
Alterations in Fecal Microbiota Linked to Environment and Sex in Red Deer (Cervus elaphus)
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Site, Subjects, and Sample Collection
2.2. Individual Recognition and Sex Identification
2.3. Fecal Microbiota DNA Extraction, Amplification, and Sequencing
2.4. Bioinformatics and Statistical Analyses
3. Results
3.1. Identification of Individuals and Sex
3.2. Diet Composition and Nutritional Composition of Wild and Captive Red Deer Winter Diets
3.3. Sequencing Analysis and Clustering
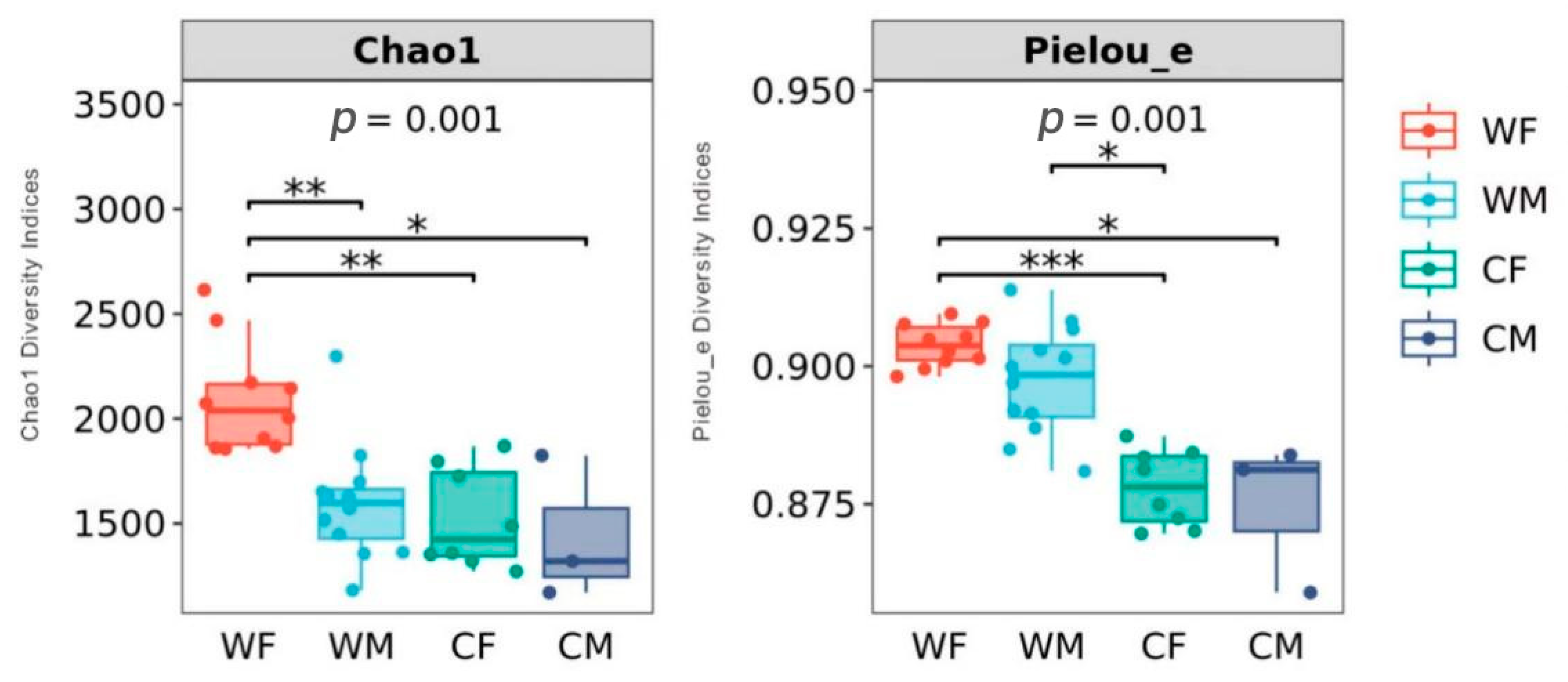
3.4. Microbial Composition and Diversity by Environment and Sex
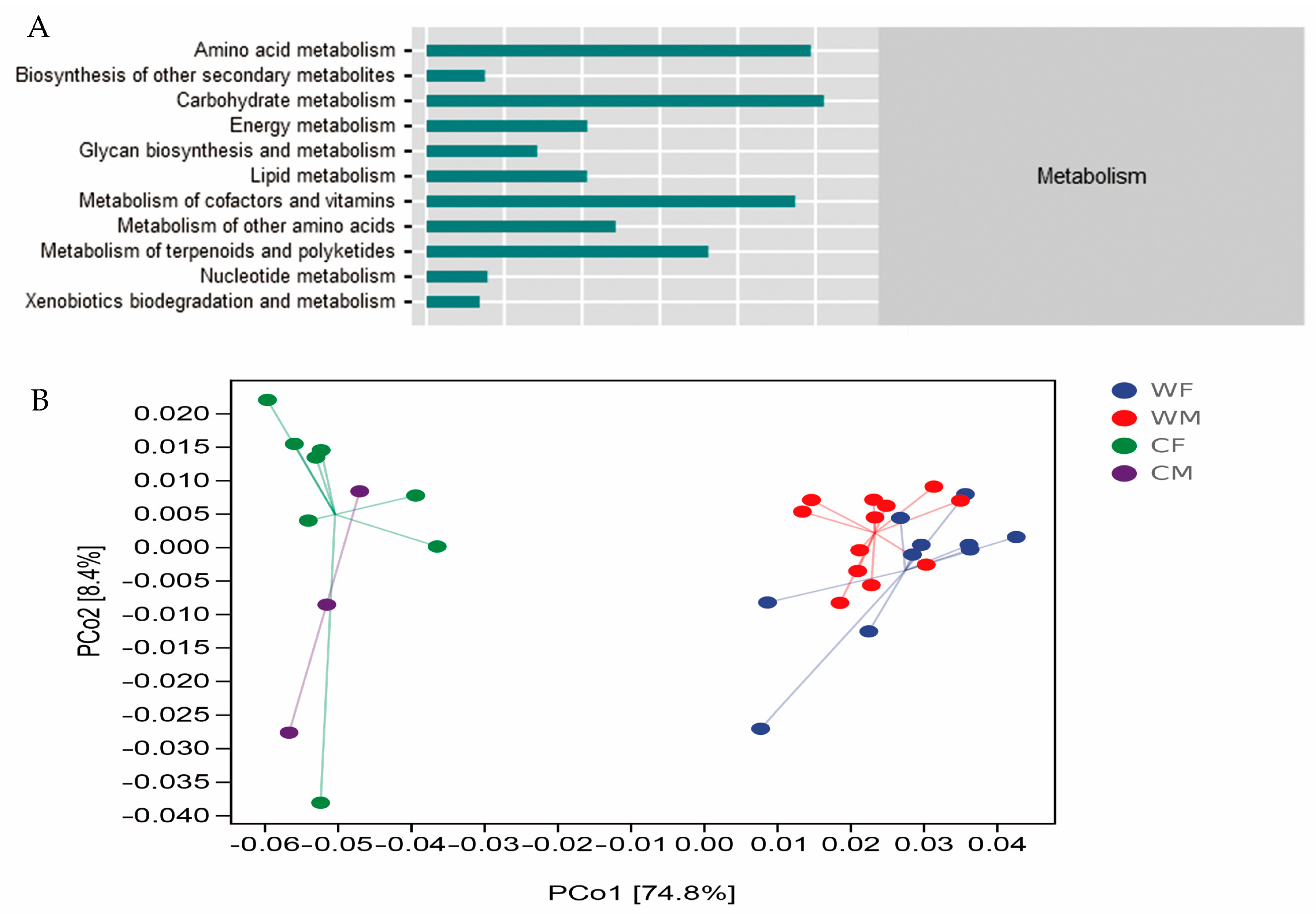
3.5. Functional Modules of Fecal Microbial Communities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, Z.; Jiang, J.; Wang, Y.; Zhang, E.; Zhang, Y.; Li, L.; Xie, F.; Cai, B.; Cao, L.; Zheng, G. Red list of China’s vertebrates. Biodivers. Sci. 2016, 24, 500–551. [Google Scholar]
- Xu, Q.X.; Zhang, M.H.; Lu, B.X. Study on the status of red deerpopulation in Heilongjiang Province. J. Econ. Anim. 2000, 4, 57–62. [Google Scholar]
- Gu, J.Y.; Yu, L.; Hua, Y.; Ning, Y.; Heng, B.; Qi, J.; Long, Z.; Yao, M.; Huang, C.; Li, Z. A comparison of food habits and prey preferences of Amur tiger (Panthera tigrisaltaica) at thesouthwestPrimorskii Krai in Russia and Hunchun in China. Integr. Zool. 2018, 13, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R. Evolution of mammals and their gut microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, E.; Minard, G.; Lindholm, J.; Saastamoinen, M. Moderate plant water stress improves larval development and impacts immunity and gut microbiota of a specialist herbivore. PLoS ONE 2019, 14, e0204292. [Google Scholar] [CrossRef]
- Cani, P.D.; Everard, A.; Duparc, T. Gut microbiota, enteroendocrine functions and metabolism. Curr. Opin. Pharmacol. 2013, 13, 935–940. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-Gut Microbiota Metabolic Interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [Green Version]
- Benson, A.K.; Kelly, S.A.; Legge, R.; Ma, F.; Low, S.J.; Kim, J.; Zhang, M.; Oh, P.L.; Nehrenberg, D.; Hua, K.; et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. USA 2010, 107, 18933–18938. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Sun, Y.; Shi, Z. Gut Microbiota of Wild and Captive Alpine Musk Deer (Moschus chrysogaster). Front. Microbiol. 2020, 10, 3156. [Google Scholar] [CrossRef]
- Sun, C.H.; Liu, H.Y.; Liu, B.; Yuan, B.D.; Lu, C.H. Analysis of the Gut Microbiome of Wild and Captive Père David’s Deer. Front. Microbiol. 2019, 10, 2331. [Google Scholar] [CrossRef]
