
An Observation Related to the Pressure Dependence of Ionic Radii


Department of Geoscience, University of Nevada Las Vegas, Las Vegas, NV 89154, USA
Geosciences 2022, 12(6), 246; https://doi.org/10.3390/geosciences12060246
Submission received: 1 May 2022
/
Revised: 8 June 2022
/
Accepted: 10 June 2022
/
Published: 14 June 2022
(This article belongs to the Section Geochemistry)
Abstract
:Here it is shown that the crystal radii of ions are represented by a simple relation rcryst = rB 3√(10 m)/N, where m and N are small integer numbers determined by the principal and orbital quantum numbers and valence, and rB is the Bohr radius. The relation holds to within 5%. This finding elucidates that despite their original definition crystal- and ionic radii are not classical but represent the limiting case of spherically symmetric spatial averages of the valence electron states and, therefore, are able to reflect changes in the valence electron configuration with pressure and temperature. The relation is used to show general pressure-effects on the radii, in particular the increase of bond coordination with pressure and metallization as limiting state. The pressure-effect is exemplified for the elements Mg and Si as major constituent cations in the Earth’s mantle, and for Ba as a large ionic lithophile element. It is found that at least to about 140 GPa the radii depend linearly on pressure. Further, if a generalization is permitted for just three elements, the pressure-dependence is lesser the higher the charge of the ion. The three elements exhibit a much weaker pressure-dependence than previously calculated non-bonding radii. For mantle geochemistry this finding implies that elements incompatible in the upper mantle remain so for the main lower mantle minerals bridgmanite and periclase and are hosted by davemaoite.
1. Introduction
The development of ab initio methods of calculating electron density distributions across electron band structures in solids pushed earlier empirical and semi-empirical approaches of understanding properties of solids nearly into oblivion. One of those earlier approaches is based on the concept of ionic radii. Ionic radii are still used in material science for ‘back of the envelope’ approximations and in inorganic chemistry for the qualitative understanding of polar solvent-solute systems. Geochemistry is one of the few disciplines in modern science that still hold on to the ionic radius concept as a quantitative means of understanding chemical and physical processes in the Earth. This faith into the ionic radius concept reflects indirectly the complexity of processes in the Earth which involve manifold changes of chemical bonding for most atomic species. Furthermore, it is related to the fact that in order to understand those processes geochemistry is guided by the distribution of minor and trace-elements rather than constituent elements of the major minerals [1]. Any trace element atom that we detect in an igneous or metamorphic rock has experienced manifold changes in its environment: solid-solid, solid-liquid transitions, transition into fluids and precipitation out of fluids. In sum these processes average the spatial differences between electron energy states and electron density distributions that adhere to each of these different bond environments and result in an apparent equivalent of a quasi-classical energy state represented by the electron kinetic energy and a spherical potential. In other words, minor and trace-element distributions of rocks derived directly or indirectly from partial melts in the Earth’s mantle may be seen as result of an enormous experiment that stretches over geologic time and the spatial scale of geologic provinces of the extend of continents and mantle convection cells and that emphasizes the spatially averaged properties of interacting atoms over the direction-dependent electronic states and density gradients which determine the state of matter in each of the many individual steps of this global scale experiment.
Hence, the application of the ionic radius model of atoms to geochemical minor and trace-element patterns is no anachronism but reflects the observable properties of atoms within the context of Earth mantle geochemistry.
However, there is one aspect within this concept that has remained nearly unexplored: Pressures in Earth range from ambient to 130 GPa at the core mantle boundary and to 360 GPa in the Earth’s inner core. A simple estimate of the effect of compression of Mg-Si-oxides over 130 GPa and of iron over 360 GPa on the energy of electrons suggests an increase in electron kinetic energy in the range of 0.5–1 eV for this range of pressure [2,3]. An increase of energy by that amount is within the range of changes in valence states of many ions and this has been observed indeed for ferrous iron which undergoes disproportionation into ferric and native iron as result of a pressure-induced high spin to low-spin transition that disfavours the larger volume of the high-spin divalent state [4,5]. It is plausible that other elements exhibit changes in their valence electron structure within the pressure-range of the Earth’s core and mantle and that these changes leave an imprint on geochemical element distributions, if they sample reservoirs at great depth in the mantle. This seems possible, in particular, for elements where the valence shell is well screened from the nucleus and where a marked increase in electron density can transfer electrons from s- to p- and d-states. This effect has been measured for metallic, dense Fe and Co [6] consistent with the pressure-induced high spin-low spin transitions oxides of these elements and it has been shown for the metallic heavier alkaline elements K, Rb and Cs [7] and for lanthanides [8]. K, Rb, Cs and lanthanides belong to the groups of LILEs (‘large ionic lithophile elements’) and the lanthanides to the REEs (‘rare earth elements’) in the context of mantle geochemistry which are both important in modelling geochemical processes. Yet, there is no direct knowledge if such transitions become effective for LILEs or REEs in the crystal-field of cation-oxide polyhedra in Earth-materials and within the pressure range of the Earth’s mantle.
Attempts to assess the effect of pressure on the spatially averaged electron distributions of atoms and ions have remained sparse [9,10] until quite recently [11,12] and have not yet considered the effect of bond coordination.
Here I report an observation that is closely related to the pressure effect on the ionic radii.
2. Method
I use the cation crystal radii listed by Shannon [13] for the coordinations 4,6,8, and 12 (henceforth I place coordination numbers in brackets {4}, {6} etc. and {X} for coordination in general). Coordination is used in the sense of bond coordination, that is: the sum of distinct radial trajectories intersecting at the cation along which finite charge transfer occurs between cat- and anion. In absence of highly accurate electron density distribution maps the distinction beween geometric and bond coordination may be ambiguous and in those cases I calculate crystal radii for different apparent coordinations (Table 1). At ambient pressure I use the radii for the coordinations specified by Shannon [13]. I derive crystal radii of Ba from crystal structure analyses of solids with Ba as constituting cation and O as anion at pressures of 0.2 to 143 GPa. In order to extract crystal radii from Ba-O interatomic distances I use an empirical equation for the pressure-effect on the O2− anion in six-fold coordination. This equation is obtained by (a) Mg-O and Si-O interatomic distances in stishovite, seifertite, akimotoite, bridgmanite, postperovskite, and periclase to 130 GPa (Table 1) and (b) the pressure-effect on the Baader radii of Si and O in the computed compression of silica polymorphs to 170 GPa [14]. The Baader radii reported by Du&Tse [14] are systematically larger than the crystal radii of Si4+ and O2−, even at ambient and low pressure: At ambient pressure the Baader radius for Si4+ in six-fold coordination is about 0.69 whereas the crystal radius is 0.54 Å [13]. This difference is owed to a different definition of the electron density cut-off in the Shannon ionic radius- and the Baader charge concept and needs no digression here (although this point is quite interesting in itself). However, the calculated pressure effect on the Baader radii of Si and O are expected to exhibit less of a systematic difference from the empirical compression than the absolute radii. The crystal radii of both elements at ambient pressure are well established [13]. Therefore, I used these 1bar crystal radii as fiducial points, and used the rescaled Si and O-radii from Du&Tse [14] for calculating the interatomic distances of Mg-O and Si-O at high-pressure and compare them with observed values (Table 1). Accordingly, the O2−-radii follow a relation r(O2−) = 1.215·P−0.0386 (radii in Å, P in GPa). Yet, by this approach the contraction of the interatomic distance Si-O are overestimated by 7% (based on Table 1) compared to the observed values and Mg-O is overestimated by ~10% at 50 GPa. The O-ion compression that Du&Tse [14] calculated serves the particular purpose of their study in reproducing the observed Si-O distances at high pressure but at the expense of an ambient pressure radius of O2− of 1.33 Å (instead of the empirical value 1.26 Å) and of overestimating the compression of Mg-O. Therefore, I corrected the relation from Du&Tse [14] for Si and O to match all Mg-Si-O and silica polymorphs but kept the shape of the functional. Hereby I obtained the relation r(O2−) = 1.269·P−0.0176 as fit equation for the pressure dependence of the crystal radius of O2− in six-fold coordination to 150 GPa, where P in GPa and r is in Å. The proper assessment of the accuracy of this equation requires a more extensive set of structure data than currently available and presently I only state that the uncertainty is better than 10%, based on the scatter of the available distances (Table 1). In the regime of 0 to 10 GPa the pressure-dependence of the O2− radius may be overestimated and this issue could be helped by considering structure data from a more extended set of compounds but this is not within the scope of the paper. It is worth noticing that the 300 K isotherm of periclase, MgO [15], where Mg-O = ½-times the unit cell axis length, is reproduced.
