

Novel Therapies in Glioblastoma Treatment: Review of Glioblastoma; Current Treatment Options; and Novel Oncolytic Viral Therapies

Abstract
:1. Introduction
2. Overview Glioblastoma
2.1. Introduction of Glioblastoma
2.2. Molecular Description
2.3. Risk Factors
2.4. Clinical Presentation and Imaging
2.5. Current Treatment Options
2.6. Role of Immunosuppressive Mechanism in Glioblastoma and Resistance to Immunotherapy
3. Novel Oncolytic Viral Therapy for Treatment of Glioblastoma
3.1. DNA Viruses
3.1.1. Herpes Simplex Virus Type I
3.1.2. Adenovirus
3.1.3. Parvoviruses
3.1.4. Myxoma Virus
3.1.5. Vaccinia Virus (VV)
3.2. RNA Viruses
3.2.1. Measles Virus
3.2.2. Vesicular Stomatitis Virus (VSV)
3.2.3. Reoviruses
3.2.4. Newcastle Disease Virus (NDV)
3.2.5. Seneca Valley Virus Isolate 001 (SVV-001)
3.2.6. Polioviruses
3.2.7. Sindbis Virus
4. Discussion
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Berger, T.R.; Wen, P.Y.; Lang-Orsini, M.; Chukwueke, U.N. World Health Organization 2021 Classification of Central Nervous System Tumors and Implications for Therapy for Adult-Type Gliomas: A Review. JAMA Oncol. 2022, 8, 1493–1501. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.E.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. Dose-dense temozolomide for newly diagnosed glioblastoma: A randomized phase III clinical trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 4085–4091. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA A Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20 (Suppl. S5), S2–S8. [Google Scholar] [CrossRef]
- Fisher, J.P.; Adamson, D.C. Current FDA-Approved Therapies for High-Grade Malignant Gliomas. Biomedicines 2021, 9, 324. [Google Scholar] [CrossRef]
- Alexander, B.M.; Cloughesy, T.F. Adult Glioblastoma. J. Clin. Oncol. 2017, 35, 2402–2409. [Google Scholar] [CrossRef]
- Moore, S.C.; Rajaraman, P.; Dubrow, R.; Darefsky, A.S.; Koebnick, C.; Hollenbeck, A.; Schatzkin, A.; Leitzmann, M.F. Height, Body Mass Index, and Physical Activity in Relation to Glioma Risk. Cancer Res. 2009, 69, 8349–8355. [Google Scholar] [CrossRef]
- Joseph, G.P.; McDermott, R.; Baryshnikova, M.A.; Cobbs, C.S.; Ulasov, I.V. Cytomegalovirus as an Oncomodulatory Agent in the Progression of Glioma. Cancer Lett. 2017, 384, 79–85. [Google Scholar] [CrossRef]
- Rice, T.; Lachance, D.H.; Molinaro, A.M.; Eckel-Passow, J.E.; Walsh, K.M.; Barnholtz-Sloan, J.; Ostrom, Q.T.; Francis, S.S.; Wiemels, J.; Jenkins, R.B.; et al. Understanding Inherited Genetic Risk of Adult Glioma—A Review. Neuro-Oncol. Pract. 2016, 3, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Razavi, S.M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune Evasion Strategies of Glioblastoma. Front. Surg. 2016, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Dutoit, V.; Migliorini, D.; Dietrich, P.Y.; Walker, P.R. Immunotherapy of Malignant Tumors in the Brain: How Different from Other Sites? Front. Oncol. 2016, 6, 256. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of Gene Expression and Chemoresistance of CD133+ Cancer Stem Cells in Glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef]
- Harder, B.G.; Blomquist, M.R.; Wang, J.; Kim, A.J.; Woodworth, G.F.; Winkles, J.A.; Loftus, J.C.; Tran, N.L. Developments in Blood-Brain Barrier Penetrance and Drug Repurposing for Improved Treatment of Glioblastoma. Front. Oncol. 2018, 8, 462. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Hodges, T.R.; Ott, M.; Xiu, J.; Gatalica, Z.; Swensen, J.; Zhou, S.; Huse, J.T.; de Groot, J.; Li, S.; Overwijk, W.W.; et al. Mutational Burden, Immune Checkpoint Expression, and Mismatch Repair in Glioma: Implications for Immune Checkpoint Immunotherapy. Neuro-Oncology 2017, 19, 1047–1057. [Google Scholar] [CrossRef]
- McLendon, R.; Friedman, A.; Bigner, D.; van Meir, E.G.; Brat, D.J.; Mastrogianakis, G.M.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; et al. Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Terrível, M.; Gromicho, C.; Matos, A.M. Oncolytic Viruses: What to Expect from Their Use in Cancer Treatment. Microbiol. Immunol. 2020, 64, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Alifieris, C.; Trafalis, D.T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol. Ther. 2015, 152, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.A.; Karajannis, M.A.; Harter, D.H. Glioblastoma multiforme: State of the art and future therapeutics. Surg. Neurol. Int. 2014, 5, 64. [Google Scholar] [CrossRef]
- Young, R.M.; Jamshidi, A.; Davis, G.; Sherman, J.H. Current trends in the surgical management and treatment of adult glioblastoma. Ann. Transl. Med. 2015, 3, 121. [Google Scholar] [CrossRef]
- Moton, S.; Elbanan, M.; Zinn, P.O.; Colen, R.R. Imaging Genomics of Glioblastoma: Biology, Biomarkers, and Breakthroughs. Top. Magn. Reson. Imaging TMRI 2015, 24, 155–163. [Google Scholar] [CrossRef]
- Cieslik, M.; Chinnaiyan, A.M. Cancer transcriptome profiling at the juncture of clinical translation. Nat. Rev. Genet. 2018, 19, 93–109. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; De Carvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 2019, 178, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Hu, Y.; Wu, F.; Guo, Q.; Qian, Z.; Hu, W.; Chen, J.; Wang, K.; Fan, X.; Wu, X.; et al. Surveying brain tumor heterogeneity by single-cell RNA-sequencing of multi-sector biopsies. Natl. Sci. Rev. 2020, 7, 1306–1318. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Levitin, H.M.; Frattini, V.; Bush, E.C.; Boyett, D.M.; Samanamud, J.; Ceccarelli, M.; Dovas, A.; Zanazzi, G.; Canoll, P.; et al. Single-cell transcriptome analysis of lineage diversity in high-grade glioma. Genome Med. 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kon, M.; De Lisi, C. Pathway-based classification of cancer subtypes. Biol. Direct 2012, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Ellor, S.V.; Pagano-Young, T.A.; Avgeropoulos, N.G. Glioblastoma: Background, standard treatment paradigms, and supportive care considerations. J. Law Med. Ethics A J. Am. Soc. Law Med. Ethics 2014, 42, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.R.; Fogh, S.E.; Giannini, C.; Kaufmann, T.J.; Raghunathan, A.; Theodosopoulos, P.V.; Clarke, J.L. Case-Based Review: Newly diagnosed glioblastoma. Neuro-Oncol. Pract. 2015, 2, 106–121. [Google Scholar] [CrossRef]
- Venkataramani, V.; Yang, Y.; Schubert, M.C.; Reyhan, E.; Tetzlaff, S.K.; Wißmann, N.; Botz, M.; Soyka, S.J.; Beretta, C.A.; Pramatarov, R.L.; et al. Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell 2022, 185, 2899–2917.e31. [Google Scholar] [CrossRef]
- Moraes, F.Y.; Lo, A.; Morgan, E.R.; Millar, B.A.; Shultz, D.B.; Maurice, C.; Harlos, C.; Kongkham, P.; Bernstein, M.; Zadeh, G.; et al. Management and Outcomes in the Oldest-Old Population with Glioblastoma. Can. J. Neurol. Sci. J. Can. Des Sci. Neurol. 2018, 45, 199–205. [Google Scholar] [CrossRef]
- Lobera, A. Imaging in Glioblastoma Multiforme. 2015. Available online: http://emedicine.medscape.com/article/340870-overview (accessed on 14 June 2022).