- Guo, J.; Jin, Y.; Tian, X.; Bao, H.; Sun, Y.; Gray, T.; Song, Y.; Zhang, M. Diet-induced microbial adaptation process of red deer (Cervus elaphus) under different introduced periods. Front. Microbiol. 2022, 13, 1033050. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, M.; Wang, S.; Han, R.; Cao, Y.; Hua, W.; Mao, Y.; Zhang, X.; Pang, X.; Wei, C.; et al. Interactions between gut microbiota, host genetics and diet relevant to development of me-tabolic syndromes in mice. ISME J. 2010, 4, 232–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Y.; Liu, W.; Tao, H.; Zhang, Y.; Liu, L.; Liu, Z.; Qiu, B.; Xu, T. Effect of industrial trans-fatty acids-enriched diet on gut microbiota of C57BL/6 mice. Eur. J. Nutr. 2019, 58, 2625–2638. [Google Scholar] [CrossRef]
- Kim, Y.S.; Unno, T.; Kim, B.Y.; Park, M.S. Sex Differences in Gut Microbiota. World J. Men’s Health 2020, 38, 48–60. [Google Scholar] [CrossRef]
- Francesco, V.; Kristina, E. How biological sex of the host shapes its gut microbiota. Science 2013, 339, 1084–1088. [Google Scholar]
- Markle, J.G.M.; Frank, D.N.; Mortin-Toth, S.; Robertson, C.E.; Feazel, L.M.; Rolle-Kampczyk, U.; von Bergen, M.; Mccoy, K.D.; Macpherson, A.J.; Danska, J.S. Sex Differences in the Gut Microbiome Drive Hormone-Dependent Regulation of Autoimmunity. Science 2013, 339, 1084–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Org, E.; Mehrabian, M.; Parks, B.W.; Shipkova, P.; Liu, X.; Drake, T.A. Sex differences and hormonal effects on gut microbiota composition in mice. Gut Microbes 2016, 7, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Kers, J.G.; Velkers, F.C.; Fischer, E.A.J.; Hermes, G.D.A.; Stegeman, J.A.; Smidt, H. Host and environmental factors affecting the intestinal microbiota in chickens. Front. Microbiol. 2018, 9, 235. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Ma, T.; Tang, W.; Li, D.; Mishra, S.K.; Xu, Z.; Wang, Q.; Jie, H. Gut Microbiome of Chinese Forest Musk Deer Examined across Gender and Age. BioMed Res. Int. 2019, 2019, 9291216. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.Y. Nutrition research status of wildlife in captivity. Chin. J. Wildl. 2011, 6, 55–58. [Google Scholar]
- Murray, M.H.; Becker, D.J.; Hall, R.J.; Hernandez, S.M. Wildlife health and supplemental feeding: A review and management recommendations. Biol. Conserv. 2016, 204, 163–174. [Google Scholar] [CrossRef]
- Wei, L.; Zhou, W.; Zhu, Z. Comparison of Changes in Gut Microbiota in Wild Boars and Domestic Pigs Using 16S rRNA Gene and Metagenomics Sequencing Technologies. Animals 2022, 12, 2270. [Google Scholar] [CrossRef]