3. Results
It was found that the examined 160 crystal radii for 79 elements that occur as cations in coordinations {4,6,8} and {12} can all be represented through a the following equation:
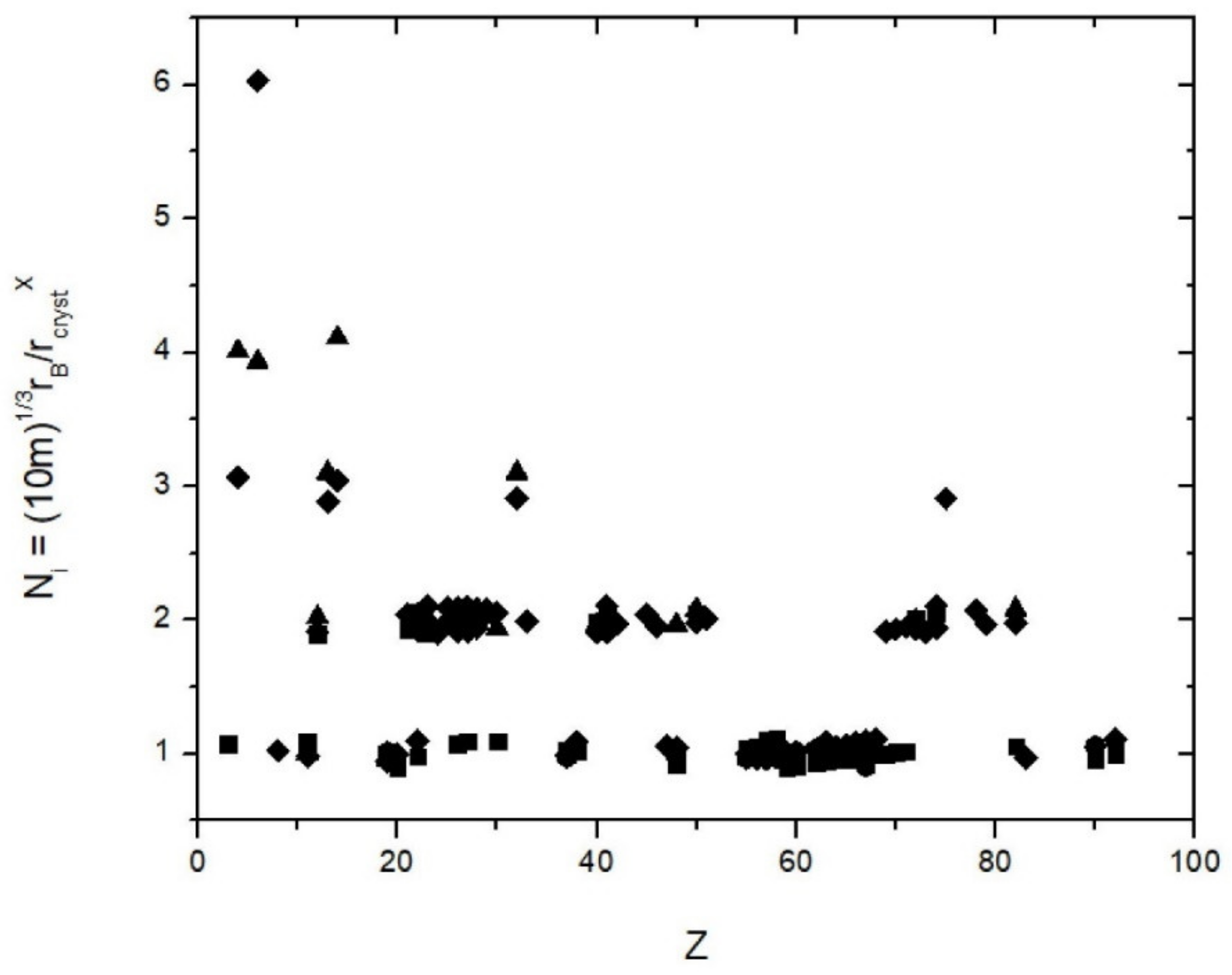
where rB is the Bohr radius (0.52177 Å), m is an integer that runs from 1 to 6 and N is an integer that runs from 1 to 4 (except for C4+ {6}, which has N = 6). This equation holds within +/− 5% (Figure 1, Table 2). First, it needs to be checked if 6 × 4 = 24 arithmetically possible combinations of integers generate the observed 160 crystal radii to within 5%. For all radii, the number before the point is either 0 or 1: the radii are either 0.ab or 1.cd with a, b, c, d running from 0 to 9 (I ignore Cs+ {12} = 2.03 Å as the only radius with a value larger than 2, Shannon [13]). Hence, for the two positions past the point there are 10 possible values each = 200 possible combinations. Within the 5% margin of uncertainty, some of these combinations are treated as equal and this reduces the number of distinct combinations to between 2·10·8 = 160 and 2·10·6 = 120 which overall gives a probability of 1/5th to 3/20th to match a radius with a combination of N and m and Equation (1) by chance. However, as shown in Figure 1 all radii are matched by Equation (1). Thus, the empirical Equation (1) is statistically meaningful. The computed radii and the Shannon radii are given in Table 2. Table 2 gives the values of N, m. One may argue that slightly different definitions of the effective ionic radii (such as by Ahrens and Pauling) give slightly different values but the 5% margin that is here allowed for accounts for almost all of these differences. It is noted that not all arithmetically possible combinations of N and m give values in the range of ionic radii and the probability of matching a radius within 5% is even less that the range estimated above.
rcryst = rB 3√(10 m)/N
Much more relevant than these arithmetic considerations is the question of the physical meaning of N and m. Equation (1) may be used to predict radii as function of N and m. Furthermore, any deviation of observed radii from Equation (1) carries a physical meaning (otherwise we may extend Equation (1) to any larger quotient of variable integers that reproduces a given radius with arbitrary precision, but this bears no relevance because these additional quotients have only arithmetic but no physical meaning). For fourfold coordination N equals the formal valence for 12 out of 19 available radii (Table 1). However, the correlation between N and valence is generally less simple.
For six-fold coordination N is equal to or smaller than the valence but remains integer (Table 2). For coordination {8} and {12} N equals valence only for univalent ions and is, thus, generally smaller than valences larger than 1+ (see Table 2). Moreover, for coordination {8} N is restrained to values of 1 or 2 and for {12} N = 1 for all available crystal radii. This point needs further examination: Based on these observations N becomes 1 if the coordination goes to infinity and rcryst = rB3√10 m. In other words: for a hypothetical very large number of bonds the distribution of charge per bond becomes accordingly very small and in the limiting case of infinitely many bonds it becomes zero and the effective radii are equivalent to non-bonding radii. Thus, 10 m represents discrete volumetric units of atomic volumes. The limiting non-bonding radii rcryst = rB3√10 m are nearly identical to, tentatively slightly smaller than atomic radii [40]. For instance for Na, K, Mg, Ca the atomic radii are 1.90, 2.43, 1.45, and 1.94 Å [40] and the corresponding rB3√10 m are 1.92, 2.42, 1.42, and 1.94 Å for m = 5, 10, 2, and 6, respectively. There are similar relations between the Wigner-Seitz radii of metals and the parameter m. This will be discussed in a separate paper. Based on what has been stated in the introduction it is plausible that m represents projections of discrete sets of valence electron states onto spherically symmetric spatial averages (and the emphasis is on the averaged spatial distribution, hence there is no violation of orthogonality for orbitals higher than s).