- Halani, S.H.; Babu, R.; Adamson, D.C. Management of Glioblastoma Multiforme in Elderly Patients: A Review of the Literature. World Neurosurg. 2017, 105, 53–62. [Google Scholar] [CrossRef]
- Perry, J.; Zinman, L.; Chambers, A.; Spithoff, K.; Lloyd, N.; Laperriere, N.; Neuro-oncology Disease Site Group; Cancer Care Ontario’s Program in Evidence-Based Care. The use of prophylactic anticonvulsants in patients with brain tumours—A systematic review. Curr. Oncol. 2006, 13, 222–229. [Google Scholar] [CrossRef]
- Schiff, D.; Lee, E.Q.; Nayak, L.; Norden, A.D.; Reardon, D.A.; Wen, P.Y. Medical management of brain tumors and the sequelae of treatment. Neuro-Oncology 2015, 17, 488–504. [Google Scholar] [CrossRef] [PubMed]
- Glantz, M.J.; Cole, B.F.; Forsyth, P.A.; Recht, L.D.; Wen, P.Y.; Chamberlain, M.C.; Grossman, S.A.; Cairncross, J.G. Practice parameter: Anticonvulsant prophylaxis in patients with newly diagnosed brain tumors. Neurology 2000, 54, 1886–1893. [Google Scholar] [CrossRef] [PubMed]
- Blissitt, P.A.; American Association of Neuroscience Nurses. Clinical practice guideline series update: Care of the adult patient with a brain tumor. J. Neurosci. Nurs. J. Am. Assoc. Neurosci. Nurses 2014, 46, 367–368. [Google Scholar] [CrossRef] [PubMed]
- Nabors, L.B.; Portnow, J.; Ammirati, M.; Baehring, J.; Brem, H.; Brown, P.; Butowski, N.; Chamberlain, M.C.; Fenstermaker, R.A.; Friedman, A.; et al. Central Nervous System Cancers, Version 1.2015. J. Natl. Compr. CancerNetw. JNCCN 2015, 13, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Nowak, B.; Rogujski, P.; Janowski, M.; Lukomska, B.; Andrzejewska, A. Mesenchymal stem cells in glioblastoma therapy and progression: How one cell does it all. Biochim. Et Biophys. Acta. Rev. Cancer 2021, 1876, 188582. [Google Scholar] [CrossRef]
- Kuhnt, D.; Becker, A.; Ganslandt, O.; Bauer, M.; Buchfelder, M.; Nimsky, C. Correlation of the extent of tumor volume resection and patient survival in surgery of glioblastoma multiforme with high-field intraoperative MRI guidance. Neuro-Oncology 2011, 13, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Roder, C.; Bisdas, S.; Ebner, F.H.; Honegger, J.; Naegele, T.; Ernemann, U.; Tatagiba, M. Maximizing the extent of resection and survival benefit of patients in glioblastoma surgery: High-field iMRI versus conventional and 5-ALA-assisted surgery. Eur. J. Surg. Oncol. J. Eur. Soc. Surg. Oncol. Br. Assoc. Surg. Oncol. 2014, 40, 297–304. [Google Scholar] [CrossRef]
- Keles, G.E.; Anderson, B.; Berger, M.S. The effect of extent of resection on time to tumor progression and survival in patients with glioblastoma multiforme of the cerebral hemisphere. Surg. Neurol. 1999, 52, 371–379. [Google Scholar] [CrossRef]
- Lacroix, M.; Abi-Said, D.; Fourney, D.R.; Gokaslan, Z.L.; Shi, W.; DeMonte, F.; Lang, F.F.; McCutcheon, I.E.; Hassenbusch, S.J.; Holland, E.; et al. A multivariate analysis of 416 patients with glioblastoma multiforme: Prognosis, extent of resection, and survival. J. Neurosurg. 2001, 95, 190–198. [Google Scholar] [CrossRef]
- Mukherjee, D.; Quiñones-Hinojosa, A. Impact of extent of resection on outcomes in patients with high-grade gliomas. In Tumors of the Central Nervous System; Hayat, M.A., Ed.; Springer: San Francisco, CA, USA, 2011; Volume 2, pp. 173–179. [Google Scholar]
- Stummer, W.; Pichlmeier, U.; Meinel, T.; Wiestler, O.D.; Zanella, F.; Reulen, H.J.; ALA-Glioma Study Group. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: A randomised controlled multicentre phase III trial. Lancet Oncol. 2006, 7, 392–401. [Google Scholar] [CrossRef]
- Wollmann, G.; Ozduman, K.; van den Pol, A.N. Oncolytic virus therapy for glioblastoma multiforme: Concepts and candidates. Cancer J. 2012, 18, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Raja, J.; Ludwig, J.M.; Gettinger, S.N.; Schalper, K.A.; Kim, H.S. Oncolytic virus immunotherapy: Future prospects for oncology. J. Immunother. Cancer 2018, 6, 140. [Google Scholar] [CrossRef] [PubMed]
- Lakomy, R.; Kazda, T.; Selingerova, I.; Poprach, A.; Pospisil, P.; Belanova, R.; Fadrus, P.; Vybihal, V.; Smrcka, M.; Jancalek, R.; et al. Real-World Evidence in Glioblastoma: Stupp’s Regimen After a Decade. Front. Oncol. 2020, 10, 840. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Cloughesy, T.; Perry, J.R.; Wick, W. Standards of care for treatment of recurrent glioblastoma—Are we there yet? Neuro-Oncology 2013, 15, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Gramatzki, D.; Dehler, S.; Rushing, E.J.; Zaugg, K.; Hofer, S.; Yonekawa, Y.; Bertalanffy, H.; Valavanis, A.; Korol, D.; Rohrmann, S.; et al. Glioblastoma in the canton of Zurich, Switzerland revisited, 2005 to 2009. Cancer 2016, 122, 2206–2215. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.M.; Kochel, C.M.; Nirschl, C.J.; Durham, N.M.; Ruzevick, J.; Alme, A.; Francica, B.J.; Elias, J.; Daniels, A.; Dubensky, T.W.; et al. Systemic tolerance mediated by melanoma brain tumors is reversible by radiotherapy and vaccination. Clin. Cancer Res. 2016, 22, 1161–1172. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Jähnisch, H.; Füssel, S.; Kiessling, A.; Wehner, R.; Zastrow, S.; Bachmann, M.; Rieber, E.P.; Wirth, M.P.; Schmitz, M. Dendritic cell-based immunotherapy for prostate cancer. Clin. Dev. Immunol. 2010, 2010, 517493. [Google Scholar] [CrossRef]
- Aurelian, L. Oncolytic viruses as immunotherapy, Progress and remaining challenges. OncoTargets Ther. 2016, 9, 2627–2637. [Google Scholar] [CrossRef]
- Jena, B.; Dotti, G.; Cooper, L.J. Redirecting t-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood 2010, 116, 1035–1044. [Google Scholar] [CrossRef]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.; et al. Tandem CAR T cells targeting HER2 and IL13R_2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [PubMed]
