller, J.; Schwartz, M.A.; Matsuda, M.; Simons, M. Suppression of RhoG activity is mediated by a syndecan 4-synectin-RhoGDI1 complex and is reversed by PKCα in a Rac1 activation pathway. J. Cell Biol. 2009, 186, 75–83. [Google Scholar] [CrossRef]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef]
- Ghossoub, R.; Lembo, F.; Rubio, A.; Gaillard, C.B.; Bouchet, J.; Vitale, N.; Slavik, J.; Machala, M.; Zimmermann, P. Syntenin-ALIX exosome biogenesis and budding into multivesicular bodies are controlled by ARF6 and PLD2. Nat. Commun. 2014, 5, 3477. [Google Scholar] [CrossRef] [Green Version]
- Friand, V.; David, G.; Zimmermann, P. Syntenin and syndecan in the biogenesis of exosomes. Biol. Cell 2015, 107, 331–341. [Google Scholar] [CrossRef]
- Roucourt, B.; Meeussen, S.; Bao, J.; Zimmermann, P.; David, G. Heparanase activates the syndecan-syntenin-ALIX exosome pathway. Cell Res. 2015, 25, 412–428. [Google Scholar] [CrossRef] [Green Version]
- Echtermeyer, F.; Streit, M.; Wilcox-Adelman, S.; Saoncella, S.; Denhez, F.; Detmar, M.; Goetinck, P. Delayed wound repair and impaired angiogenesis in mice lacking syndecan-4. J. Clin. Investig. 2001, 107, R9–R14. [Google Scholar] [CrossRef] [Green Version]
- Matsui, Y.; Ikesue, M.; Danzaki, K.; Morimoto, J.; Sato, M.; Tanaka, S.; Kojima, T.; Tsutsui, H.; Uede, T. Syndecan-4 prevents cardiac rupture and dysfunction after myocardial infarction. Circ. Res. 2011, 108, 1328–1339. [Google Scholar] [CrossRef]
- Chen, Y.; Shi-Wen, X.; van Beek, J.; Kennedy, L.; McLeod, M.; Renzoni, E.A.; Bou-Gharios, G.; Wilcox-Adelman, S.; Goetinck, P.F.; Eastwood, M.; et al. Matrix contraction by dermal fibroblasts requires transforming growth factor-beta/activin-linked kinase 5, heparan sulfate-containing proteoglycans, and MEK/ERK: Insights into pathological scarring in chronic fibrotic disease. Am. J. Pathol. 2005, 167, 1699–1711. [Google Scholar] [CrossRef]
- Chen, Y.; Leask, A.; Abraham, D.J.; Pala, D.; Shiwen, X.; Khan, K.; Liu, S.; Carter, D.E.; Wilcox-Adelman, S.; Goetinck, P.; et al. Heparan sulfate-dependent ERK activation contributes to the overexpression of fibrotic proteins and enhanced contraction by scleroderma fibroblasts. Arthritis Rheumatol. 2008, 58, 577–585. [Google Scholar] [CrossRef]
- Schellings, M.W.; Vanhoutte, D.; van Almen, G.C.; Swinnen, M.; Leenders, J.J.; Kubben, N.; van Leeuwen, R.E.; Hofstra, L.; Heymans, S.; Pinto, Y.M. Syndecan-1 amplifies angiotensin II-induced cardiac fibrosis. Hypertension 2010, 55, 249–256. [Google Scholar] [CrossRef]
- Lunde, I.G.; Herum, K.M.; Carlson, C.C.; Christensen, G. Syndecans in heart fibrosis. Cell Tissue Res. 2016, 365, 539–552. [Google Scholar] [CrossRef]
- Kliment, C.R.; Englert, J.M.; Gochuico, B.R.; Yu, G.; Kaminski, N.; Rosas, I.; Oury, T.D. Oxidative stress alters syndecan-1 distribution in lungs with pulmonary fibrosis. J. Biol. Chem. 2009, 284, 3537–3545. [Google Scholar] [CrossRef]
- Morita, H.; David, G.; Mizutani, A.; Shinzato, T.; Habuchi, H.; Maeda, K.; Kimata, K. Heparan sulfate proteoglycans in the human sclerosing and scarring kidney. Changes in heparan sulfate moiety. Contrib. Nephrol. 1994, 107, 174–179. [Google Scholar]