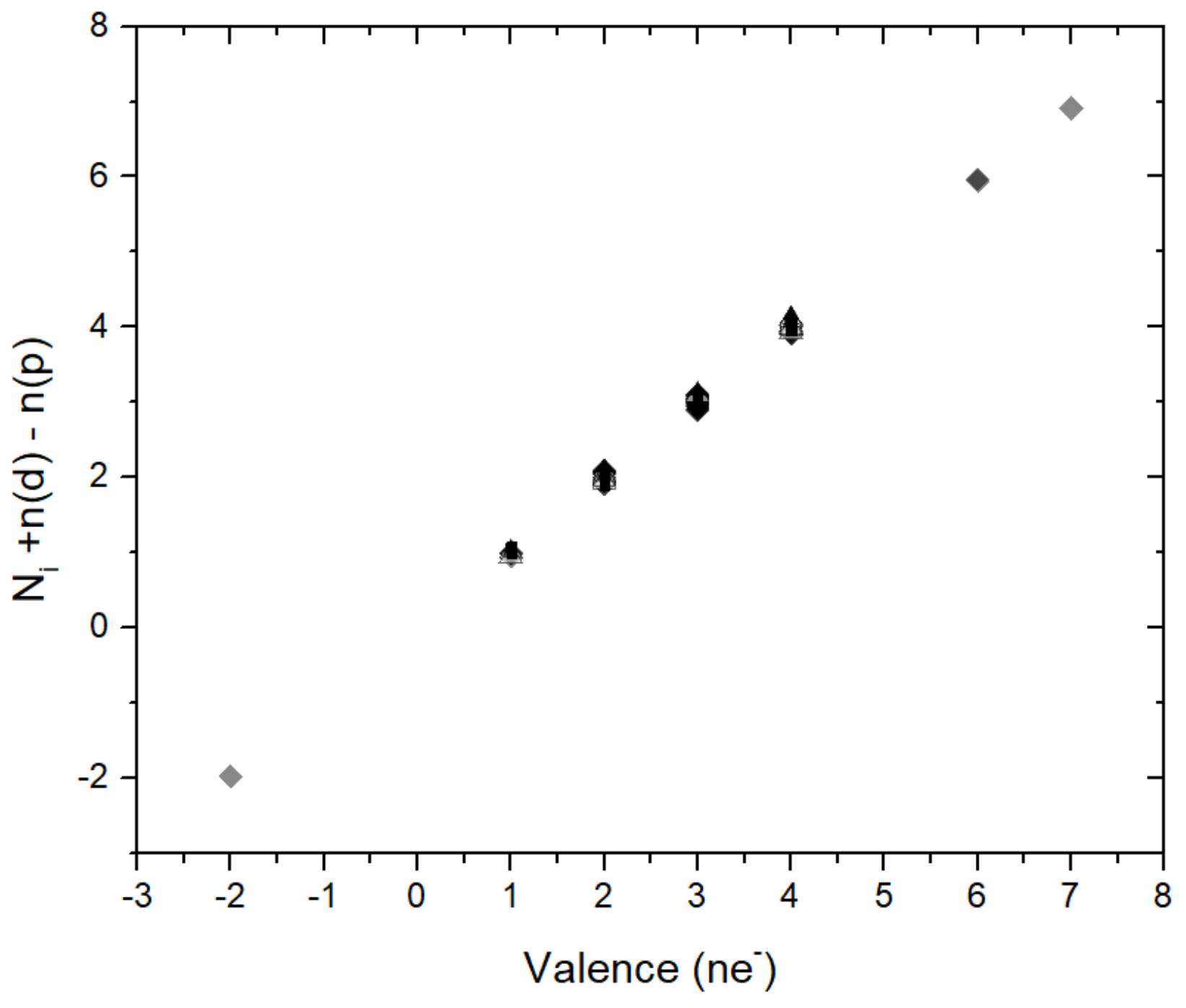
For the parameter N I observe a very simple relation with the valence electron states: As shown in Figure 2 the parameter N is related to the valence of the ions through a simple relation equation Ni + n(d) − n(p) = valence where n(d), n(p) are the numbers of occupied (ionic) valence p- and d-states and where Ni assumes the values 1,…, 6 given in Table 2. The small observeable deviations from this relations is mostly within 5%, in few cases up to 10% (Figure 2) and is intepreted as the difference between the actual and the formal integer valences in solids where partial charge transfer between ions is possible. With this parametrization Equation (1) is reformulated as:
where L = principal quantum number of the valence shell, n(pL) and n(dL−1) the occupancies of the valence p- and d-states. Hence, rB 3√(10 m) is the radius of the non-bonding atoms and Ni = Valence + n(pL) − n(dL−1) modifies these radii according to their bond states.
rcryst = rB 3√(10 m)/N = rB 3√(10 m)/[Valence + n(pL) − n(dL−1)]
N, m, and their correlation with valence electron configuration and valence imply that that crystal- and ionic radii are not equivalent to Thomas-Fermi-type quasiclassical, spherical electron density distributions where excess charge is added or removed. Instead, the occupation of electronic states is intrinsic to the ionic radii (despite their original classical concept). This is consistent with the fact that the radii depend on valence spin states where high- and low-spin states are possible. Independent on that it is known that Thomas-Fermi-atoms with addition or removal of electron charge cannot be used for modeling chemical bonds or yield convincing representations of bonding solids [41].
The observed minor deviations from (1) and (2) are likely not just empirical uncertainties but represent actual physical properties such as partial charge transfer between ions, partially occupied states, or the effect of f-electrons for high-Z elements or relativistic corrections for higher Z ions. For instance, Equation (1) predicts equal radii for Zr and Hf and Nb and Ta, respectively, and the observed lanthanide crystal radii [13] show a minor but systematic deviation from the calculated ones (Figure 1).
Taking Equations (1) and (2) and Figure 1 and Figure 2 together it can be stated that crystal (ionic) radii are not quasi-classical approximations of electron densities but projections of discrete sets of valence electron states onto spherically symmetric spatial averages. The parameters m and N and the correlation in Figure 2 tentatively constrain the sets of allowed states under this symmetry condition. It is noted that the orthogonality of the orbital states is not consistent with spherical symmetry. Hence, the spherical symmetry is not applied to the energy states but to a spatial integration of manifold projections of these states into the configurational space.
4. Effect of Pressure on Ionic Radii
Because of the relation between crystal (and ionic) radii and electronic states, it is expected that the radii not only show a continuous contraction over larger pressure ranges, but also discrete contractive transitions. This effect has been shown theoretically for non-bonding radii in confining continuous fields [12]. Here, we use empirical data from Table 1 and Equation (1) to examine the effect of pressure on crystal radii. Equation (1) is reformulated as
where (rcryst/rB)3 is a relative volume with limiting case r = rB ⇔ 10 m = N3. Since m is related to non-bonding radii (see above), the limit is interpreted as the transition to a metallic state. The trend toward pressure-induced metallization is empirically well confirmed [3]. Equation (3) provides a quantitative parametrization of ionic volume compression with metallization as limiting case. Based on the correlation between N and bond coordination (see above) a balance between the decrease of (rcryst/rB)3, and the increase of bond coordination is expected and ionic radii of elements which can increase bond coordination by involving orbitals into binding states that are empty at low pressure are expected to exhibit weaker compression than ions which remain with a fixed bond coordination. The correlation between pressure and coordination also has been inferred from observation [42,43]. Before testing this hypothesis with radii that are obtained from experimentally measured interatomic distances, it should be noted that Shannon crystal radii of highly charged low-Z ions in tetrahedral coordination like Si4+ are almost equal, for C4+ even smaller than rB but this observation simply reflects that the bonds that involve tetravalent C and Si in tetrahedral coordination are predominantly not ionic and the radius model not well applicable. With this limitation in mind it is also clear that r approaching rB does not strictly imply a state where electrons gradually enter the state of a metal or a degenerate Fermi plasma but rather marks a transition to other types of bonding which no longer obey Equation (3). This could be covalent bonding [42], but recent experimental studies indicate that at extreme pressure transitions from ionic to covalent are accompanied by transfer of electron density to non-bonding states centered on interstitial lattice sites (‘high-pressure electrides’, [44,45]). In addition, there are actual pressure-driven valence transition for some transition metal elements such as Fe and in some cases inner-shell electrons become involved in the valence shell such as in the case of Li+ [46], Cl− [47], Mg2+ [48]. More relevant for Earth is the regime in between this extreme pressure limit and the ambient pressure state. How does the balance between m and N change upon compression? In the present paper Ba2+ is discussed as example of a LILE along with Mg2+ and Si4+ as the two major constituent cations in Earth mantle material (since O2− is used to obtain the radii of these three cations it is not an independent parameter—see Section 2). The pressure-dependent crystal radii of these three ions, Ba, Mg, and Si are given in Table 1 and shown in Figure 3.
(rcryst/rB)3 = 10 m/N3
The radii were obtained from available experimental data (Table 1) and the pressure-dependent radius of O2− (see Methods). The coordination of Ba varies between {6} and {18} (Table 1) and, therefore, shows much variation. Equation (1) can be used to assess bond coordination under pressure by checking which radii for the different geometric coordinations give m-parameters closest to integer values and this was used to extract radii for Ba in six-fold bond coordination.