- Dapash, M.; Hou, D.; Castro, B.; Lee-Chang, C.; Lesniak, M.S. The Interplay between Glioblastoma and Its Microenvironment. Cells 2021, 10, 2257. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yang, S.; Zhang, F.; Cheng, F.; Wang, X.; Rao, J. Influence of the Hippo-YAP signaling pathway on tumor associated macrophages (TAMs) and its implications on cancer immunosuppressive microenvironment. Ann. Transl. Med. 2020, 8, 399. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, D.L.; Liu, Z.; Sathaiah, M.; Ravindranathan, R.; Guo, Z.; He, Y.; Guo, Z.S. Oncolytic viruses as therapeutic cancer vaccines. Mol. Cancer 2013, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Gujar, S.; Bell, J.; Diallo, J.S. SnapShot: Cancer Immunotherapy with Oncolytic Viruses. Cell 2019, 176, 1240–1240.e1. [Google Scholar] [CrossRef] [PubMed]
- Angom, R.S.; Nakka, N.M.R.; Bhattacharya, S. Advances in Glioblastoma Therapy: An Update on Current Approaches. Brain Sci. 2023, 13, 1536. [Google Scholar] [CrossRef] [PubMed]
- Asija, S.; Chatterjee, A.; Yadav, S.; Chekuri, G.; Karulkar, A.; Jaiswal, A.K.; Goda, J.S.; Purwar, R. Combinatorial approaches to effective therapy in glioblastoma (GBM): Current status and what the future holds. Int. Rev. Immunol. 2022, 41, 582–605. [Google Scholar] [CrossRef] [PubMed]
- Tahir, M.; Ahmad, N.; Lei, D.; Ali, S. Emerging role of oncolytic viruses and stem cells in gene therapy: Should they be integrated? Drug Discov. Today 2022, 27, 2244–2251. [Google Scholar] [CrossRef]
- Webb, M.J.; Sener, U.; Vile, R.G. Current Status and Challenges of Oncolytic Virotherapy for the Treatment of Glioblastoma. Pharmaceuticals 2023, 16, 793. [Google Scholar] [CrossRef]
- Huang, B.; Li, X.; Li, Y.; Zhang, J.; Zong, Z.; Zhang, H. Current Immunotherapies for Glioblastoma Multiforme. Front. Immunol. 2021, 11, 603911. [Google Scholar] [CrossRef]
- Mahmoud, A.B.; Ajina, R.; Aref, S.; Darwish, M.; Alsayb, M.; Taher, M.; AlSharif, S.A.; Hashem, A.M.; Alkayyal, A.A. Advances in immunotherapy for glioblastoma multiforme. Front. Immunol. 2022, 13, 944452. [Google Scholar] [CrossRef] [PubMed]
- Mihelson, N.; McGavern, D.B. Viral Control of Glioblastoma. Viruses 2021, 13, 1264. [Google Scholar] [CrossRef] [PubMed]
- Shoaf, M.L.; Desjardins, A. Oncolytic Viral Therapy for Malignant Glioma and Their Application in Clinical Practice. Neurotherapeutics 2022, 19, 1818–1831. [Google Scholar] [CrossRef] [PubMed]
- Kamynina, M.; Tskhovrebova, S.; Fares, J.; Timashev, P.; Laevskaya, A.; Ulasov, I. Oncolytic Virus-Induced Autophagy in Glioblastoma. Cancers 2021, 13, 3482. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Long, X.; Liu, J.; Cheng, P. Glioblastoma microenvironment and its reprogramming by oncolytic virotherapy. Front. Cell. Neurosci. 2022, 16, 819363. [Google Scholar] [CrossRef] [PubMed]
- Haddad, A.F.; Young, J.S.; Mummaneni, N.V.; Kasahara, N.; Aghi, M.K. Immunologic aspects of viral therapy for glioblastoma and implications for interactions with immunotherapies. J. Neuro-Oncol. 2021, 152, 1–13. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Wang, Y.; Kong, Z.; Chen, W.; Li, J.; Chen, W.; Tong, Y.; Ma, W.; Wang, Y. Effects of oncolytic viruses and viral vectors on immunity in glioblastoma. Gene Ther. 2022, 29, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Xia, Q.; Muhammad, T.; Liu, L.; Meng, X.; Bars-Cortina, D.; Khan, A.A.; Huang, Y.; Dong, L. Glioblastoma Therapy: Rationale for a Mesenchymal Stem Cell-based Vehicle to Carry Recombinant Viruses. Stem Cell Rev. Rep. 2022, 18, 523–543. [Google Scholar] [CrossRef]
- Blitz, S.E.; Kappel, A.D.; Gessler, F.A.; Klinger, N.V.; Arnaout, O.; Lu, Y.; Peruzzi, P.P.; Smith, T.R.; Chiocca, E.A.; Friedman, G.K.; et al. Tumor-Associated Macrophages/Microglia in Glioblastoma Oncolytic Virotherapy: A Double-Edged Sword. Int. J. Mol. Sci. 2022, 23, 1808. [Google Scholar] [CrossRef]
- Zhou, C.; Chen, Q.; Chen, Y.; Qin, C.F. Oncolytic Zika Virus: New Option for Glioblastoma Treatment. DNA Cell Biol. 2023, 42, 267–273. [Google Scholar] [CrossRef]
- Marelli, G.; Howells, A.; Lemoine, N.R.; Wang, Y. Oncolytic Viral Therapy and the Immune System: A Double-Edged Sword Against Cancer. Front. Immunol. 2018, 9, 866. [Google Scholar] [CrossRef] [PubMed]
- Markert, J.M.; Medlock, M.D.; Rabkin, S.D.; Gillespie, G.Y.; Todo, T.; Hunter, W.D.; Palmer, C.A.; Feigenbaum, F.; Tornatore, C.; Tufaro, F.; et al. Conditionally Replicating Herpes Simplex Virus Mutant, G207 for the Treatment of Malignant Glioma: Results of a Phase I Trial. Gene Ther. 2000, 7, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Markert, J.M.; Liechty, P.G.; Wang, W.; Gaston, S.; Braz, E.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Lakeman, A.D.; Palmer, C.A.; et al. Phase Ib Trial of Mutant Herpes Simplex Virus G207 Inoculated Pre-and Post-Tumor Resection for Recurrent GBM. Mol. Ther. 2009, 17, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Markert, J.M.; Razdan, S.N.; Kuo, H.C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Agee, B.S.; Coleman, J.M.; et al. A Phase 1 Trial of Oncolytic HSV-1, G207, given in Combination with Radiation for Recurrent GBM Demonstrates Safety and Radiographic Responses. Mol. Ther. 2014, 22, 1048–1055. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Abbed, K.M.; Tatter, S.; Louis, D.N.; Hochberg, F.H.; Barker, F.; Kracher, J.; Grossman, S.A.; Fisher, J.D.; Carson, K.; et al. A Phase I Open-Label, Dose-Escalation, Multi-Institutional Trial of Injection with an E1B-Attenuated Adenovirus, ONYX-015, into the Peritumoral Region of Recurrent Malignant Gliomas, in the Adjuvant Setting. Mol. Ther. 2004, 10, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Kicielinski, K.P.; Chiocca, E.A.; Yu, J.S.; Gill, G.M.; Coffey, M.; Markert, J.M. Phase 1 Clinical Trial of Intratumoral Reovirus Infusion for the Treatment of Recurrent Malignant Gliomas in Adults. Mol. Ther. 2014, 22, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Paraskevakou, G.; Iankov, I.; Giannini, C.; Schroeder, M.; Sarkaria, J.; Puri, R.K.; Russell, S.J.; Galanis, E. Interleukin-13 Displaying Retargeted Oncolytic Measles Virus Strains Have Significant Activity Against Gliomas with Improved Specificity. Mol. Ther. 2008, 16, 1556–1564. [Google Scholar] [CrossRef]
