
Appraising the Genetic Makeup of an Allochthonous Southern Pike Population: An Opportunity to Predict the Evolution of Introgressive Hybridization in Isolated Populations?

 , ,
, ,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
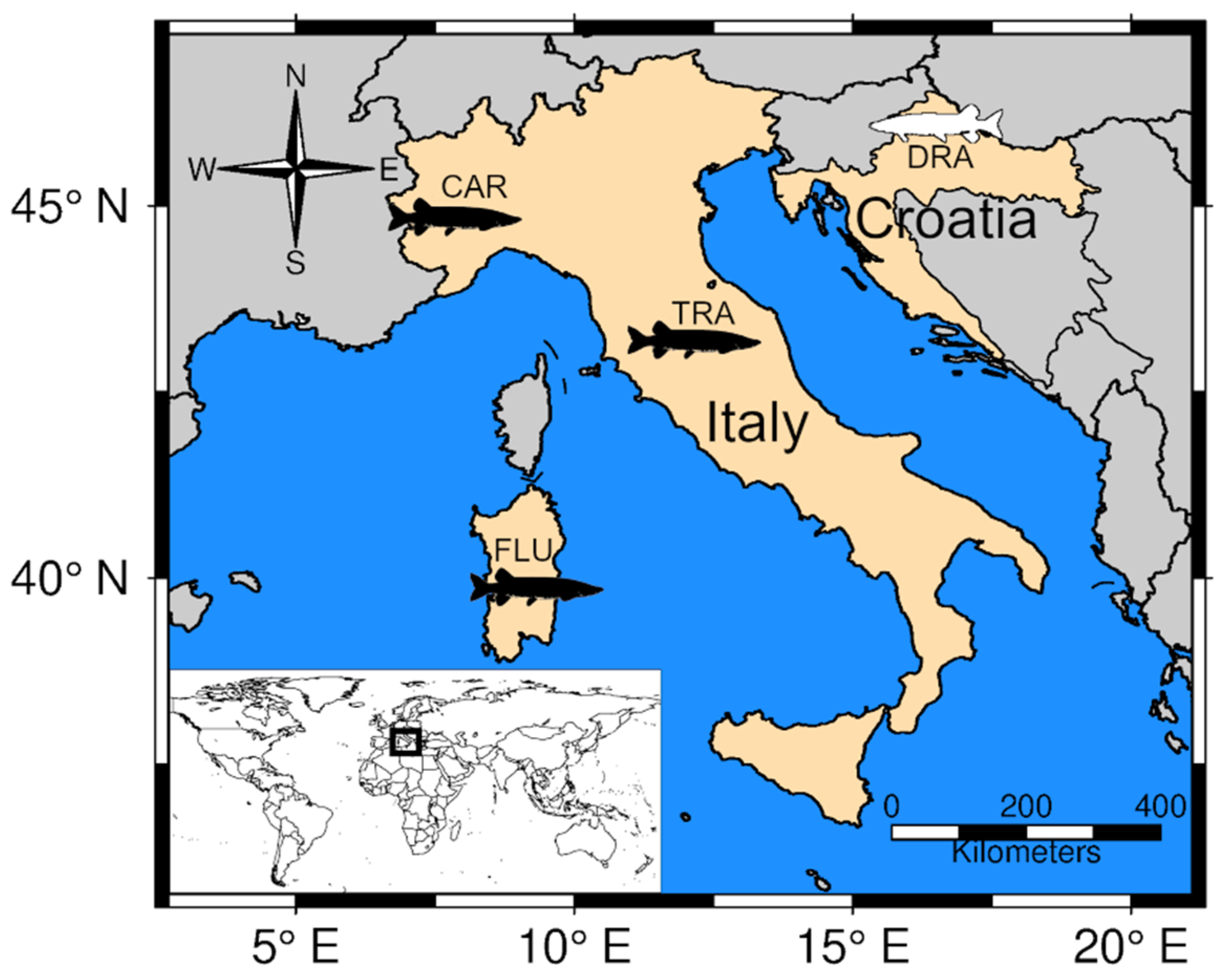
2.1. Study Area
2.2. Sampling, DNA Extraction, and PCR Protocols
2.3. Marker Validation
2.4. Patterns of Genetic Variation
2.5. Hybrid Detection
3. Results
3.1. Marker Validation
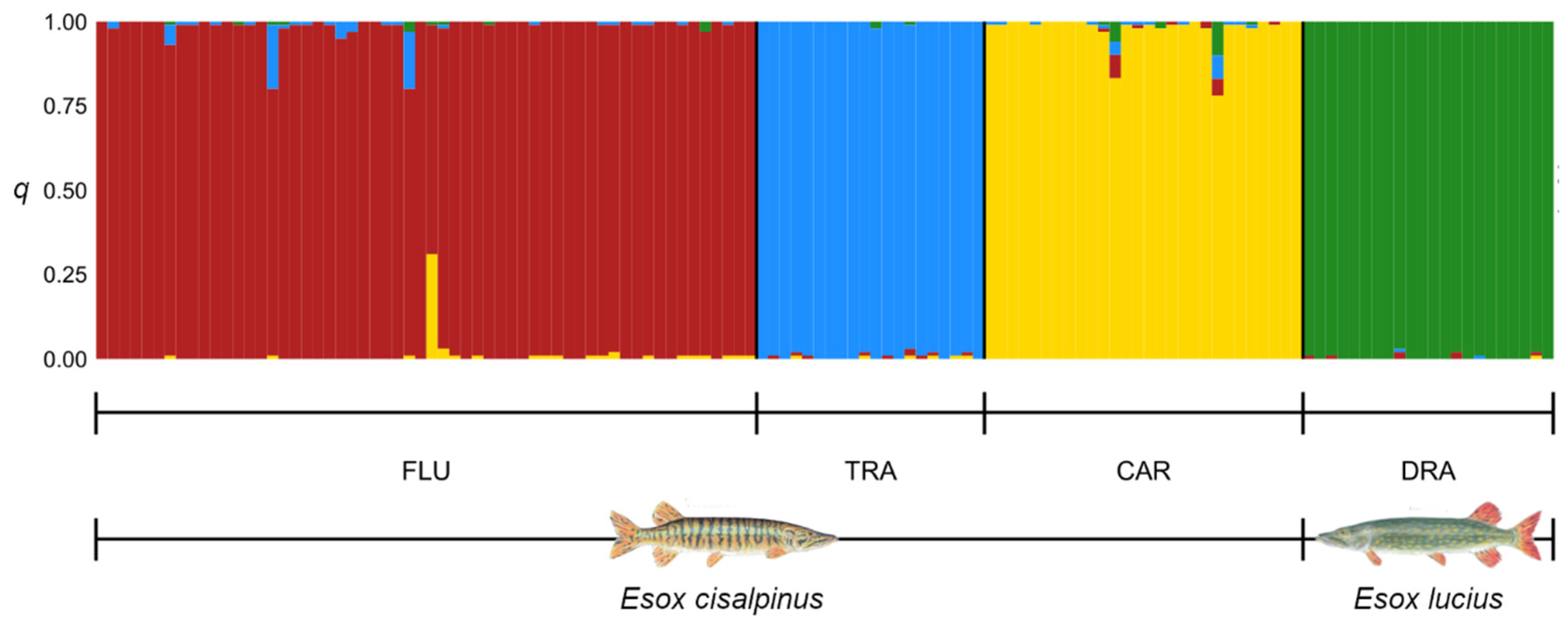
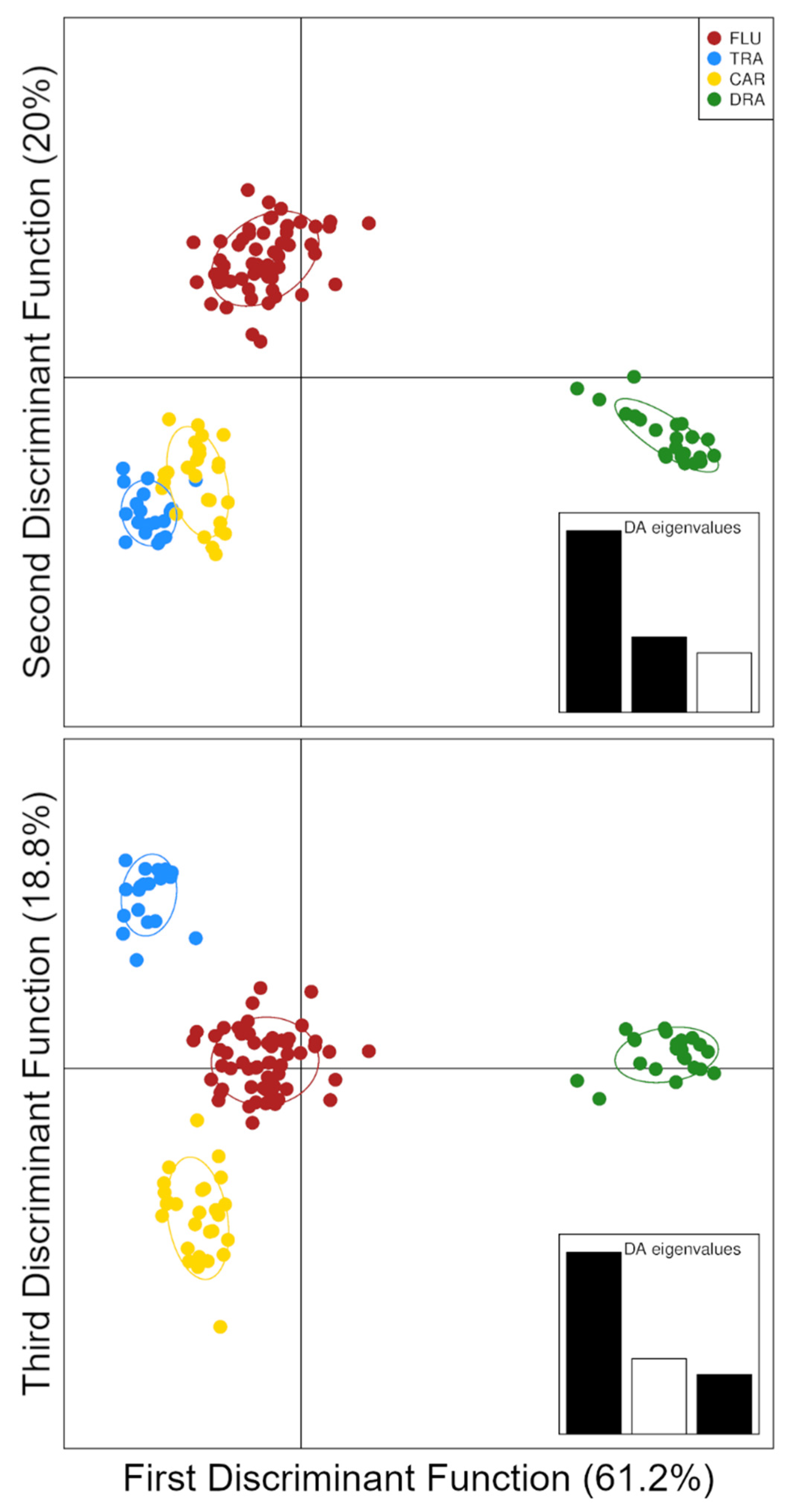
3.2. Patterns of Genetic Variation
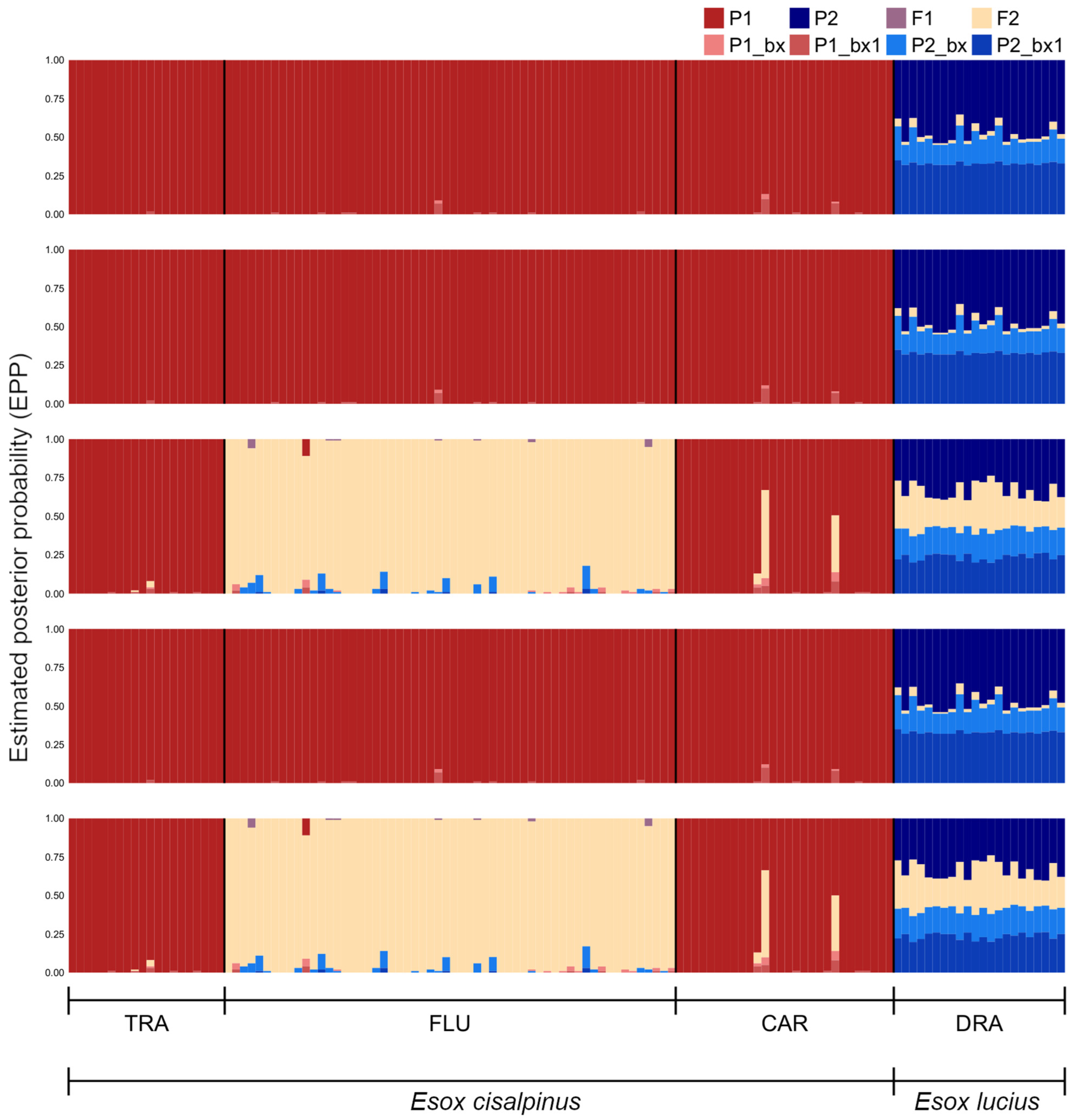
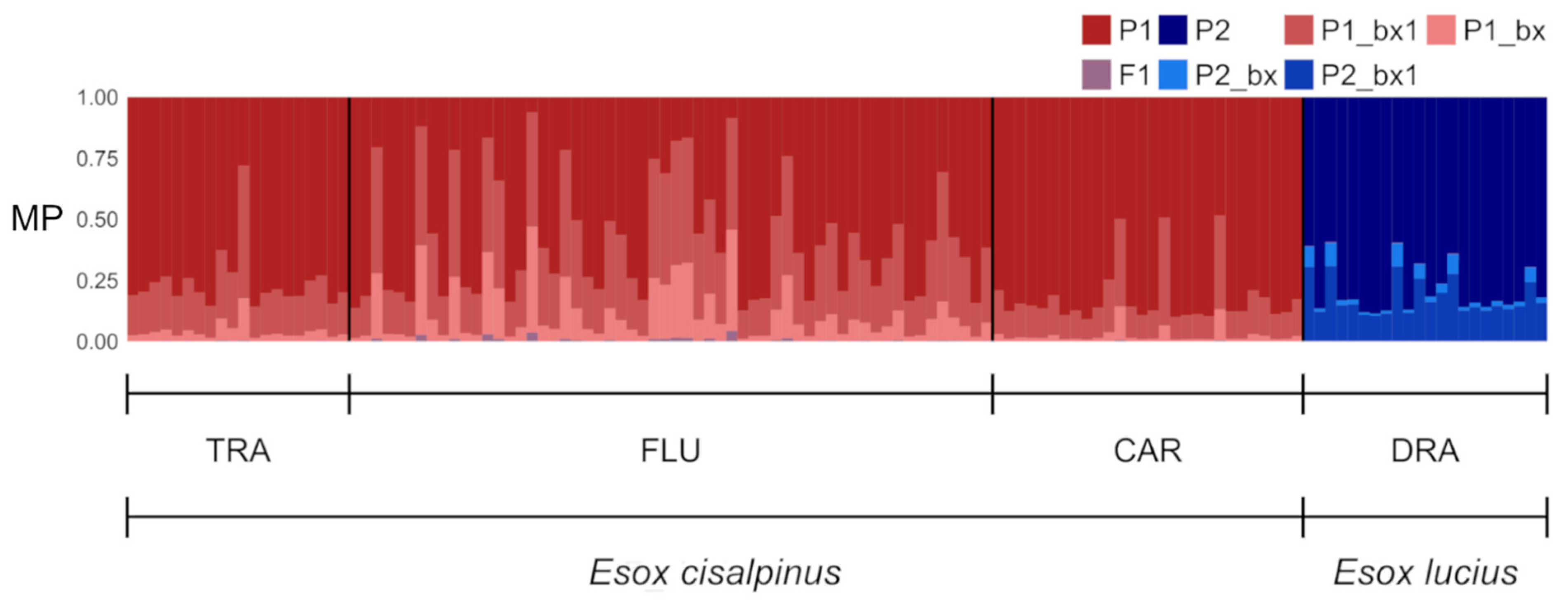
3.3. Hybrid Detection
4. Discussion
4.1. Marker Validation
4.2. Overall Patterns of Genetic Diversity