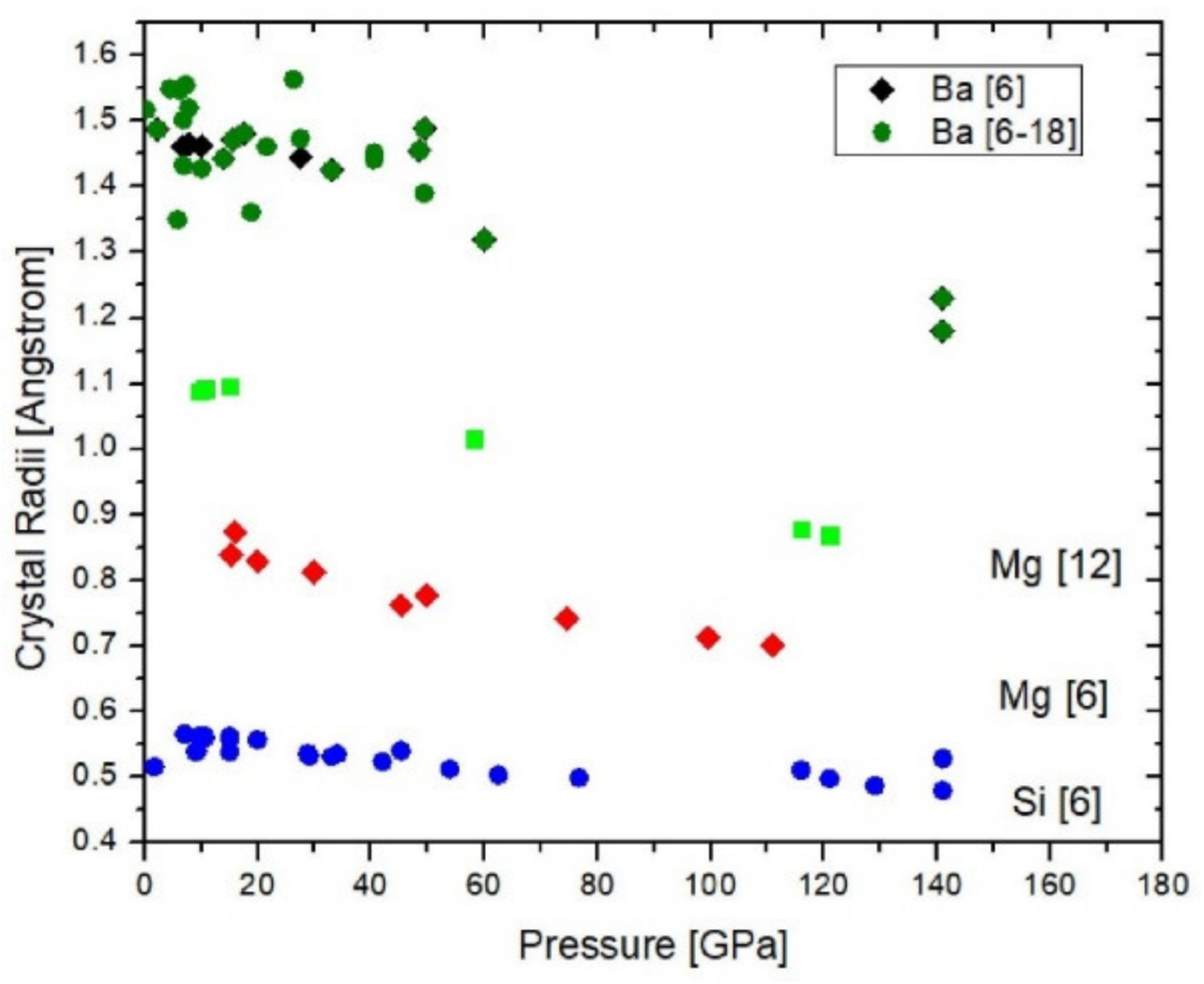
Overall, Figure 3 shows that all examined crystal radii compress linearly within the given uncertainties with their radial compressibilities on the order of 0.4 to 2 × 10−23 m3/N. Si [6] is the least compressible ion with rcryst(Si) = 0.552(5) − 0.000425(72)·P. Ba{6} (as well as the average of all rBa(X)) and Mg {12} exhibit nearly the same dependence on pressure: rcryst(Ba) = 1.50(1) − 0.0020(2)·P and rcryst(Mg) = 1.119(7) − 0.00204(10)·P for Mg{12}, whereas Mg{6} with rcryst (Mg) = 0.864(11) − 0.0016(2)·P is slightly less compressible but overlaps with rBa within uncertainties. All radii are in Å and pressure is in GPa. It is worth noting that the fitted zero pressure radii are equal to the Shannon radii within uncertainties. The apparent linear pressure-dependence for Ba does not suggest any pressure-induced change in valence. In fact, based on a formal valence of 2+ and Equation (2) the valence configuration is 6s15d16p0 where 0.91 < N < 1.05 gives the deviation of the actual from these formal values of valence and electron configuration. The observation that Si4+ is ~4.7-times less compressible than Mg2+ and Ba2+, independent on their coordination, suggests that it is not coordination but charge that controls the ionic compressibility, if three elements permit any generalization.
The fact that the compression of twelve- and six-coordinated Mg2+ is both linear over more than 100 GPa and that the radial compressibilities are somewhat but not markedly different from each other does not support a trend towards coordinating changes over narrow pressure intervals for this element. For Ba there are not enough data for coordinations other than {6} to make a similar clear statement but the average compression of Ba2+ for all coordinations (Table 1) does not deviate from the compression of Ba2+{6}. This observation suggests that the increase of coordination with pressure is not as effective a mechanism of compression as inferred above. The observed crystal radii of Mg, Si and Ba exhibit up to 120–140 GPa a much weaker dependence on pressure as the calculated non-bonding radii (see ref. [12], more precisely for Ba2+ the ratio of the calculated non-bonding over the empirical crystal radii in Fig. 3 decreases from about 1.6 to 1.25 times between 0 and 50 GPa).
If the compression behaviour of Ba2+ is representative for other LILEs, it may be stated that LILEs that are geochemically incompatible in the upper mantle remain so over the pressure range of the lower mantle. This does not imply that they are as easily mobilized in the lower mantle as they are in the upper mantle because they are predominantly hosted in davemaoite, CaSiO3-perovskite [49,50], which has accordingly low solubility in bridgmanite and which is solidus phase during partial melting of lower mantle rock [51]. Hence, the LILEs remain in the residual of partial melt events (such as in an early magma ocean) rather than entering an ascending melt.
5. Summary
In sum we have shown that the crystal radii of cations are represented through a simple Equation (1) with the Bohr radius and two discrete integer parameters which are related to valence electron state of the cation in an oxide crystal field. Therefore, crystal (ionic) radii are not quasi-classical approximations of electron densities but projections of discrete sets of valence electron states onto spherically symmetric spatial averages. This relation does not violate the orthogonality of the orbital states because the spherical symmetry is not applied to the energy states but the result of the spatial integration of manifold projections of these states into the configurational space. This statement defines the crystal and ionic radii, respectively. The discrete integer parameters m and N are functions of the sets of allowed states under this symmetry condition (Figure 2).
Based on the new relation (1) it can be shown that pressure increases coordination and, in absence of decomposition or a valence transition, drives toward pressure-induced metallization. In the 0–140 GPa regime Ba exhibits a pressure dependence equal to Mg in {12}-fold coordination and from the limited available data it appears that ionic compressibility is more determined by the cation charge rather than the nuclear charge number and coordination. The observed ionic compressibilities of Mg, Si, and Ba are markedly less that those predicted for non-bonding radii [12]. Overall, the observed ionic radial compressibilities range around 0.4 to 2·10−23 m3/N.
For mantle geochemistry these findings imply that incompatible elements remain so for the deep mantle main minerals bridgmanite and periclase. Consequently, these elements are predominantly hosted by davemaoite, which is solidus phase for lower mantle peridotite and contains Ca as constituent element. Hence, davemaoite retains Ba and probably other high-Z cations of equivalent behaviour rather that they enter an ascending melt. The scheme of upper mantle geochemistry where incompatibility equals depletion upon partial melting is therefore reversed for the lower mantle.
Funding
This research received no external funding.
Data Availability Statement
All used data are given in the paper.
Acknowledgments
The author thanks two anonymous reviewers and his colleague Shichun Huang for their very helpful comments.
Conflicts of Interest
The author declares no conflict of interest.