- Allen, C.; Opyrchal, M.; Aderca, I.; Schroeder, M.A.; Sarkaria, J.N.; Domingo, E.; Federspiel, M.J.; Galanis, E. Oncolytic Measles Virus Strains Have Significant Antitumor Activity against Glioma Stem Cells. Gene Ther. 2013, 20, 444–449. [Google Scholar] [CrossRef]
- Freeman, A.I.; Zakay-Rones, Z.; Gomori, J.M.; Linetsky, E.; Rasooly, L.; Greenbaum, E.; Rozenman-Yair, S.; Panet, A.; Libson, E.; Irving, C.S.; et al. Phase I/II Trial of Intravenous NDV-HUJ Oncolytic Virus in Recurrent Glioblastoma Multiforme. Mol. Ther. 2006, 13, 221–228. [Google Scholar] [CrossRef]
- Gromeier, M.; Lachmann, S.; Rosenfeld, M.R.; Gutin, P.H.; Wimmer, E. Intergeneric Poliovirus Recombinants for the Treatment of Malignant Glioma. Proc. Natl. Acad. Sci. USA 2000, 97, 6803–6808. [Google Scholar] [CrossRef]
- Desjardins, A.; Gromeier, M.; Herndon, J.E., 2nd; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.M.; Sonabend, A.M.; Bruce, J.N. Convection-Enhanced Delivery. Neurotherapeutics 2017, 14, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kim, D.W.; Deraffele, G.; Mitcham, J.; Coffin, R.S.; Kim-Schulze, S. Local and Distant Immunity Induced by Intralesional Vaccination with an Oncolytic Herpes Virus Encoding GM-CSF in Patients with Stage IIIc and IV Melanoma. Ann. Surg. Oncol. 2010, 17, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Hsu, E.; Keene, D.; Ventureyra, E.; Matzinger, M.A.; Jimenez, C.; Wang, H.S.; Grimard, L. Bone Marrow Metastasis in Astrocytic Gliomata. J. Neuro-Oncol. 1998, 37, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Zadeh, G.; Lang, F.; Daras, M.; Cloughesy, T.; Colman, H.; Ong, S.; Ramakrishna, R.; Vogelbaum, M.; Groves, M.; Nassiri, F.; et al. ATIM-24. Interim results of a phase II multicenter study of the conditionally replicative oncolytic adenovirus DNX-2401 with pembrolizumab (keytruda) for recurrent glioblastoma; captive study (KEYNOTE-192). Neuro-Oncology 2018, 20, vi6. [Google Scholar] [CrossRef]
- Hamad, A.; Yusubalieva, G.M.; Baklaushev, V.P.; Chumakov, P.M.; Lipatova, A.V. Recent Developments in Glioblastoma Therapy: Oncolytic Viruses and Emerging Future Strategies. Viruses 2023, 15, 547. [Google Scholar] [CrossRef]
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef]
- Krummenacher, C.; Nicola, A.V.; Whitbeck, J.C.; Lou, H.; Hou, W.; Lambris, J.D.; Geraghty, R.J.; Spear, P.G.; Cohen, G.H.; Eisenberg, R.J. Herpes Simplex Virus Glycoprotein D Can Bind to Poliovirus Receptor-Related Protein 1 or Herpesvirus Entry Mediator, Two Structurally Unrelated Mediators of Virus Entry. J. Virol. 1998, 72, 7064–7074. [Google Scholar] [CrossRef]
- Bommareddy, P.K.; Patel, A.; Hossain, S.; Kaufman, H.L. Talimogene Laherparepvec (T-VEC) and Other Oncolytic Viruses for the Treatment of Melanoma. Am. J. Clin. Dermatol. 2017, 18, 1–15. [Google Scholar] [CrossRef]
- Rehman, H.; Silk, A.W.; Kane, M.P.; Kaufman, H.L. Into the Clinic: Talimogene Laherparepvec (T-VEC), a First-in-Class Intratumoral Oncolytic Viral Therapy. J. Immunother. Cancer 2016, 4, 53. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Alexander, D.; Tallóczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 Confers Neurovirulence by Targeting the Beclin 1 Autophagy Protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Holman, H.A.; MacLean, A.R. Neurovirulent Factor ICP34.5 Uniquely Expressed in the Herpes Simplex Virus Type 1 Δγ134.5 Mutant 1716. J. Neurovirol. 2008, 14, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Mineta, T.; Rabkin, S.D.; Yazaki, T.; Hunter, W.D.; Martuza, R.L. Attenuated Multi–Mutated Herpes Simplex Virus-1 for the Treatment of Malignant Gliomas. Nat. Med. 1995, 1, 938–943. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, S.; Fukuhara, H.; Todo, T. Oncolytic Virus Therapy in Japan: Progress in Clinical Trials and Future Perspectives. Jpn. J. Clin. Oncol. 2019, 49, 201–209. [Google Scholar] [CrossRef]
- Todo, T.; Martuza, R.L.; Rabkin, S.D.; Johnson, P.A. Oncolytic Herpes Simplex Virus Vector with Enhanced MHC Class I Presentation and Tumor Cell Killing. Proc. Natl. Acad. Sci. USA 2001, 98, 6396–6401. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral Oncolytic Herpes Virus G47∆ for Residual or Recurrent Glioblastoma: A Phase 2 Trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef]
- Todo, T.; Ino, Y.; Ohtsu, H.; Shibahara, J.; Tanaka, M. A Phase I/II Study of Triple-Mutated Oncolytic Herpes Virus G47∆ in Patients with Progressive Glioblastoma. Nat. Commun. 2022, 13, 4119. [Google Scholar] [CrossRef]
- Kambara, H.; Okano, H.; Chiocca, E.A.; Saeki, Y. An Oncolytic HSV-1 Mutant Expressing ICP34.5 under Control of a Nestin Promoter Increases Survival of Animals Even When Symptomatic from a Brain Tumor. Cancer Res. 2005, 65, 2832–2839. [Google Scholar] [CrossRef]
- Trinchieri, G. Interleukin-12 and the Regulation of Innate Resistance and Adaptive Immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef]
- Roth, J.C.; Cassady, K.A.; Cody, J.J.; Parker, J.N.; Price, K.H.; Coleman, J.M.; Peggins, J.O.; Noker, P.E.; Powers, N.W.; Grimes, S.D.; et al. Evaluation of the Safety and Biodistribution of M032, an Attenuated Herpes Simplex Virus Type 1 Expressing HIL-12, After Intracerebral Administration to Aotus Nonhuman Primates. Hum. Gene Ther. Clin. Dev. 2014, 25, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, H.; Nguyen, T.; Kasai, K.; Passaro, C.; Ito, H.; Goins, W.F.; Shaikh, I.; Erdelyi, R.; Nishihara, R.; Nakano, I.; et al. Toxicity and Efficacy of a Novel Gadd34-Expressing Oncolytic Hsv-1 for the Treatment of Experimental Glioblastoma. Clin. Cancer Res. 2018, 24, 2574–2584. [Google Scholar] [CrossRef] [PubMed]