- Yung, S.; Woods, A.; Chan, T.M.; Davies, M.; Williams, J.D.; Couchman, J.R. Syndecan-4 up-regulation in proliferative renal disease is related to microfilament organization. FASEB J. 2001, 15, 1631–1633. [Google Scholar] [CrossRef]
- Fan, Q.; Shike, T.; Shigihara, T.; Tanimoto, M.; Gohda, T.; Makita, Y.; Wang, L.N.; Horikoshi, S.; Tomino, Y. Gene expression profile in diabetic KK/Ta mice. Kidney Int. 2003, 64, 1978–1985. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Klass, C.; Woods, A. Syndecan-2 regulates transforming growth factor-beta signaling. J. Biol. Chem. 2004, 279, 15715–15718. [Google Scholar] [CrossRef]
- Cevikbas, F.; Schaefer, L.; Uhlig, P.; Robenek, H.; Theilmeier, G.; Echtermeyer, F.; Bruckner, P. Unilateral nephrectomy leads to up-regulation of syndecan-2- and TGF-β-mediated glomerulosclerosis in syndecan-4 deficient male mice. Matrix Biol. 2008, 27, 42–52. [Google Scholar] [CrossRef]
- Padberg, J.S.; Wiesinger, A.; di Marco, G.S.; Reuter, S.; Grabner, A.; Kentrup, D.; Lukasz, A.; Oberleithner, H.; Pavenstadt, H.; Brand, M.; et al. Damage of the endothelial glycocalyx in chronic kidney disease. Atherosclerosis 2014, 234, 335–343. [Google Scholar] [CrossRef]
- Ruiz, X.D.; Mlakar, L.R.; Yamaguchi, Y.; Su, Y.; Larregina, A.T.; Pilewski, J.M.; Feghali-Bostwick, C.A. Syndecan-2 is a novel target of insulin-like growth factor binding protein-3 and is over-expressed in fibrosis. PLoS ONE 2012, 7, e43049. [Google Scholar] [CrossRef]
- Tsoyi, K.; Chu, S.G.; Patino-Jaramillo, N.G.; Wilder, J.; Villalba, J.; Doyle-Eisele, M.; McDonald, J.; Liu, X.; El-Chemaly, S.; Perrella, M.A.; et al. Syndecan-2 Attenuates Radiation-induced Pulmonary Fibrosis and Inhibits Fibroblast Activation by Regulating PI3K/Akt/ROCK Pathway via CD148. Am. J. Respir. Cell Mol. Biol. 2018, 58, 208–215. [Google Scholar] [CrossRef]
- Scarpellini, A.; Huang, L.; Burhan, I.; Schroeder, N.; Funck, M.; Johnson, T.S.; Verderio, E.A. Syndecan-4 knockout leads to reduced extracellular transglutaminase-2 and protects against tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2014, 25, 1013–1027. [Google Scholar] [CrossRef]
- Alhasan, A.A.; Spielhofer, J.; Kusche-Gullberg, M.; Kirby, J.A.; Ali, S. Role of 6-O-sulfated heparan sulfate in chronic renal fibrosis. J. Biol. Chem. 2014, 289, 20295–20306. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Sanderson, R.D. Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J. Cell. Mol. Med. 2011, 15, 1013–1031. [Google Scholar] [CrossRef] [Green Version]
- Nagarajan, A.; Malvi, P.; Wajapeyee, N. Heparan Sulfate and Heparan Sulfate Proteoglycans in Cancer Initiation and Progression. Front. Endocrinol. (Lausanne) 2018, 9, 483. [Google Scholar] [CrossRef]
- Baburajeev, C.P.; Mohan, C.D.; Rangappa, S.; Mason, D.J.; Fuchs, J.E.; Bender, A.; Barash, U.; Vlodavsky, I.; Basappa; Rangappa, K.S. Identification of Novel Class of Triazolo-Thiadiazoles as Potent Inhibitors of Human Heparanase and their Anticancer Activity. BMC Cancer 2017, 17, 235. [Google Scholar] [CrossRef]