References
- Goldschmidt, V.M. The principles of distribution of chemical elements in minerals and rocks. The seventh Hugo Muller Lecture, delivered before the Chemical Society on March 17th, 1937. J. Chem. Soc. 1937, 655–673. [Google Scholar] [CrossRef]
- Bukowinski, M.S.T. Quantum Geophysics. Annu. Rev. Earth Planet. Sci. 1994, 22, 167–205. [Google Scholar] [CrossRef]
- Mao, H.K.; Chen, X.J.; Ding, Y.; Li, B.; Wang, L. Solids, liquids, and gases under high pressure. Rev. Mod. Phys. 2018, 90, 015007. [Google Scholar] [CrossRef] [Green Version]
- Sturhahn, W.; Jackson, J.M.; Lin, J.F. The spin state of iron in minerals of Earth’s lower mantle. Geophys. Res. Lett. 2005, 32, L12307. [Google Scholar] [CrossRef] [Green Version]
- Frost, D.J.; McCammon, C.A. The redox state of Earth’s mantle. Annu. Rev. Earth Planet. Sci. 2008, 36, 389–420. [Google Scholar] [CrossRef]
- Iota, V.; Klepeis, J.-H.P.; Yoo, C.-S.; Lang, J.; Haskel, D.; Srajer, G. Electronic structure and magnetism in compressed 3d transition metals. Appl. Phys. Lett. 2007, 90, 042505. [Google Scholar] [CrossRef]
- Fabbris, G.; Lim, J.; Veiga, L.; Haskel, D.; Schilling, J.S. Electronic and structural ground state of heavy alkali metals at high pressure. Phys. Rev. B 2015, 91, 085111. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, O.; Brooks, M.S.S.; Johansson, B. Orbital polarization in narrow-band systems: Application to volume collapses in light lanthanides. Phys. Rev. B 1990, 41, 7311–7314. [Google Scholar] [CrossRef]
- Royce, E.B. Stability of the Electronic Configuration and Compressibility of Electron Orbitals in Metals under Shock-Wave Compression. Phys. Rev. Ser. I 1967, 164, 929–943. [Google Scholar] [CrossRef]
- Batsanov, S.S. Cationic radii from structures of extremely compressed solids. Acta Cryst. 2013, B69, 563–569. [Google Scholar] [CrossRef]
- Cammi, R.; Rahm, M.; Hoffmann, R.; Ashcroft, N.W. Varying Electronic Configurations in Compressed Atoms: From the Role of the Spatial Extension of Atomic Orbitals to the Change of Electronic Configuration as an Isobaric Transformation. J. Chem. Theory Comput. 2020, 16, 5047–5056. [Google Scholar] [CrossRef] [PubMed]
- Rahm, M.; Ångqvist, M.; Rahm, J.M.; Erhart, P.; Cammi, R. Non-Bonded Radii of the Atoms Under Compression. ChemPhysChem 2020, 21, 2441–2453. [Google Scholar] [CrossRef] [PubMed]
- Shannon, R.D. Revised effective ionicradii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Du, X.; Tse, J.S. Oxygen Packing Fraction and the Structure of Silicon and Germanium Oxide Glasses. J. Phys. Chem. B 2017, 121, 10726–10732. [Google Scholar] [CrossRef]
- Jacobsen, S.D.; Holl, C.M.; Adams, K.A.; Fischer, R.A.; Martin, E.S.; Bina, C.R.; Lin, J.-F.; Prakapenka, V.B.; Kubo, A.; Dera, P. Compression of single-crystal magnesium oxide to 118 GPa and a ruby pressure gauge for helium pressure media. Am. Miner. 2008, 93, 1823–1828. [Google Scholar] [CrossRef]
- Weir, S.T.; Vohra, Y.K.; Ruoff, A.L. High-pressure phase transitions and the equations of state of BaS and BaO. Phys. Rev. B 1986, 33, 4221–4226. [Google Scholar] [CrossRef] [PubMed]
- Liu, L. A Dense Modification of BaO and Its Crystal Structure. J. Appl. Phys. 1971, 42, 3702–3704. [Google Scholar] [CrossRef]
- Crichton, W.A.; Merlini, M.; Hanfland, M.; Müller, H. The crystal structure of barite, BaSO4, at high pressure. Am. Miner. 2011, 96, 364–367. [Google Scholar] [CrossRef]
- Santamaría-Pérez, D.; Chulia-Jordan, R. Compression of mineral barite, BaSO4: A structural study. High Press. Res. 2012, 32, 81–88. [Google Scholar] [CrossRef]
- Errandonea, D.; Pellicer-Porres, J.; Manjón, F.J.; Segura, A.; Ferrer-Roca, C.; Kumar, R.S.; Tschauner, O.; López-Solano, J.; Rodríguez-Hernández, P.; Radescu, S.; et al. Determination of the high-presure crystal structure of BaWO4 and PbWO4. Phys. Rev. B Condens. Matter Mater. Phys. 2006, 73, 224103. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.; Xiao, W.; Zhou, W.; Chen, M.; Zhou, W.; Li, X.; Li, Y.; Liu, J. High pressure X-ray diffraction study on BaWO4-II. High Press. Res. 2012, 32, 262–269. [Google Scholar] [CrossRef]
- Yusa, H.; Sata, N.; Ohishi, Y. Rhombohedral (9R) and hexagonal (6H) perovskites in barium silicates under high pressure. Am. Miner. 2007, 92, 648–654. [Google Scholar] [CrossRef]
- Efthimiopoulos, I.; Kunc, K.; Karmakar, S.; Syassen, K.; Hanfland, M.; Vajenine, G. Structural transformation and vibrational properties of BaO2 at high pressures. Phys. Rev. B 2010, 82, 134125. [Google Scholar] [CrossRef]
- Friese, K.; Kanke, Y.; Grzechnik, A. Characterization of the pressure-induced second-order phase transition in the mixed-valence vanadate BaV6O11. Acta Crystallogr. Sect. B Struct. Sci. 2009, 65, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Curetti, N.; Benna, P.; Bruno, E. High-pressure structural configuration and phase transition in celsian, BaAl2Si2O8. Phys. Chem. Miner. 2016, 44, 181–192. [Google Scholar] [CrossRef]
- Panchal, V.; Garg, N.; Sharma, S.M. Raman and X-ray diffraction investigations on BaMoO4 under high pressures. J. Phys. Condens. Matter 2006, 18, 3917–3929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazen, R.M.; Finger, L.W. High-pressure and high-temperature crystallographic study of the gillespite I–II phase transition. Am. Mineral. 1983, 68, 595–603. [Google Scholar]
- Andrault, D.; Angel, R.J.; Mosenfelder, J.L.; Le Bihan, T. Equation of state of stishovite to lower mantle pressures. Am. Miner. 2003, 88, 301–307. [Google Scholar] [CrossRef]
- Zhang, L.; Popov, D.; Meng, Y.; Wang, J.; Ji, C.; Li, B.; Mao, H.-K. In-situ crystal structure determination of seifertite SiO2 at 129 GPa: Studying a minor phase near Earth’s core–mantle boundary. Am. Miner. 2016, 101, 231–234. [Google Scholar] [CrossRef] [Green Version]
- Ross, N.L.; Shu, J.-F.; Hazen, R.M.; Gasparik, T. High-pressure crystal chemistry of stishovite. Am. Mineral. 1990, 75, 739–747. [Google Scholar]
- Yamanaka, T.; Fukuda, T.; Mimaki, J. Bonding character of SiO2 stishovite under high pressures up to 30 Gpa. Phys. Chem. Miner. 2002, 29, 633–641. [Google Scholar] [CrossRef]
- Oganov, A.R.; Ono, S. Theoretical and experimental evidence for a post-perovskite phase of MgSiO3 in Earth’s D″ layer. Nature 2004, 430, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Hirose, K.; Kawamura, K.; Sata, N.; Ohishi, Y. Post-Perovskite Phase Transition in MgSiO3. Science 2004, 304, 855–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugahara, M.; Yoshiasa, A.; Komatsu, Y.; Yamanaka, T.; Bolfan-Casanova, N.; Nakatsuka, A.; Sasaki, S.; Tanaka, M. Reinvestigation of the MgSiO3 perovskite structure at high pressure. Am. Miner. 2006, 91, 533–536. [Google Scholar] [CrossRef]
- Yamanaka, T.; Komatsu, Y.; Sugahara, M.; Nagai, T. Structure change of MgSiO3, MgGeO3, and MgTiO3 ilmenites under compression. Am. Miner. 2005, 90, 1301–1307. [Google Scholar] [CrossRef]
- Ross, N.; Hazen, R.M. High-pressure crystal chemistry of MgSiO3 perovskite. Phys. Chem. Miner. 1990, 17, 228–237. [Google Scholar] [CrossRef]
- Kudoh, Y.; Ito, E.; Takeda, H. Effect of pressure on the crystal structure of perovskite-type MgSiO3. Phys. Chem. Miner. 1987, 14, 350–354. [Google Scholar] [CrossRef]
- Finkelstein, G.J.; Dera, P.K.; Jahn, S.; Oganov, A.R.; Holl, C.M.; Meng, Y.; Duffy, T.S. Phase transitions and equation of state of forsterite to 90 GPa from single-crystal X-ray diffraction and molecular modeling. Am. Miner. 2014, 99, 35–43. [Google Scholar] [CrossRef]