- Studebaker, A.W.; Hutzen, B.J.; Pierson, C.R.; Haworth, K.B.; Cripe, T.P.; Jackson, E.M.; Leonard, J.R. Oncolytic Herpes Virus RRp450 Shows Efficacy in Orthotopic Xenograft Group 3/4 Medulloblastomas and Atypical Teratoid/Rhabdoid Tumors. Mol. Ther. Oncolytics 2017, 6, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Ogbomo, H.; Zemp, F.J.; Lun, X.; Zhang, J.; Stack, D.; Rahman, M.M.; Mcfadden, G.; Mody, C.H.; Forsyth, P.A. Myxoma Virus Infection Promotes NK Lysis of Malignant Gliomas In Vitro and In Vivo. PLoS ONE 2013, 8, e66825. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Ma, R.; Russell, L.; Yoo, J.Y.; Han, J.; Cui, H.; Yi, P.; Zhang, J.; Nakashima, H.; Dai, H.; et al. An Oncolytic Herpesvirus Expressing E-Cadherin Improves Survival in Mouse Models of Glioblastoma. Nat. Biotechnol. 2018, 37, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Sette, P.; Amankulor, N.; Li, A.; Marzulli, M.; Leronni, D.; Zhang, M.; Goins, W.F.; Kaur, B.; Bolyard, C.; Cripe, T.P.; et al. GBM-Targeted OHSV Armed with Matrix Metalloproteinase 9 Enhances Anti-Tumor Activity and Animal Survival. Mol. Ther. Oncolytics 2019, 15, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Mazzacurati, L.; Marzulli, M.; Reinhart, B.; Miyagawa, Y.; Uchida, H.; Goins, W.F.; Li, A.; Kaur, B.; Caligiuri, M.; Cripe, T.; et al. Use of MiRNA Response Sequences to Block Off-Target Replication and Increase the Safety of an Unattenuated, Glioblastoma-Targeted Oncolytic HSV. Mol. Ther. 2015, 23, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.Y.; Jeong, M.; Mushiake, H.; Kim, B.M.; Kim, W.B.; Ko, J.P.; Kim, M.H.; Kim, M.; Kim, T.H.; Robbins, P.D.; et al. Cancer Gene Therapy Using a Novel Secretable Trimeric TRAIL. Gene Ther. 2005, 13, 330–338. [Google Scholar] [CrossRef]
- Tamura, K.; Wakimoto, H.; Agarwal, A.S.; Rabkin, S.D.; Bhere, D.; Martuza, R.L.; Kuroda, T.; Kasmieh, R.; Shah, K. Multimechanistic Tumor Targeted Oncolytic Virus Overcomes Resistance in Brain Tumors. Mol. Ther. 2013, 21, 68–77. [Google Scholar] [CrossRef]
- Passaro, C.; Alayo, Q.; de Laura, I.; McNulty, J.; Grauwet, K.; Ito, H.; Bhaskaran, V.; Mineo, M.; Lawler, S.E.; Shah, K.; et al. Arming an Oncolytic Herpes Simplex Virus Type 1 with a Single-Chain Fragment Variable Antibody against PD-1 for Experimental Glioblastoma Therapy. Clin. Cancer Res. 2019, 25, 290–299. [Google Scholar] [CrossRef]
- Sharma, A.; Li, X.; Bangari, D.S.; Mittal, S.K. Adenovirus Receptors and Their Implications in Gene Delivery. Virus Res. 2009, 143, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Kiyokawa, J.; Wakimoto, H. Preclinical and Clinical Development of Oncolytic Adenovirus for the Treatment of Malignant Glioma. Oncolytic Virotherapy 2019, 8, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Aguilar, L.K.; Bell, S.D.; Kaur, B.; Hardcastle, J.; Cavaliere, R.; McGregor, J.; Lo, S.; Ray-Chaudhuri, A.; Chakravarti, A.; et al. Phase IB Study of Gene-Mediated Cytotoxic Immunotherapy Adjuvant to up-Front Surgery and Intensive Timing Radiation for Malignant Glioma. J. Clin. Oncol. 2011, 29, 3611–3619. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, L.A.; Manzanera, A.G.; Bell, S.D.; Cavaliere, R.; McGregor, J.M.; Grecula, J.C.; Newton, H.B.; Lo, S.S.; Badie, B.; Portnow, J.; et al. Phase II Multicenter Study of Gene-Mediated Cytotoxic Immunotherapy as Adjuvant to Surgical Resection for Newly Diagnosed Malignant Glioma. Neuro-Oncology 2016, 18, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Liang, M. Oncorine, the World First Oncolytic Virus Medicine and Its Update in China. Curr. Cancer Drug Targets 2018, 18, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. China Approves World’s First Oncolytic Virus Therapy for Cancer Treatment. J. Natl. Cancer Inst. 2006, 98, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Grand, R.J.A. Adenovirus E1B 55-Kilodalton Protein: Multiple Roles in Viral Infection and Cell Transformation. J. Virol. 2009, 83, 4000–4012. [Google Scholar] [CrossRef]
- Jiang, H.; Gomez-Manzano, C.; Aoki, H.; Alonso, M.M.; Kondo, S.; McCormick, F.; Xu, J.; Kondo, Y.; Bekele, B.N.; Colman, H.; et al. Examination of the Therapeutic Potential of Delta-24-RGD in Brain Tumor Stem Cells: Role of Autophagic Cell Death. JNCI J. Natl. Cancer Inst. 2007, 99, 1410–1414. [Google Scholar] [CrossRef]
- Avβ3 and Avβ5 Integrin Expression in Glioma Periphery: Neurosurgery. Available online: https://journals.lww.com/neurosurgery/Abstract/2001/08000/_v_3_and__v_5_Integrin_Expression_in_Glioma.22.aspx (accessed on 15 November 2022).
- Asaoka, K.; Tada, M.; Sawamura, Y.; Ikeda, J.; Abe, H. Dependence of Efficient Adenoviral Gene Delivery in Malignant Glioma Cells on the Expression Levels of the Coxsackievirus and Adenovirus Receptor. J. Neurosurg. 2000, 92, 1002–1008. [Google Scholar] [CrossRef]
- Martínez-Vélez, N.; Garcia-Moure, M.; Marigil, M.; González-Huarriz, M.; Puigdelloses, M.; Pérez-Larraya, J.G.; Zalacaín, M.; Marrodán, L.; Varela-Guruceaga, M.; Laspidea, V.; et al. The Oncolytic Virus Delta-24-RGD Elicits an Antitumor Effect in Pediatric Glioma and DIPG Mouse Models. Nat. Commun. 2019, 10, 2235. [Google Scholar] [CrossRef]
- Jiang, H.; Shin, D.H.; Nguyen, T.T.; Fueyo, J.; Fan, X.; Henry, V.; Carrillo, C.C.; Yi, Y.; Alonso, M.M.; Collier, T.L.; et al. Localized Treatment with Oncolytic Adenovirus Delta-24-RGDOX Induces Systemic Immunity against Disseminated Subcutaneous and Intracranial Melanomas. Clin. Cancer Res. 2019, 25, 6801–6814. [Google Scholar] [CrossRef] [PubMed]