- Yang, Y.; Macleod, V.; Miao, H.Q.; Theus, A.; Zhan, F.; Shaughnessy, J.D., Jr.; Sawyer, J.; Li, J.P.; Zcharia, E.; Vlodavsky, I.; et al. Heparanase enhances syndecan-1 shedding: A novel mechanism for stimulation of tumor growth and metastasis. J. Biol. Chem. 2007, 282, 13326–13333. [Google Scholar] [CrossRef]
- Knelson, E.H.; Gaviglio, A.L.; Nee, J.C.; Starr, M.D.; Nixon, A.B.; Marcus, S.G.; Blobe, G.C. Stromal heparan sulfate differentiates neuroblasts to suppress neuroblastoma growth. J. Clin. Investig. 2014, 124, 3016–3031. [Google Scholar] [CrossRef]
- Bloushtain, N.; Qimron, U.; Bar-Ilan, A.; Hershkovitz, O.; Gazit, R.; Fima, E.; Korc, M.; Vlodavsky, I.; Bovin, N.V.; Porgador, A. Membrane-associated heparan sulfate proteoglycans are involved in the recognition of cellular targets by NKp30 and NKp46. J. Immunol. 2004, 173, 2392–2401. [Google Scholar] [CrossRef]
- Brennan, T.V.; Lin, L.; Brandstadter, J.D.; Rendell, V.R.; Dredge, K.; Huang, X.; Yang, Y. Heparan sulfate mimetic PG545-mediated antilymphoma effects require TLR9-dependent NK cell activation. J. Clin. Investig. 2016, 126, 207–219. [Google Scholar] [CrossRef]
- Douglass, S.; Goyal, A.; Iozzo, R.V. The role of perlecan and endorepellin in the control of tumor angiogenesis and endothelial cell autophagy. Connect. Tissue Res. 2015, 56, 381–391. [Google Scholar] [CrossRef] [Green Version]
- Folkman, J. Antiangiogenesis in cancer therapy—Endostatin and its mechanisms of action. Exp. Cell Res. 2006, 312, 594–607. [Google Scholar] [CrossRef]
- Gubbiotti, M.A.; Neill, T.; Iozzo, R.V. A current view of perlecan in physiology and pathology: A mosaic of functions. Matrix Biol. 2017, 57–58, 285–298. [Google Scholar] [CrossRef]
- Maiza, A.; Chantepie, S.; Vera, C.; Fifre, A.; Huynh, M.B.; Stettler, O.; Ouidja, M.O.; Papy-Garcia, D. The role of heparan sulfates in protein aggregation and their potential impact on neurodegeneration. FEBS Lett. 2018, 592, 3806–3818. [Google Scholar] [CrossRef]
- Signorini, M.; Bortolotti, F.; Poltronieri, L.; Bergamini, C.M. Human erythrocyte transglutaminase: Purification and preliminary characterisation. Biol. Chem. Hoppe Seyler 1988, 369, 275–281. [Google Scholar] [CrossRef]
- Scarpellini, A.; Germack, R.; Lortat-Jacob, H.; Muramatsu, T.; Billett, E.; Johnson, T.; Verderio, E.A. Heparan sulfate proteoglycans are receptors for the cell-surface trafficking and biological activity of transglutaminase-2. J. Biol. Chem. 2009, 284, 18411–18423. [Google Scholar] [CrossRef]
- Verderio, E.A.; Scarpellini, A.; Johnson, T.S. Novel interactions of TG2 with heparan sulfate proteoglycans: Reflection on physiological implications. Amino Acids 2009, 36, 671–677. [Google Scholar] [CrossRef]
- Schuksz, M.; Fuster, M.M.; Brown, J.R.; Crawford, B.E.; Ditto, D.P.; Lawrence, R.; Glass, C.A.; Wang, L.; Tor, Y.; Esko, J.D. Surfen, a small molecule antagonist of heparan sulfate. Proc. Natl. Acad. Sci. USA 2008, 105, 13075–13080. [Google Scholar] [CrossRef] [Green Version]
- Dierker, T.; Dreier, R.; Migone, M.; Hamer, S.; Grobe, K. Heparan sulfate and transglutaminase activity are required for the formation of covalently cross-linked hedgehog oligomers. J. Biol. Chem. 2009, 284, 32562–32571. [Google Scholar] [CrossRef]