- Jacobsen, S.D.; Demouchy, S.; Frost, D.J.; Ballaran, T.B.; Kung, J. A systematic study of OH in hydrous wadsleyite from polarized FTIR spectroscopy and single-crystal X-ray diffraction: Oxygen sites for hydrogen storage in Earth’s interior. Am. Miner. 2005, 90, 61–70. [Google Scholar] [CrossRef]
- Clementi, E.; Raimondi, D.L.; Reinhardt, W.P. Atomic Screening Constants from SCF Functions. II. Atoms with 37 to 86 Electrons. J. Chem. Phys. 1967, 47, 1300–1307. [Google Scholar] [CrossRef]
- Spruch, L. Pedagogic notes on Thomas-Fermi theory (and on some improvements)—Atoms, stars and the stability of bulk matter. Rev. Mod. Phys. 1991, 63, 151–209. [Google Scholar] [CrossRef]
- Shannon, R.D.; Prewitt, C.T. Coordination and volume changes accompanying high-pressure phase trans-formations of oxides. Mater. Res. Bull. 1969, 4, 57–59. [Google Scholar] [CrossRef]
- Tschauner, O. High-pressure minerals. Am. Miner. 2019, 104, 1701–1731. [Google Scholar] [CrossRef]
- Neaton, J.B.; Ashcroft, N.W. On the Constitution of Sodium at Higher Densities. Phys. Rev. Lett. 2001, 86, 2830–2833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, M.-S.; Hoffmann, R. High Pressure Electrides: A Predictive Chemical and Physical Theory. Acc. Chem. Res. 2014, 47, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Kim, D.Y.; Yang, L.; Li, N.; Tang, L.; Amine, K.; Mao, H.K. Oxygen-Rich Lithium Oxide Phases Formed at High Pressure for Potential Lithium-Air Battery Electrode. Adv. Sci. 2017, 4, 1600453. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Oganov, A.R.; Goncharov, A.F.; Zhu, Q.; Boulfelfel, S.E.; Lyakhov, A.O.; Stavrou, E.; Somayazulu, M.; Prakapenka, V.B.; Konôpková, Z. Unexpected Stable Stoichiometries of Sodium Chlorides. Science 2013, 342, 1502–1505. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Oganov, A.R.; Lyakhov, A.O. Novel stable compounds in the Mg–O system under high pressure. Phys. Chem. Chem. Phys. 2013, 15, 7696–7700. [Google Scholar] [CrossRef]
- Hirose, K.; Shimizu, N.; van Westrenen, W.; Fei, Y.W. Trace element partitioning in Earth’s lower mantle and implications for geochemical consequences of partial melting at the core-mantle boundary. Phys. Earth Planet. Inter. 2004, 146, 249–260. [Google Scholar] [CrossRef]
- Tschauner, O.; Huang, S.; Yang, S.; Humayun, M.; Liu, W.; Corder, S.N.G.; Bechtel, H.A.; Tischler, J.; Rossman, G.R. Discovery of davemaoite, CaSiO3 -perovskite, as a mineral from the lower mantle. Science 2021, 374, 891–894. [Google Scholar] [CrossRef]
- Hirose, K.; Sinmyo, R.; Hernlund, J. Perovskite in Earth’s deep interior. Science 2017, 358, 734–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
The ratio of rB 3√(10 m)//r (X), where r (x) are the crystal radii by Shannon (1976) and X = 4, 6, 8 and 12, the bond coordination. This ratio gives the parameter N in Equation (1), which is independent on the nuclear charge number Z and assumes within uncertainties integer values N = 1, 2, 3, 4 and 6 for C4+ {6}. Symbols: Triangles = {4}, diamonds = {6}, squares = {8} and hexagons = {12}.
Figure 1.
The ratio of rB 3√(10 m)//r (X), where r (x) are the crystal radii by Shannon (1976) and X = 4, 6, 8 and 12, the bond coordination. This ratio gives the parameter N in Equation (1), which is independent on the nuclear charge number Z and assumes within uncertainties integer values N = 1, 2, 3, 4 and 6 for C4+ {6}. Symbols: Triangles = {4}, diamonds = {6}, squares = {8} and hexagons = {12}.

Figure 2.
The figure shows that the parameter N in Equation (1) is related to formal valence through the Equation Ni + n(d) − n(p) = valence. n(p) and n(d) give the occupation of the valence p- and d-states, respectively. Ni runs through the values 1 to 6 that are given in Table 2.
Figure 2.
The figure shows that the parameter N in Equation (1) is related to formal valence through the Equation Ni + n(d) − n(p) = valence. n(p) and n(d) give the occupation of the valence p- and d-states, respectively. Ni runs through the values 1 to 6 that are given in Table 2.

Figure 3.
Crystal radii of Ba (black = coordination {6}, green: coordination {6} to {18}), Mg (green squares = coordination {12}, red diamonds = coordination {6}), and Si (blue) as function of pressure.
Figure 3.
Crystal radii of Ba (black = coordination {6}, green: coordination {6} to {18}), Mg (green squares = coordination {12}, red diamonds = coordination {6}), and Si (blue) as function of pressure.


{kind=link}
{kind=link}
{kind=link}
Table 1.
(a): Crystal radii, pressures and coordination for Ba2+; (b): crystal radii, pressures and measured average cation-oxygen bond distances for Mg and Si. The bond coordination for Si is [6], above 100 GPa possibly [8]; for Mg, the two bond coordinations [6,12] are clearly distinguished by their difference.
Table 1.
(a): Crystal radii, pressures and coordination for Ba2+; (b): crystal radii, pressures and measured average cation-oxygen bond distances for Mg and Si. The bond coordination for Si is [6], above 100 GPa possibly [8]; for Mg, the two bond coordinations [6,12] are clearly distinguished by their difference.
| a. Barium | |||||||
| Compound | Coordination | Pressure (GPa) | Ba-O(Å) | r (Å) | References | ||
| BaO_III | 8 | 18.8 | 2.566 | 1.361 | [16] | ||
| BaO_III | 8 | 60 | 2.50 | 1.32 | [16] | ||
| BaO, NiAs-type | 6 | 13.9 | 2.654 | 1.443 | [17] | ||
| Baryte | 8 | 17.5 | 2.760 | 1.5 | [18] | ||
| Baryte | 6 | 17.5 | 2.688 | 1.482 | [18] | ||
| Baryte | 8 | 2.15 | 2.815 | 1.479 | [18] | ||
| Baryte | 6 | 2.15 | 2.739 | 1.487 | [18] | ||
| Baryte | 8 | 0.22 | 2.82 | 1.563 | [18] | ||
| Baryte-II | 8 | 0 | 2.85 | 1.59 | [19] | ||
| Baryte-II | 8 | 40.5 | 2.63 | 1.441 | [19] | ||
| Baryte-II | 12 | 40.5 | 2.653 | 1.46 | [19] | ||
| BaWO4, fergusonite-type | 12 | 15.6 | 2.708 | 1.52 | [20,21] | ||
| BaSiO3, BaFeO3-type | 6 | 48.5 | 2.64 | 1.455 | [22] | ||
| BaSiO3, BaRuO3-type | 18 | 27.9 | 2.766 | 1.569 | [22] | ||
| BaSiO3, BaTiO3-type | 6 | 141 | 2.393 | 1.23 | [22] | ||
| BaO2 | 8 | 49.4 | 2.57 | 1.39 | [23] | ||
| BaAlSi3O8 -II | 14 | 7.81 | 2.75 | 1.52 | [24] | ||
| BaV6O11 | 12 | 5.82 | 1.52 | [25] | |||
| BaMoO4 | 12 | 7.2 | 1.41 * | [26] | |||
| Gillespite, BaFeSi4O10 | 8 | 4.5 | 2.784 | 1.549 | [27] | ||
| BaO2_CuCl2-typ | 12 | 6.8 | 2.729 | 1.50 | [23] | ||
| b. Mg-Silicates, Silica, Periclase | |||||||
| Compound | P [GPa] | Mg-O | Si-O | rSi (Å) {6,7} | rMg (Å) {6,12} | ||
| Periclase, MgO | 15.3 | 2.0485 | 0.839 | [15] | |||
| 29.9 | 2.0085 | 0.813 | [15] | ||||
| 49.7 | 1.962 | 0.778 | [15] | ||||
| 74.6 | 1.918 | 0.742 | [15] | ||||
| 99.6 | 1.8825 | 0.712 | [15] | ||||
| 111 | 1.869 | 0.701 | [15] | ||||
| Stishovite, SiO2 | 7 | 1.792 | 0.566 | [28] | |||
| 10.5 | 1.781 | 0.564 | [28] | ||||
| 15 | 1.772 | 0.562 | [28] | ||||
| 34 | 1.728 | 0.536 | [28] | ||||
| 76.75 | 1.675 | 0.500 | [28] | ||||
| 1.7 | 1.773 | 0.516 | [29] | ||||
| 9 | 1.76 | 0.539 | [29] | ||||
| 15 | 1.749 | 0.539 | [29] | ||||
| 9.26 | 1.7603 | 0.540 | [30] | ||||
| 29.1 | 1.7287 | 0.533 | [30] | ||||
| Seifertite, SiO2 | 129 | 1.652 | 0.487 | [31] | |||
| Akimotoite, MgSiO3 | 19.91 | 2.033 | 1.761 | 0.557 | 0.829 | [32] | |
| Bridgmanite, MgSiO3 | 10.6 | 2.31 | 1.777 | 0.560 | 1.093 | [33] | |
| 9.6 | 2.3075 | 1.782 | 0.563 | 1.088 | [34] | ||
| 15 | 2.3057 | 1.769 | 0.559 | 1.096 | [35] | ||
| postperovskite, MgSiO3 | 121 | 2.035 | 1.664 | 0.498 | 0.869 | [36] | |
| 116 | 2.045 | 1.678 | 0.511 | 0.878 | [37] | ||
| Forsterite-II, Mg2SiO4 | 45.3 | 1.949 | 1.727 | 0.541 | 0.763 | [38] | |
| 45.3 | 1.949 | (1.866) | (0.68) | 0.763 | [38] | ||
| Forsterite-III, Mg2SiO4 | 58.2 | 2.198 | 1.815 | 0.634 | 1.017 | [38] | |
| Wadsleyite, Mg2SiO4 | 16 | 2.082 | ** | ** | 0.874 | [39] | |
* Value for rBa is unusual. ** Si [4], not considered here.