- Puigdelloses, M.; Garcia-Moure, M.; Labiano, S.; Laspidea, V.; Gonzalez-Huarriz, M.; Zalacain, M.; Marrodan, L.; Martinez-Velez, N.; de La Nava, D.; Ausejo, I.; et al. CD137 and PD-L1 Targeting with Immunovirotherapy Induces a Potent and Durable Antitumor Immune Response in Glioblastoma Models. J. Immunother. Cancer 2021, 9, e002644. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Molina, Y.; Jiang, H.; Fueyo, J.; Nguyen, T.; Shin, D.H.; Youssef, G.; Fan, X.; Gumin, J.; Alonso, M.M.; Phadnis, S.; et al. GITRL-Armed Delta-24-RGD Oncolytic Adenovirus Prolongs Survival and Induces Anti-Glioma Immune Memory. Neurooncol. Adv. 2019, 1, vdz009. [Google Scholar] [CrossRef] [PubMed]
- Marchini, A.; Bonifati, S.; Scott, E.M.; Angelova, A.L.; Rommelaere, J. Oncolytic Parvoviruses: From Basic Virology to Clinical Applications. Virol. J. 2015, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Angelova, A.L.; Barf, M.; Geletneky, K.; Unterberg, A.; Rommelaere, J. Immunotherapeutic Potential of Oncolytic H-1 Parvovirus: Hints of Glioblastoma Microenvironment Conversion towards Immunogenicity. Viruses 2017, 9, 382. [Google Scholar] [CrossRef] [PubMed]
- di Piazza, M.; Mader, C.; Geletneky, K.; Herrero y Calle, M.; Weber, E.; Schlehofer, J.; Deleu, L.; Rommelaere, J. Cytosolic Activation of Cathepsins Mediates Parvovirus H-1-Induced Killing of Cisplatin and TRAIL-Resistant Glioma Cells. J. Virol. 2007, 81, 4186–4198. [Google Scholar] [CrossRef]
- Geletneky, K.; Kiprianova, I.; Ayache, A.; Koch, R.; Herrero, Y.; Calle, M.; Deleu, L.; Sommer, C.; Thomas, N.; Rommelaere, J.; et al. Regression of Advanced Rat and Human Gliomas by Local or Systemic Treatment with Oncolytic Parvovirus H-1 in Rat Models. Neuro-Oncology 2010, 12, 804–814. [Google Scholar] [CrossRef]
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.J.; Neumann, J.O.; Schöning, T.; Hüsing, J.; Beelte, B.; et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol. Ther. 2017, 25, 2620–2634. [Google Scholar] [CrossRef]
- Geletneky, K.; Hartkopf, A.D.; Krempien, R.; Rommelaere, J.; Schlehofer, J.R. Improved Killing of Human High-Grade Glioma Cells by Combining Ionizing Radiation with Oncolytic Parvovirus H-1 Infection. J. Biomed. Biotechnol. 2010, 2010, 350748. [Google Scholar] [CrossRef]
- Geletneky, K.; Angelova, A.; Leuchs, B.; Bartsch, A.; Capper, D.; Hajda, J.; Rommelaere, J. Atnt-07favorable Response of Patients with Glioblastoma at Second or Third Recurrence to Repeated Injection of Oncolytic Parvovirus H-1 in Combination with Bevacicumab. Neuro-Oncology 2015, 17, v11. [Google Scholar] [CrossRef]
- Angelova, A.; Ferreira, T.; Bretscher, C.; Rommelaere, J.; Marchini, A. Parvovirus-Based Combinatorial Immunotherapy: A Reinforced Therapeutic Strategy against Poor-Prognosis Solid Cancers. Cancers 2021, 13, 342. [Google Scholar] [CrossRef] [PubMed]
- Idbaih, A.; Erbs, P.; Foloppe, J.; Chneiweiss, H.; Kempf, J.; Homerin, M.; Schmitt, C.; Them, L.N.; Delattre, J.-Y. TG6002: A Novel Oncolytic and Vectorized Gene pro-Drug Therapy Approach to Treat Glioblastoma. J. Clin. Oncol. 2017, 35, e13510. [Google Scholar] [CrossRef]
- Wang, F.; Ma, Y.; Barrett, J.W.; Gao, X.; Loh, J.; Barton, E.; Virgin, H.W., IV; McFadden, G. Disruption of Erk-Dependent Type I Interferon Induction Breaks the Myxoma Virus Species Barrier. Nat. Immunol. 2004, 5, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Lun, X.; Yang, W.; Alain, T.; Shi, Z.Q.; Muzik, H.; Barrett, J.W.; McFadden, G.; Bell, J.; Hamilton, M.G.; Senger, D.L.; et al. Myxoma Virus Is a Novel Oncolytic Virus with Significant Antitumor Activity against Experimental Human Gliomas. Cancer Res. 2005, 65, 9982–9990. [Google Scholar] [CrossRef] [PubMed]
- Pisklakova, A.; McKenzie, B.; Zemp, F.; Lun, X.; Kenchappa, R.S.; Etame, A.B.; Rahman, M.M.; Reilly, K.; Pilon-Thomas, S.; McFadden, G.; et al. M011L-Deficient Oncolytic Myxoma Virus Induces Apoptosis in Brain Tumor-Initiating Cells and Enhances Survival in a Novel Immunocompetent Mouse Model of Glioblastoma. Neuro-Oncology 2016, 18, 1088–1098. [Google Scholar] [CrossRef]
- Al Yaghchi, C.; Zhang, Z.; Alusi, G.; Lemoine, N.R.; Wang, Y. Vaccinia Virus, a Promising New Therapeutic Agent for Pancreatic Cancer. Immunotherapy 2015, 7, 1249–1258. [Google Scholar] [CrossRef]
- Lam, P.Y.; Breakefield, X.O. Potential of gene therapy for brain tumors. Hum. Mol. Genet. 2001, 10, 777–787. [Google Scholar] [CrossRef]
- Foloppe, J.; Kempf, J.; Futin, N.; Kintz, J.; Cordier, P.; Pichon, C.; Findeli, A.; Vorburger, F.; Quemeneur, E.; Erbs, P. The Enhanced Tumor Specificity of TG6002, an Armed Oncolytic Vaccinia Virus Deleted in Two Genes Involved in Nucleotide Metabolism. Mol. Ther. Oncolytics 2019, 14, 1–14. [Google Scholar] [CrossRef]
- Geletneky, K.; Weiss, C.; Bernhard, H.; Capper, D.; Leuchs, B.; Marchini, A. Rommelaere, J. ATIM-29. First clinical observation of improved anti-tumor effects of viro-immunotherapy with oncolytic parvovirus H-1 in combination with PD-1 checkpoint blockade and bevacicumab in patients with recurrent glioblastoma. Neuro-Oncology 2016, 18, vi24. [Google Scholar] [CrossRef]
- Anderson, B.D.; Nakamura, T.; Russell, S.J.; Peng, K.W. High CD46 Receptor Density Determines Preferential Killing of Tumor Cells by Oncolytic Measles Virus. Cancer Res. 2004, 64, 4919–4926. [Google Scholar] [CrossRef]
- McDonald, C.J.; Erlichman, C.; Ingle, J.N.; Rosales, G.A.; Allen, C.; Greiner, S.M.; Harvey, M.E.; Zollman, P.J.; Russell, S.J.; Galanis, E. A Measles Virus Vaccine Strain Derivative as a Novel Oncolytic Agent against Breast Cancer. Breast Cancer Res. Treat. 2006, 99, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Blechacz, B.; Splinter, P.L.; Greiner, S.; Myers, R.; Peng, K.W.; Federspiel, M.J.; Russell, S.J.; LaRusso, N.F. Engineered Measles Virus as a Novel Oncolytic Viral Therapy System for Hepatocellular Carcinoma. Hepatology 2006, 44, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Sarkaria, J.N.; Petell, C.A.; Paraskevakou, G.; Zollman, P.J.; Schroeder, M.; Carlson, B.; Decker, P.A.; Wu, W.; James, C.D.; et al. Combination of Measles Virus Virotherapy and Radiation Therapy Has Synergistic Activity in the Treatment of Glioblastoma Multiforme. Clin. Cancer Res. 2007, 13, 7155–7165. [Google Scholar] [CrossRef] [PubMed]