- Ebert, A.D.; Laussmann, M.; Wegehingel, S.; Kaderali, L.; Erfle, H.; Reichert, J.; Lechner, J.; Beer, H.D.; Pepperkok, R.; Nickel, W. Tec-kinase-mediated phosphorylation of fibroblast growth factor 2 is essential for unconventional secretion. Traffic 2010, 11, 813–826. [Google Scholar] [CrossRef]
- La Venuta, G.; Wegehingel, S.; Sehr, P.; Muller, H.M.; Dimou, E.; Steringer, J.P.; Grotwinkel, M.; Hentze, N.; Mayer, M.P.; Will, D.W.; et al. Small Molecule Inhibitors Targeting Tec Kinase Block Unconventional Secretion of Fibroblast Growth Factor 2. J. Biol. Chem. 2016, 291, 17787–17803. [Google Scholar] [CrossRef]
- Nickel, W. Unconventional secretion: An extracellular trap for export of fibroblast growth factor 2. J. Cell Sci. 2007, 120, 2295–2299. [Google Scholar] [CrossRef]
- Nickel, W. The unconventional secretory machinery of fibroblast growth factor 2. Traffic 2011, 12, 799–805. [Google Scholar] [CrossRef]
- Steringer, J.P.; Bleicken, S.; Andreas, H.; Zacherl, S.; Laussmann, M.; Temmerman, K.; Contreras, F.X.; Bharat, T.A.; Lechner, J.; Muller, H.M.; et al. Phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2)-dependent oligomerization of fibroblast growth factor 2 (FGF2) triggers the formation of a lipidic membrane pore implicated in unconventional secretion. J. Biol. Chem. 2012, 287, 27659–27669. [Google Scholar] [CrossRef]
- Zehe, C.; Engling, A.; Wegehingel, S.; Schafer, T.; Nickel, W. Cell-surface heparan sulfate proteoglycans are essential components of the unconventional export machinery of FGF-2. Proc. Natl. Acad. Sci. USA 2006, 103, 15479–15484. [Google Scholar] [CrossRef] [Green Version]
- Skogberg, G.; Gudmundsdottir, J.; van der Post, S.; Sandstrom, K.; Bruhn, S.; Benson, M.; Mincheva-Nilsson, L.; Baranov, V.; Telemo, E.; Ekwall, O. Characterization of human thymic exosomes. PLoS ONE 2013, 8, e67554. [Google Scholar] [CrossRef]
- Piacentini, M.; D’Eletto, M.; Farrace, M.G.; Rodolfo, C.; Del Nonno, F.; Ippolito, G.; Falasca, L. Characterization of distinct sub-cellular location of transglutaminase type II: Changes in intracellular distribution in physiological and pathological states. Cell Tissue Res. 2014, 358, 793–805. [Google Scholar] [CrossRef]
- Diaz-Hidalgo, L.; Altuntas, S.; Rossin, F.; D’Eletto, M.; Marsella, C.; Farrace, M.G.; Falasca, L.; Antonioli, M.; Fimia, G.M.; Piacentini, M. Transglutaminase type 2-dependent selective recruitment of proteins into exosomes under stressful cellular conditions. Biochim. Biophys. Acta 2016, 1863, 2084–2092. [Google Scholar] [CrossRef]
- Santhanam, L.; Berkowitz, D.E.; Belkin, A.M. Nitric oxide regulates non-classical secretion of tissue transglutaminase. Commun. Integr. Biol. 2011, 4, 584–586. [Google Scholar] [CrossRef]
- Zemskov, E.A.; Mikhailenko, I.; Hsia, R.C.; Zaritskaya, L.; Belkin, A.M. Unconventional secretion of tissue transglutaminase involves phospholipid-dependent delivery into recycling endosomes. PLoS ONE 2011, 6, e19414. [Google Scholar] [CrossRef]
- Jandu, S.K.; Webb, A.K.; Pak, A.; Sevinc, B.; Nyhan, D.; Belkin, A.M.; Flavahan, N.A.; Berkowitz, D.E.; Santhanam, L. Nitric oxide regulates tissue transglutaminase localization and function in the vasculature. Amino Acids 2013, 44, 261–269. [Google Scholar] [CrossRef]