Table 2.
(a): Parameters n = the ratios of the calculated crystal radii according to Equation (1) and the radii reported by Shannon [13] labeled as robs. All values are in Å. The ratios are given for coordinations {4},{6},{8} and {12}. The values of the integer parameter m that minimizes the ratios rB 3√(10 m)/r (X) are given for each coordination in a separate column. (b): Crystal radii in Å calculated based on Equation (1) with the parameters m as listed in Table 2 rclc and the radii robs reported by Shannon [13] in Table 2.
Table 2.
(a): Parameters n = the ratios of the calculated crystal radii according to Equation (1) and the radii reported by Shannon [13] labeled as robs. All values are in Å. The ratios are given for coordinations {4},{6},{8} and {12}. The values of the integer parameter m that minimizes the ratios rB 3√(10 m)/r (X) are given for each coordination in a separate column. (b): Crystal radii in Å calculated based on Equation (1) with the parameters m as listed in Table 2 rclc and the radii robs reported by Shannon [13] in Table 2.
| a | ||||||||
| Ion | n(4) = rB 3√(10 m)/r(4)obs | m | n(6) = rB 3√(10 m)/r(6)obs | m | n(8) = rB 3√(10 m)/r(8)obs | m | n(12) = rB 3√(10 m)/r(12)obs | M |
| Li+ | 1.07547 | 1 | ||||||
| Na+ | 1.00885 | 1 | 0.98276 | 1 | 1.08808 | 2 | 1.07457 | 3 |
| K+ | 0.95117 | 2 | 0.94492 | 2 | 0.99642 | 3 | 1.0166 | 4 |
| Rb+ | 0.99042 | 3 | 1.03403 | 4 | 0.97288 | 4 | ||
| Cs+ | 0.99975 | 4 | 1.03684 | 5 | 0.96498 | 5 | ||
| Be2+ | 4.01 | 3 | 3.06703 | 4 | ||||
| Mg2+ | 2.02292 | 2 | 1.91174 | 3 | 1.89249 | 5 | ||
| Ca2+ | 1 | 1 | 0.90476 | 1 | 0.97045 | 2 | ||
| Sr2+ | 1.08808 | 2 | 1.02591 | 2 | 1.04057 | 3 | ||
| Ba2+ | 0.96394 | 2 | 1.05391 | 3 | 1.03403 | 4 | ||
| Al3+ | 3.10207 | 3 | ||||||
| Si4+ | 4.11025 | 3 | 3.04463 | 3 | ||||
| Sn4+ | 2.08155 | 2 | 1.98084 | 3 | 2.05185 | 5 | ||
| Pb4+ | 2.08114 | 3 | 1.97765 | 4 | 1.05555 | 1 | ||
| Ge4+ | 3.10207 | 3 | 2.90935 | 5 | ||||
| C4+ | 3.93102 | 1 | 6.03183 | 4 | ||||
| Sc3+ | 2.04469 | 4 | 1.92996 | 5 | ||||
| Y3+ | 1.09615 | 1 | 0.9836 | 1 | ||||
| La3+ | 0.97269 | 1 | 1.10482 | 2 | 0.95751 | 2 | ||
| Ce3+ | 0.9913 | 1 | 1.11946 | 2 | 0.97045 | 2 | ||
| Pr3+ | 1.00885 | 1 | 0.90047 | 1 | ||||
| Nd3+ | 1.01513 | 1 | 0.91273 | 1 | 1.01863 | 1 | ||
| Sm3+ | 1.03825 | 1 | 0.93519 | 1 | 1.04078 | 2 | ||
| Eu3+ | 1.04875 | 1 | 0.94527 | 1 | ||||
| Eu2+ | 1.09639 | 2 | 1.03329 | 2 | ||||
| Gd3+ | 1.05751 | 1 | 0.95557 | 1 | ||||
| Tb3+ | 1.07243 | 1 | 0.9661 | 1 | ||||
| Dy3+ | 1.08365 | 1 | 0.97686 | 1 | ||||
| Ho3+ | 1.0951 | 1 | 0.98701 | 1 | 0.90476 | 1 | ||
| Er3+ | 1.10679 | 1 | 0.9965 | 1 | ||||
| Tm3+ | 1.91104 | 5 | 1.00529 | 1 | ||||
| Yb3+ | 1.93379 | 5 | 1.01333 | 1 | ||||
| Lu3+ | 1.94731 | 5 | 1.02059 | 1 | ||||
| Th4+ | 1.05555 | 1 | 0.95798 | 1 | 1.06391 | 2 | ||
| U4+ | 1.10679 | 1 | 1 | 1 | 1.09639 | 2 | ||
| Ti2+ | 1.94926 | 5 | ||||||
| Ti3+ | 2.02975 | 3 | ||||||
| Ti4+ | 2.03571 | 1 | 1.92788 | 2 | 2.0563 | 4 | ||
| Zr4+ | 1.9675 | 2 | 1.91174 | 3 | 1.98904 | 5 | ||
| Hf4+ | 1.99482 | 2 | 1.93423 | 3 | 2.00955 | 5 | ||
| V2+ | 1.94575 | 4 | ||||||
| V3+ | 2.10782 | 3 | ||||||
| V4+ | 1.99482 | 2 | 1.91174 | 3 | ||||
| Nb3+ | 1.91174 | 3 | ||||||
| Nb5+ | 2.10782 | 3 | 2.0563 | 4 | ||||
| Ta3+ | 1.91174 | 3 | ||||||
| Ta5+ | 2.10782 | 3 | 2.0563 | 4 | ||||
| Cr3+ | 1.90235 | 2 | ||||||
| Mo3+ | 1.98084 | 3 | ||||||
| Mo6+ | 1.9675 | 2 | ||||||
| W6+ | 1.94091 | 2 | ||||||
| Mn3+HS | 2.09439 | 3 | ||||||
| Mn3+LS | 1.99482 | 2 | ||||||
| Re7+ | 2.90935 | 5 | ||||||
| Fe2+HS | 1.9669 | 4 | 1.07547 | 1 | ||||
| Fe2+LS | 1.91503 | 2 | ||||||
| Fe3+HS | 2.09439 | 3 | 1.9669 | 4 | ||||
| Fe3+LS | 2.08155 | 2 | ||||||
| Ru4+ | ||||||||
| Os4+ | ||||||||
| Co2+HS | 1.99482 | 2 | 2.04469 | 4 | 1.09615 | 1 | ||
| Co2+LS | 2.08114 | 3 | ||||||
| Co3+HS | 1.91503 | 2 | ||||||
| Co3+LS | 2.09675 | 2 | ||||||
| Rh3+ | 2.04236 | 3 | ||||||
| Ir4+ | ||||||||
| Ni2+ | 2.08155 | 2 | 1.98084 | 3 | ||||
| Ni3+HS | 1.94091 | 2 | ||||||
| Ni3+LS | 2.05182 | 2 | ||||||
| Pd2+ | 1.94926 | 5 | ||||||
| Pt2+ | 2.07368 | 5 | ||||||
| Cu2+ | 2.02292 | 2 | 2.07994 | 4 | ||||
| Ag2+ | 1.05555 | 1 | ||||||
| Au3+ | 1.96895 | 5 | ||||||
| Zn2+ | 1.94091 | 2 | 2.0563 | 4 | 1.09615 | 1 | ||
| Cd2+ | 1.9669 | 4 | 1.04587 | 1 | 0.91935 | 1 | 0.99053 | 2 |