- Opyrchal, M.; Allen, C.; Iankov, I.; Aderca, I.; Schroeder, M.; Sarkaria, J.; Galanis, E. Effective Radiovirotherapy for Malignant Gliomas by Using Oncolytic Measles Virus Strains Encoding the Sodium Iodide Symporter (MV-NIS). Hum. Gene Ther. 2011, 23, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Iankov, I.D.; Allen, C.; Aderca, I.; Federspiel, M.J.; Tindall, D.J.; Morris, J.C.; Koutsilieris, M.; Russell, S.J.; Galanis, E. Noninvasive Imaging and Radiovirotherapy of Prostate Cancer Using an Oncolytic Measles Virus Expressing the Sodium Iodide Symporter. Mol. Ther. 2009, 17, 2041–2048. [Google Scholar] [CrossRef]
- Nikolic, J.; Belot, L.; Raux, H.; Legrand, P.; Gaudin, Y.; Albertini, A.A. Structural Basis for the Recognition of LDL-Receptor Family Members by VSV Glycoprotein. Nat. Commun. 2018, 9, 1029. [Google Scholar] [CrossRef]
- Nikitina, A.S.; Lipatova, A.V.; Goncharov, A.O.; Kliuchnikova, A.A.; Pyatnitskiy, M.A.; Kuznetsova, K.G.; Hamad, A.; Vorobyev, P.O.; Alekseeva, O.N.; Mahmoud, M.; et al. Multiomic Profiling Identified EGF Receptor Signaling as a Potential Inhibitor of Type I Interferon Response in Models of Oncolytic Therapy by Vesicular Stomatitis Virus. Int. J. Mol. Sci. 2022, 23, 5244. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, T.; Davis, J.N.; Marzi, A.; Marchese, A.M.; Robek, M.D.; van den Pol, A.N. Mucin-Like Domain of Ebola Virus Glycoprotein Enhances Selective Oncolytic Actions against Brain Tumors. J. Virol. 2020, 94, e01967-19. [Google Scholar] [CrossRef]
- Muik, A.; Stubbert, L.J.; Jahedi, R.Z.; Geib, Y.; Kimpel, J.; Dold, C.; Tober, R.; Volk, A.; Klein, S.; Dietrich, U.; et al. Re-Engineering Vesicular Stomatitis Virus to Abrogate Neurotoxicity, Circumvent Humoral Immunity, and Enhance Oncolytic Potency. Cancer Res. 2014, 74, 3567–3578. [Google Scholar] [CrossRef]
- Wilcox, M.E.; Yang, W.Q.; Senger, D.; Rewcastle, N.B.; Morris, D.G.; Brasher, P.M.A.; Shi, Z.Q.; Johnston, R.N.; Nishikawa, S.; Lee, P.W.K.; et al. Reovirus as an Oncolytic Agent Against Experimental Human Malignant Gliomas. JNCI J. Natl. Cancer Inst. 2001, 93, 903–912. [Google Scholar] [CrossRef]
- Ganar, K.; Das, M.; Sinha, S.; Kumar, S. Newcastle Disease Virus: Current Status and Our Understanding. Virus Res. 2014, 184, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Chen, Y.; Hong, X.; Liu, X.; Su, X.; Li, S.; Dong, X.; Zhao, G.; Li, Y. Newcastle Disease Virus Enhances the Growth-Inhibiting and Proapoptotic Effects of Temozolomide on Glioblastoma Cells in vitro and in vivo. Sci. Rep. 2018, 8, 11470. [Google Scholar] [CrossRef] [PubMed]
- García-Romero, N.; Palacín-Aliana, I.; Esteban-Rubio, S.; Madurga, R.; Rius-Rocabert, S.; Carrión-Navarro, J.; Presa, J.; Cuadrado-Castano, S.; Sánchez-Gómez, P.; García-Sastre, A.; et al. Newcastle Disease Virus (NDV) Oncolytic Activity in Human Glioma Tumors Is Dependent on CDKN2A-Type I IFN Gene Cluster Codeletion. Cells 2020, 9, 1405. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Chen, G.Z. Comment to “Recurrent Glioblastoma Treated with Recombinant Poliovirus”. Chin. Med. J. 2018, 131, 2645–2646. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, J.M.; Mustafa, Z.; Ideris, A. Newcastle Disease Virus Interaction in Targeted Therapy against Proliferation and Invasion Pathways of Glioblastoma Multiforme. Biomed. Res. Int. 2014, 2014, 386470. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.J. Oncolytic Seneca Valley Virus: Past Perspectives and Future Directions. Oncolytic Virotherapy 2016, 5, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.J.; Wasinger, A.M.; Brey, R.N.; Dunleavey, J.M.; Croix, B.S.; Bann, J.G. Seneca Valley Virus Exploits TEM8, a Collagen Receptor Implicated in Tumor Growth. Front. Oncol. 2018, 8, 506. [Google Scholar] [CrossRef]
- Corbett, V.; Hallenbeck, P.; Rychahou, P.; Chauhan, A. Evolving Role of Seneca Valley Virus and Its Biomarker TEM8/ANTXR1 in Cancer Therapeutics. Front. Mol. Biosci. 2022, 9, 868. [Google Scholar] [CrossRef]
- Xu, J.; Yang, X.; Deng, Q.; Yang, C.; Wang, D.; Jiang, G.; Yao, X.; He, X.; Ding, J.; Qiang, J.; et al. TEM8 Marks Neovasculogenic Tumor-Initiating Cells in Triple-Negative Breast Cancer. Nat. Commun. 2021, 12, 4413. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, X.; Mao, H.; Baxter, P.A.; Huang, Y.; Yu, L.; Wadhwa, L.; Su, J.M.; Adesina, A.; Perlaky, L.; et al. Intravenous Injection of Oncolytic Picornavirus SVV-001 Prolongs Animal Survival in a Panel of Primary Tumor–Based Orthotopic Xenograft Mouse Models of Pediatric Glioma. Neuro-Oncology 2013, 15, 1173–1185. [Google Scholar] [CrossRef]
- Goetz, C.; Dobrikova, E.; Shveygert, M.; Dobrikov, M.; Gromeier, M. Oncolytic Poliovirus against Malignant Glioma. Future Virol. 2011, 6, 1045–1058. [Google Scholar] [CrossRef] [PubMed]
- Sloan, K.E.; Stewart, J.K.; Treloar, A.F.; Matthews, R.T.; Jay, D.G. CD155/PVR Enhances Glioma Cell Dispersal by Regulating Adhesion Signaling and Focal Adhesion Dynamics. Cancer Res. 2005, 65, 10930–10937. [Google Scholar] [CrossRef] [PubMed]
- Gromeier, M.; Alexander, L.; Wimmer, E. Internal Ribosomal Entry Site Substitution Eliminates Neurovirulence in Intergeneric Poliovirus Recombinants. Proc. Natl. Acad. Sci. USA 1996, 93, 2370–2375. [Google Scholar] [CrossRef] [PubMed]
- Gromeier, M.; Bossert, B.; Arita, M.; Nomoto, A.; Wimmer, E. Dual Stem Loops within the Poliovirus Internal Ribosomal Entry Site Control Neurovirulence. J. Virol. 1999, 73, 958–964. [Google Scholar] [CrossRef]
- PVSRIPO in Recurrent Malignant Glioma—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02986178 (accessed on 9 November 2022).