- Beckouche, N.; Bignon, M.; Lelarge, V.; Mathivet, T.; Pichol-Thievend, C.; Berndt, S.; Hardouin, J.; Garand, M.; Ardidie-Robouant, C.; Barret, A.; et al. The interaction of heparan sulfate proteoglycans with endothelial transglutaminase-2 limits VEGF165-induced angiogenesis. Sci. Signal 2015, 8, ra70. [Google Scholar] [CrossRef]
- Teesalu, K.; Panarina, M.; Uibo, O.; Uibo, R.; Utt, M. Autoantibodies from patients with celiac disease inhibit transglutaminase 2 binding to heparin/heparan sulfate and interfere with intestinal epithelial cell adhesion. Amino Acids 2012, 42, 1055–1064. [Google Scholar] [CrossRef]
- Teesalu, K.; Uibo, O.; Uibo, R.; Utt, M. Kinetic and functional characterisation of the heparin-binding peptides from human transglutaminase 2. J. Pept. Sci. 2012, 18, 350–356. [Google Scholar] [CrossRef]
- Wang, Z.; Collighan, R.J.; Pytel, K.; Rathbone, D.L.; Li, X.; Griffin, M. Characterization of heparin-binding site of tissue transglutaminase: Its importance in cell surface targeting, matrix deposition, and cell signaling. J. Biol. Chem. 2012, 287, 13063–13083. [Google Scholar] [CrossRef]
- Gambetti, S.; Dondi, A.; Cervellati, C.; Squerzanti, M.; Pansini, F.S.; Bergamini, C.M. Interaction with heparin protects tissue transglutaminase against inactivation by heating and by proteolysis. Biochimie 2005, 87, 551–555. [Google Scholar] [CrossRef]
- Nadella, V.; Wang, Z.; Johnson, T.S.; Griffin, M.; Devitt, A. Transglutaminase 2 interacts with syndecan-4 and CD44 at the surface of human macrophages to promote removal of apoptotic cells. Biochim. Biophys. Acta 2015, 1853, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Badarau, E.; Wang, Z.; Rathbone, D.L.; Costanzi, A.; Thibault, T.; Murdoch, C.E.; El Alaoui, S.; Bartkeviciute, M.; Griffin, M. Development of Potent and Selective Tissue Transglutaminase Inhibitors: Their Effect on TG2 Function and Application in Pathological Conditions. Chem. Biol. 2015, 22, 1347–1361. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Griffin, M. The role of TG2 in regulating S100A4-mediated mammary tumour cell migration. PLoS ONE 2013, 8, e57017. [Google Scholar] [CrossRef]
- Szondy, Z.; Sarang, Z.; Molnar, P.; Nemeth, T.; Piacentini, M.; Mastroberardino, P.G.; Falasca, L.; Aeschlimann, D.; Kovacs, J.; Kiss, I.; et al. Transglutaminase 2−/− mice reveal a phagocytosis-associated crosstalk between macrophages and apoptotic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 7812–7817. [Google Scholar] [CrossRef]
- Wipff, P.J.; Hinz, B. Integrins and the activation of latent transforming growth factor β1—An intimate relationship. Eur. J. Cell Biol. 2008, 87, 601–615. [Google Scholar] [CrossRef]
- Worthington, J.J.; Klementowicz, J.E.; Travis, M.A. TGFβ: A sleeping giant awoken by integrins. Trends Biochem. Sci. 2011, 36, 47–54. [Google Scholar] [CrossRef]
- Hinz, B. The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef]