- Zhong, L.; Zhi, X.; Sun, Y.; Liu, X.; Sun, B.; Zhou, S.; Zhang, W.; Zhang, M. Winter for aging of sympatric red deer and sika deer in northeast China: Diet composition, forage selection, bite diameter and browse intensity. J. For. Res. 2020, 25, 276–284. [Google Scholar] [CrossRef]
- Li, B.; Bai, S.Y.; Xu, Y.C.; Zhang, W.; Ma, J.Z. Identification of sika deer and red deer using partial cytochrome b and 12s ribosomal rna genes. J. For. Res. 2006, 17, 160–162. [Google Scholar] [CrossRef]
- Kuehn, R.; Schroeder, W.; Pirchner, F.; Rottmann, O. Genetic diversity, gene flow and drift in bavarian red deer populations (cervus elaphus). Conserv. Genet. 2003, 4, 157–166. [Google Scholar] [CrossRef]
- Riedemann, N.C.; Guo, R.F.; Sarma, V.J.; Laudes, I.J.; Huber-Lang, M.; Warner, R.L.; Albrecht, E.A.; Speyer, C.L.; Ward, P.A. Expression and function of the c5a receptor in rat alveolar epithelial cells. J. Immunol. 2002, 168, 1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.M.; Zhang, M.H. Population size and sex ratio of wapiti (Cervus elephusxanthopygus) as revealed by fecal DNA. Acta Ecol. 2010, 30, 6249–6254. [Google Scholar]
- Bellemain, E.; Swenson, J.E.; Tallmon, D.; Brunberg, S.; Taberlet, P. Estimating population size of elusive animals with DNA from hunter-collected feces: Four methods for brown bears. Conserv. Biol. 2005, 19, 150–161. [Google Scholar] [CrossRef]
- Tian, X.M.; Zhang, M.H.; Zhang, H.; Yang, C.W.; Jin, Z.M. Genetic diversity of wapiti population in easterWandashan Mountains of Heilongjiang Province., China based on microsatellite analysis. Chin. J. Ecol. 2010, 29, 543–548. [Google Scholar]
- Park, S.D.E. Trypanotolerance in West African Cattle and the Population Genetic Effects of Selection; University of Dublin: Dublin, Ireland, 2001. [Google Scholar]
- Kamler, J.F.; Jędrzejewska, B.; Jędrzejewski, W. Factors affecting daily ranges of red deer Cervus elaphus in Białowieża Primeval Forest, Poland. Acta Theriol. 2007, 52, 113–118. [Google Scholar] [CrossRef]
- Huber, S.; Bruns, U.; Arnold, W. Sex determination of red deer using polymerase chain reaction of DNA from feces. Wildl. Soc. Bull. 2002, 30, 208–212. [Google Scholar]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.; Al-Ghalith, G.A.; Caporaso, J.G. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Prepr. 2018, 6, e27295v2. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. Dada2: High-resolution sample inference from illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Kaehler, B.D.; Ram, R.J.; Matthew, D.; Evan, B.; Rob, K. Optimizing taxonomic classification of marker-gene amplicon sequences with qiime 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Chao, A. Non-parametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Pielou, E.C. The measurement of diversity in different types of biological collections. J. Theor. Biol. 1966, 13, 131–144. [Google Scholar] [CrossRef]
- Gruninger, R.J. Diversity of rumen bacteria in Canadian cervids. PLoS ONE 2014, 9, e89682. [Google Scholar] [CrossRef]
- Li, J. The gut bacterial community composition of wild Cervus albirostris (white-lipped deer) detected by the 16S ribosomal RNA gene sequencing. Curr. Microbiol. 2017, 74, 1100–1107. [Google Scholar] [CrossRef]
- Jewell, K.A.; McCormick, C.A.; Odt, C.L.; Weimer, P.J.; Suen, G. Ruminal bacterial community composition in dairy cows is dynamic over the course of two lactations and correlates with feed efficiency. Appl. Environ. Microbiol. 2015, 81, 4697–4710. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Yang, H.T.; Han, S.Y. Comparison of the gut microbiota composition between wild and captive sika deer (Cervus nipponhortulorum) from feces by high-throughput sequencing. AMB Express. 2017, 7, 212. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Y.; Cao, P.H.; Wang, L.; Zhao, Z.Y.; Chen, Y.; Yang, Y. Bacterial community diversity associated with different levels of dietary nutrition in the rumen of sheep. Appl. Microbiol. Biotechnol. 2017, 101, 3717–3728. [Google Scholar] [CrossRef]
- Maslowski, K.M.; Mackay, C.R. Diet, gut microbiota and immune responses. Nat. Immunol. 2011, 1, 5–9. [Google Scholar] [CrossRef]
- Chi, X.; Gao, H.; Wu, G.; Qin, W.; Song, P.; Wang, L.; Chen, J.; Cai, Z.; Zhang, T. Comparison of gut microbiota diversity betweenwild and captive bharals (Pseudoisnayaur). BMC Vet. Res. 2019, 15, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Chi, X.; Qin, W.; Wang, L.; Song, P.; Cai, Z.; Zhang, J.; Zhang, T. Comparison of the gut microbiota composition betweenthe wild and captive Tibetan wild ass (Equus kiang). J. Appl. Microbiol. 2019, 126, 1869–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Chi, X.; Li, G.; Qin, W.; Song, P.; Jiang, F.; Liu, D.; Zhang, J.; Zhou, X.; Li, S. Gut microbial diversity andstabilizingfunctions enhance the plateau adaptability of Tibetan wild ass (Equus kiang). MicrobiologyOpen 2020, 9, 1150–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, G.; Xia, T.; Wei, Q.; Dong, Y.; Zhao, C.; Yang, X.; Zhang, L.; Wang, X.; Sha, W.; Zhang, H. Analysis of gut microbiotain three species belonging to different genera (Hemitragus, Pseudois, and Ovis) from the subfamily Caprinae in the absence of environmental variance. Ecol. Evol. 2021, 11, 12129–12140. [Google Scholar] [CrossRef]