- Hamad, A.; Soboleva, A.V.; Vorobyev, P.O.; Mahmoud, M.; Vasilenko, K.V.; Chumakov, P.M.; Lipatova, A.V. Development of a recombinant oncolytic poliovirus type 3 strain with altered cell tropism. Bull. Russ. State Med. Univ. 2022, 4, 5–10. [Google Scholar] [CrossRef]


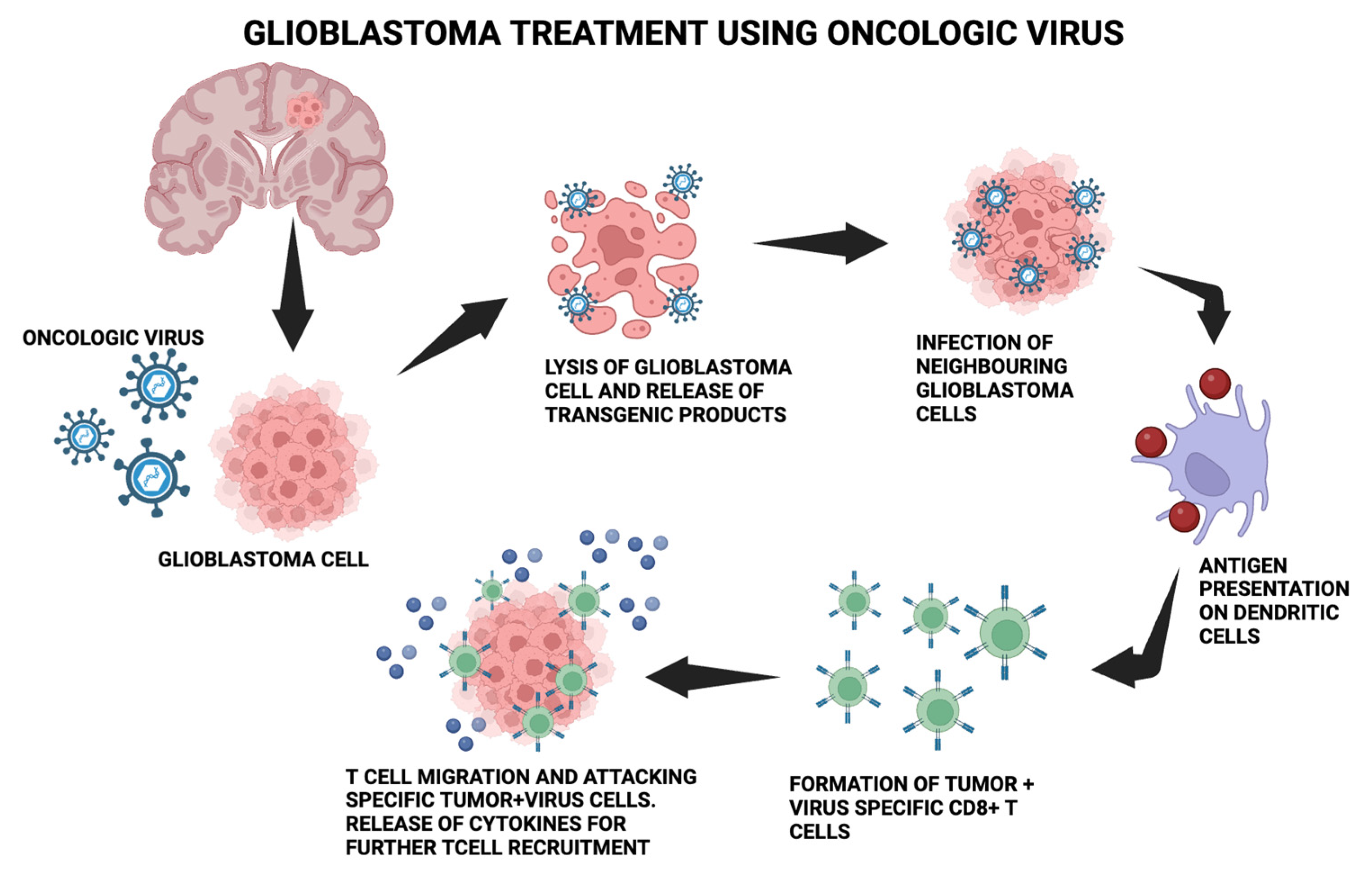{kind=link}
| New Onset Glioblastoma | Recurrent Gliobastoma |
|---|---|
| Patients with freshly diagnosed glioblastoma, IDH-wildtype, should be offered concomitant TMZ and RT. | Magnetic resonance imaging (MRI) augmented with gadolinium contrast is advised. |
| Patients who have undergone concurrent RT with TMZ should be offered adjuvant TMZ for six months. | 18-FDG is not advised for use in regular diagnostic procedures. |
| After chemoradiation treatment, individuals may receive adjuvant TMZ in addition to alternating electric field therapy. | Patients with symptomatic pGBM are advised to undergo cytoreductive surgery. |
| Treatment with bevacizumab is not advised. | It is not recommended to reevaluate the methylation status of 06-methylguanine-DNA methyltransferase (MGMT) and the state of isocitrate dehydrogenase (IDH). |
| Hypofractionated radiation therapy combined with TMZ is a suitable choice when the projected survival benefits of a six-week radiation treatment combined with TMZ may not exceed the hazards. | The activity of the mismatch repair enzyme (MRE) 1/programmed death ligand (PDL) is not a helpful part of routine diagnostic testing. |
| When treating patients who are older, in poor performance status, or who have concerns regarding toxicity or prognosis, hypofractionated RT alone or TMZ alone are reasonable options. | Epidermal growth factor receptor (EGFR) amplification may be useful for diagnosis if it has never been tested before. |
| No therapeutic approach is recommended or discouraged; instead, if at all feasible, these patients should be directed to take part in a clinical study. | Patients who are interested in or eligible for clinical trials or molecularly guided treatment may have their large panel sequencing needs taken into consideration. |
| Treatment with TMZ may be beneficial, particularly if it is continued for more than five months after stopping the medication. | |
| When an aged patient’s MGMT promoter status is methylated, fotemustine is recommended. | |
| For adult patients, tumor treatment fields (TTFs) with additional chemotherapy may be taken into consideration. |
| Oncolytic Virus | Outcomes |
|---|---|
| HSV-1 MVR-C5252 (C5252) that has been genetically altered | Safety and tolerability dose-limiting toxicities (DLT) and maximum tolerated dose (MTD) |
| Modified genetically HSV-1 M032 | MTD |
| G207 administered once via catheters into tumors | Safety and tolerability |
| Oncolytic viral vector rQNestin34.5v.2 HSV | MTD |
| DNX-2440, a genetically modified adenovirus | Safety, overall survival, and objective response rate |
| Adenoviral Nsc-crad-s-pk7 | - |
| Adenovirus DNX-2401 | MTD and incidence of adverse event |
| H-1 parvovirus (H-1PV) | Safety and tolerability |
| PVSRIPO, a recombinant nonpathogenic polio-rhinovirus chimera, is injected into a tumor via CED | MTD, dose-limiting toxicities, recommended phase 2 dose |
| PVSRIPO | 14 days following PVSRIPO therapy, toxicity |
| PVSRIPO delivered into a tumor via CED | objective radiographic response, duration of objective radiographic response at 24 and 36 months |
| Live, replication-competent wild-type reovirus REOLYSIN | MTD, DLTs, and six-month response rate |
| Combination of modified vaccinia virus TG6002 and 5-FC | DLTs and the six-month course of the tumor |
| Oncolytic Virus | Advantages |
|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, S. Novel Therapies in Glioblastoma Treatment: Review of Glioblastoma; Current Treatment Options; and Novel Oncolytic Viral Therapies. Med. Sci. 2024, 12, 1. https://doi.org/10.3390/medsci12010001
Shah S. Novel Therapies in Glioblastoma Treatment: Review of Glioblastoma; Current Treatment Options; and Novel Oncolytic Viral Therapies. Medical Sciences. 2024; 12(1):1. https://doi.org/10.3390/medsci12010001
Chicago/Turabian StyleShah, Siddharth. 2024. "Novel Therapies in Glioblastoma Treatment: Review of Glioblastoma; Current Treatment Options; and Novel Oncolytic Viral Therapies" Medical Sciences 12, no. 1: 1. https://doi.org/10.3390/medsci12010001