- Badarau, E.; Mongeot, A.; Collighan, R.; Rathbone, D.; Griffin, M. Imidazolium-based warheads strongly influence activity of water-soluble peptidic transglutaminase inhibitors. Eur. J. Med. Chem. 2013, 66, 526–530. [Google Scholar] [CrossRef]
- Erdem, M.; Erdem, S.; Sanli, O.; Sak, H.; Kilicaslan, I.; Sahin, F.; Telci, D. Up-regulation of TGM2 with ITGB1 and SDC4 is important in the development and metastasis of renal cell carcinoma. Urol. Oncol. 2014, 32, 25.e13–25.e20. [Google Scholar] [CrossRef]
- Cardin, A.D.; Weintraub, H.J. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 1989, 9, 21–32. [Google Scholar] [CrossRef]
- Kreuger, J.; Spillmann, D.; Li, J.P.; Lindahl, U. Interactions between heparan sulfate and proteins: The concept of specificity. J. Cell Biol. 2006, 174, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Pinkas, D.M.; Strop, P.; Brunger, A.T.; Khosla, C. Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 2007, 5, e327. [Google Scholar] [CrossRef] [PubMed]
- Jang, T.H.; Lee, D.S.; Choi, K.; Jeong, E.M.; Kim, I.G.; Kim, Y.W.; Chun, J.N.; Jeon, J.H.; Park, H.H. Crystal structure of transglutaminase 2 with GTP complex and amino acid sequence evidence of evolution of GTP binding site. PLoS ONE 2014, 9, e107005. [Google Scholar] [CrossRef] [PubMed]
- Phatak, V.M.; Croft, S.M.; Rameshaiah Setty, S.G.; Scarpellini, A.; Hughes, D.C.; Rees, R.; McArdle, S.; Verderio, E.A. Expression of transglutaminase-2 isoforms in normal human tissues and cancer cell lines: Dysregulation of alternative splicing in cancer. Amino Acids 2013, 44, 33–44. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Evidence of TG2-HSPGs Interaction | References |
|---|---|
| TG2-HS binding studies | [10,63,161,162,178,179,180,181,182] |
| Mapping of TG2-heparin binding site | [10,180,181] |
| Co-precipitation of TG2 and Sdc4 | [9,33,34,35,63,162,183,184] |
| Co-localisation of TG2 and HS/Sdc4 | [63,148,162] |
| Interaction between HS and TG2-FN heterocomplex | [10,32,33,34,35,180,181,185] |
| Studies of TG2 in Sdc4 knockout models | [9,33,148,162] |
| Approach | Proposed Heparin Binding Site(s) of TG2 | Reference |
|---|---|---|
| Surface plasmon resonance (SPR) to test heparin-affinity of TG2 peptides | LRRWKNHGCQRVKY 261-274 (peptide P2) KFLKNAGRDCSRRS 202-215 (peptide P1) | [180] |
| Heparin sepharose column to test the affinity of cell lysates of HEK293/T17 cells transfected with human TG2 mutant cDNAs | NPKFLKNAGRDCSRRSS 200-216 (peptide P1) | [181] |
| Surface plasmon resonance (SPR) to test heparin-affinity of recombinant human TG2 mutants | RRWK 262-265 (mutant M1) KQKRK 598-602 (mutant M3) R19 (mutant M4), R28 (mutant M5) and K634 (mutant M7) | [10] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furini, G.; Verderio, E.A.M. Spotlight on the Transglutaminase 2-Heparan Sulfate Interaction. Med. Sci. 2019, 7, 5. https://doi.org/10.3390/medsci7010005
Furini G, Verderio EAM. Spotlight on the Transglutaminase 2-Heparan Sulfate Interaction. Medical Sciences. 2019; 7(1):5. https://doi.org/10.3390/medsci7010005
Chicago/Turabian StyleFurini, Giulia, and Elisabetta A.M. Verderio. 2019. "Spotlight on the Transglutaminase 2-Heparan Sulfate Interaction" Medical Sciences 7, no. 1: 5. https://doi.org/10.3390/medsci7010005