| As3+ | 1.99482 | 2 | ||||||
| Sb3+ | 2.01061 | 4 | ||||||
| Bi3+ | 0.97436 | 1 | ||||||
| O2− | 1.02756 | 3 | ||||||
| b | ||||||||
| Ion | r (4)obs (Å) | r (4)clc (Å) | r (6)obs (Å) | r (6)clc (Å) | r (8)obs (Å) | r (8)clc (Å) | r (12)obs (Å) | r (12)clc (Å) |
| Li+ | 0.73 | 0.72 | 0.9 | 0.900 | 1.06 | 1.060 | ||
| Na+ | 1.13 | 1.145 | 1.16 | 1.160 | 1.32 | 1.380 | 1.53 | 1.527 |
| K+ | 1.51 | 1.523 | 1.52 | 1.527 | 1.65 | 1.647 | 1.78 | 1.785 |
| Rb+ | 1.66 | 1.706 | 1.75 | 1.785 | 1.86 | 1.837 | ||
| Cs+ | 1.81 | 1.834 | 1.88 | 1.837 | 2.02 | 2.132 | ||
| Be2+ | 0.41 | 0.411 | 0.59 | 0.595 | ||||
| Mg2+ | 0.71 | 0.711 | 0.86 | 0.853 | 1.03 | 1.020 | ||
| Ca2+ | 1.14 | 1.145 | 1.26 | 1.219 | 1.48 | 1.482 | ||
| Sr2+ | 1.32 | 1.377 | 1.4 | 1.378 | 1.58 | 1.527 | ||
| Ba2+ | 1.49 | 1.485 | 1.56 | 1.482 | 1.75 | 1.785 | ||
| Al3+ | 0.53 | 0.533 | 0.675 | 0.689 | ||||
| Si4+ | 0.4 | 0.411 | 0.54 | 0.551 | ||||
| Sn4+ | 0.69 | 0.689 | 0.83 | 0.853 | 0.95 | 1.020 | ||
| Pb4+ | 0.79 | 0.793 | 0.915 | 0.916 | 1.08 | 1.102 | ||
| Ge4+ | 0.53 | 0.533 | 0.67 | 0.689 | ||||
| C4+ | 0.29 | 0.288 | 0.3 | 0.296 | ||||
| Sc3+ | 0.885 | 0.892 | 1.01 | 1.020 | ||||
| Y3+ | 1.04 | 0.96 | 1.159 | 1.145 | ||||
| La3+ | 1.172 | 1.19 | 1.3 | 1.377 | 1.5 | 1.482 | ||
| Ce3+ | 1.15 | 1.14 | 1.283 | 1.219 | 1.48 | 1.482 | ||
| Pr3+ | 1.13 | 1.14 | 1.266 | 1.219 | ||||
| Nd3+ | 1.123 | 1.19 | 1.249 | 1.219 | 1.41 | 1.422 | ||
| Sm3+ | 1.098 | 1.14 | 1.219 | 1.219 | 1.38 | 1.378 | ||
| Eu3+ | 1.087 | 1.14 | 1.206 | 1.219 | ||||
| Eu2+ | 1.31 | 1.43 | 1.39 | 1.377 | ||||
| Gd3+ | 1.078 | 1.14 | 1.193 | 1.218 | ||||
| Tb3+ | 1.063 | 1.14 | 1.18 | 1.22 | ||||
| Dy3+ | 1.052 | 1.14 | 1.167 | 1.22 | ||||
| Ho3+ | 1.041 | 1.14 | 1.155 | 1.14 | 1.26 | 1.219 | ||
| Er3+ | 1.03 | 0.98 | 1.144 | 1.14 | ||||
| Tm3+ | 1.02 | 0.98 | 1.134 | 1.14 | ||||
| Yb3+ | 1.008 | 0.98 | 1.125 | 1.14 | ||||
| Lu3+ | 1.001 | 0.96 | 1.117 | 1.14 | ||||
| Th4+ | 1.08 | 1.190 | 1.19 | 1.218 | 1.35 | 1.377 | ||
| U4+ | 1.03 | 1.190 | 1.14 | 1.145 | 1.31 | 1.377 | ||
| Ti2+ | 1 | 0.960 | ||||||
| Ti3+ | 0.81 | 0.793 | ||||||
| Ti4+ | 0.56 | 0.552 | 0.745 | 0.740 | 0.88 | 0.893 | ||
| Zr4+ | 0.73 | 0.720 | 0.86 | 0.853 | 0.98 | 0.960 | ||
| Hf4+ | 0.72 | 0.720 | 0.85 | 0.853 | 0.97 | 0.960 | ||
| V2+ | 0.93 | 0.918 | ||||||
| V3+ | 0.78 | 0.787 | ||||||
| V4+ | 0.72 | 0.720 | 0.86 | 0.853 | ||||
| Nb3+ | 0.86 | 0.853 | ||||||
| Nb5+ | 0.78 | 0.775 | 0.88 | 0.892 | ||||
| Ta3+ | 0.86 | 0.853 | ||||||
| Ta5+ | 0.78 | 0.787 | 0.88 | 0.892 | ||||
| Cr3+ | 0.755 | 0.763 | ||||||
| Mo3+ | 0.83 | 0.793 | ||||||
| Mo6+ | 0.73 | 0.720 | ||||||
| W6+ | 0.74 | 0.740 | ||||||
| Mn3+HS | 0.785 | 0.787 | ||||||
| Mn3+LS | 0.72 | 0.720 | ||||||
| Re7+ | 0.67 | 0.689 | ||||||
| Fe2+HS | 0.77 | 0.775 | 0.92 | 0.916 | 1.06 | 1.066 | ||
| Fe2+LS | 0.75 | 0.740 | ||||||
| Fe3+HS | 0.785 | 0.763 | 0.92 | 0.918 | ||||
| Fe3+LS | 0.69 | 0.689 | ||||||
| Ru4+ | 0.76 | 0.763 | ||||||
| Os4+ | 0.77 | 0.775 | ||||||
| Co2+HS | 0.72 | 0.720 | 0.885 | 0.892 | 1.04 | 1.020 | ||
| Co2+LS | 0.79 | 0.793 | ||||||
| Co3+HS | 0.75 | 0.740 | ||||||
| Co3+LS | 0.685 | 0.689 | ||||||
| Rh3+ | 0.805 | 0.763 | ||||||
| Ir4+ | 0.765 | 0.763 | ||||||
| Ni2+ | 0.69 | 0.720 | 0.83 | 0.793 | ||||
| Ni3+HS | 0.74 | 0.74 | ||||||
| Ni3+LS | 0.7 | 0.689 | ||||||
| Pd2+ | 1 | 0.960 | ||||||
| Pt2+ | 0.94 | 0.960 | ||||||
| Cu2+ | 0.71 | 0.720 | 0.87 | 0.916 | ||||
| Ag2+ | 1.08 | 1.102 | ||||||
| Au3+ | 0.99 | 0.916 | ||||||
| Zn2+ | 0.74 | 0.720 | 0.88 | 0.916 | ||||
| Cd2+ | 0.92 | 1.102 | 1.09 | 1.102 | 1.24 | 1.219 | 1.45 | 1.440 |
| As3+ | 0.72 | 0.720 | ||||||
| Sb3+ | 0.9 | 0.893 | ||||||
| Bi3+ | 1.17 | 1.19 | ||||||
| O2− | 1.60 | 1.64 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tschauner, O. An Observation Related to the Pressure Dependence of Ionic Radii. Geosciences 2022, 12, 246. https://doi.org/10.3390/geosciences12060246
AMA Style
Tschauner O. An Observation Related to the Pressure Dependence of Ionic Radii. Geosciences. 2022; 12(6):246. https://doi.org/10.3390/geosciences12060246
Chicago/Turabian StyleTschauner, Oliver. 2022. "An Observation Related to the Pressure Dependence of Ionic Radii" Geosciences 12, no. 6: 246. https://doi.org/10.3390/geosciences12060246
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.