- Shang, Z.; Kong, Q.; Li, J.; Liu, S.; Tan, Z.; Shang, P.; Wang, H. Characterization of Bacterial Microbial Diversity in Wild Yak and Domestic Yak in Qiangtang Region of Tibet. Pak. J. Zool. 2021, 54, 1001–1009. [Google Scholar]
- Zhang, M.; Shi, M.; Fan, M.; Xu, S.; Li, Y.; Zhang, T.; Cha, M.; Liu, Y.; Guo, X.; Chen, Q. Comparative analysis of gutmicrobiota changes in Père David’s deer populations in Beijing Milu Park and Shishou, Hubei Province in China. Front. Microbiol. 2018, 9, 1258. [Google Scholar] [CrossRef]
- Menke, S.; Heurich, M.; Henrich, M.; Wilhelm, K.; Sommer, S. Impact of winter enclosures on the gut bacterial microbiota of red deer in the Bavarian Forest National Park. Wildl. Biol. 2019, 2019, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Mooring, M.S.; Fitzpatrick, T.A.; Benjamin, J.E.; Fraser, I.C.; Nishihira, T.T.; Reisig, D.D.; Rominger, E.M. Sexual segregation in desert bighorn sheep (Ovis candensismexicana). Behaviour 2003, 140, 183–207. [Google Scholar]
- Bonenfant, C.; Gaillard, J.M.; Klein, F. Variation in harem size of red deer (Cervus elaphus L.): The effects of adult sex ratio and age-structure. J. Zool. 2004, 264, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Antwis, R.E.; Edwards, K.L.; Unwin, B.; Walker, S.L.; Shultz, S. Rare gut microbiota associated with breeding success, hormone metabolites and ovarian cycle phase in the critically endangered eastern black rhino. Microbiome 2019, 7, 1–12. [Google Scholar] [CrossRef]
- Dominianni, C.; Sinha, R.; James, J. Goedert, Sex, Body Mass Index, and Dietary Fiber Intake Infuence the Human Gut Microbiome. PLoS ONE 2015, 10, e0124599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, R.; Shi, J.; Fuhrman, B.; Xu, X.; Veenstra, T.D.; Gail, M.H.; Gajer, P.; Ravel, J.; Goedert, J.J. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolites: A cross-sectional study. J. Transl. Med. 2012, 10, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, S.M.; Cunningham, S.A.; Dunlop, A.L.; Corwin, E.J. The Maternal Gut Microbiome During Pregnancy. MCN Am. J. Matern. Child. Nurs. 2017, 42, 310–317. [Google Scholar] [CrossRef]
- Phillips, C.D.; John, H.; Wilkinson, J.E.; Koenig, L.; Rees, E.; Webala, P. Microbiome structural and functional interactions across host dietary niche space. Integr. Comp. Biol. 2017, 57, 743–755. [Google Scholar] [CrossRef] [Green Version]
- Dai, Z.L.; Wu, G.; Zhu, W.Y. Amino acid metabolism in intestinal bacteria: Links between gut ecology and host health. Front. Biosci. 2011, 16, 1768–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapira, M. Gut microbiotas and host evolution: Scaling up symbiosis. Trends Ecol. Evol. 2016, 31, 539–549. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Fndriks, L. Roles of the gut in the metabolic syndrome: An overview. J. Int. Med. 2017, 81, 319–336. [Google Scholar] [CrossRef]
- Dill-Mcfarland, K.A.; Weimer, P.J.; Pauli, J.N.; Peery, M.Z.; Suen, G. Diet specialization selects for an unusual and simplified gut microbiota in two and three-toed sloths. Environ. Microbiol. 2015, 18, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Bergman, E.N. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 1990, 70, 567–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Z.; Zhang, W.; Wang, L.; Hou, R.; Zhang, M.; Fei, L. The bamboo-eating giant panda harbors a carnivore-like gut microbiota, with excessive seasonal variations. mBio 2015, 6, e00022-15. [Google Scholar] [CrossRef] [Green Version]
- Borbón-García, A.; Reyes, A.; Vives-Flórez, M.; Caballero, S. Captivity Shapes the Gut Microbiota of Andean Bears: Insights into Health Surveillance. Front. Microbiol. 2017, 8, 1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tax_Name | WF | WM | CF | CM |
|---|---|---|---|---|
| Oscillospira | 4.71 ± 0.79 | 4.55 ± 0.73 | 4.55 ± 0.41 | 2.89 ± 0.31 |
| 5-7N15 | 3.81 ± 1.4 | 4.51 ± 1.53 | 4.51 ± 1.71 | 4.01 ± 1.29 |
| Ruminococcus | 3.05 ± 0.5 | 3.1 ± 0.56 | 3.1 ± 0.37 | 2.03 ± 0.6 |
| Roseburia | 2.27 ± 1.11 | 2.17 ± 1.08 | 2.17 ± 0.63 | 0.37 ± 0.35 |
| Clostridium | 1.55 ± 0.59 | 1.67 ± 1.01 | 1.67 ± 0.67 | 0.88 ± 0.68 |
| Prevotella | 1.32 ± 0.11 | 1.25 ± 0.12 | 1.25 ± 0.08 | 1.31 ± 0.31 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Yu, Y.; Guo, J.; Zhong, L.; Zhang, M. Alterations in Fecal Microbiota Linked to Environment and Sex in Red Deer (Cervus elaphus). Animals 2023, 13, 929. https://doi.org/10.3390/ani13050929
Sun Y, Yu Y, Guo J, Zhong L, Zhang M. Alterations in Fecal Microbiota Linked to Environment and Sex in Red Deer (Cervus elaphus). Animals. 2023; 13(5):929. https://doi.org/10.3390/ani13050929
Chicago/Turabian StyleSun, Yue, Yanze Yu, Jinhao Guo, Linqiang Zhong, and Minghai Zhang. 2023. "Alterations in Fecal Microbiota Linked to Environment and Sex in Red Deer (Cervus elaphus)" Animals 13, no. 5: 929. https://doi.org/10.3390/ani13050929