4.3. Hybrid Detection
4.4. Genetic Makeup of the Alto Flumendosa Lake Population
4.5. Fate of an Introduced Population: Should the Pike Go or Should They Stay?
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Simberloff, D.; Martin, J.-L.; Genovesi, P.; Maris, V.; Wardle, D.A.; Aronson, J.; Courchamp, F.; Galil, B.; García-Berthou, E.; Pascal, M.; et al. Impacts of Biological Invasions: What’s What and the Way Forward. Trends Ecol. Evol. 2013, 28, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, B.T.; Irvine, R.L.; Howald, G.R.; Russello, M.A. The Promise of Genetics and Genomics for Improving Invasive Mammal Management on Islands. Front. Ecol. Evol. 2021, 9, 704809. [Google Scholar] [CrossRef]
- Aguilar, A.; Banks, J.D.; Levine, K.F.; Wayne, R.K. Population Genetics of Northern Pike (Esox Lucius) Introduced into Lake Davis, California. Can. J. Fish. Aquat. Sci. 2005, 62, 1589–1599. [Google Scholar] [CrossRef]
- Dörr, A.J.M.; Scoparo, M.; Cardinali, I.; La Porta, G.; Caldaroni, B.; Magara, G.; Pallottini, M.; Selvaggi, R.; Cenci-Goga, B.; Goretti, E.; et al. Population Ecology and Genetic Diversity of the Invasive Alien Species Procambarus clarkii in Lake Trasimeno (Italy). Biology 2021, 10, 1059. [Google Scholar] [CrossRef]
- Sanna, D.; Azzena, I.; Scarpa, F.; Cossu, P.; Pira, A.; Gagliardi, F.; Casu, M. First Record of the Alien Species Procambarus Virginalis Lyko, 2017 in Fresh Waters of Sardinia and Insight into Its Genetic Variability. Life 2021, 11, 606. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.J.; Duarte, G.; Segurado, P.; Branco, P. Major Threats to European Freshwater Fish Species. Sci. Total Environ. 2021, 797, 149105. [Google Scholar] [CrossRef]
- García-Berthou, E.; Alcaraz, C.; Pou-Rovira, Q.; Zamora, L.; Coenders, G.; Feo, C. Introduction Pathways and Establishment Rates of Invasive Aquatic Species in Europe. Can. J. Fish. Aquat. Sci. 2005, 62, 453–463. [Google Scholar] [CrossRef]
- Splendiani, A.; Ruggeri, P.; Giovannotti, M.; Caputo Barucchi, V. Role of Environmental Factors in the Spread of Domestic Trout in Mediterranean Streams. Freshw. Biol. 2013, 58, 2089–2101. [Google Scholar] [CrossRef]
- Larsen, P.F.; Hansen, M.M.; Nielsen, E.E.; Jensen, L.F.; Loeschcke, V. Stocking Impact and Temporal Stability of Genetic Composition in a Brackish Northern Pike Population (Esox Lucius L.), Assessed Using Microsatellite DNA Analysis of Historical and Contemporary Samples. Heredity 2005, 95, 136–143. [Google Scholar] [CrossRef] [Green Version]
- Grant, W.S.; Jasper, J.; Bekkevold, D.; Adkison, M. Responsible Genetic Approach to Stock Restoration, Sea Ranching and Stock Enhancement of Marine Fishes and Invertebrates. Rev. Fish Biol. Fish. 2017, 27, 615–649. [Google Scholar] [CrossRef]
- Meraner, A.; Venturi, A.; Ficetola, G.F.; Rossi, S.; Candiotto, A.; Gandolfi, A. Massive Invasion of Exotic Barbus Barbus and Introgressive Hybridization with Endemic Barbus Plebejus in Northern Italy: Where, How and Why? Mol. Ecol. 2013, 22, 5295–5312. [Google Scholar] [CrossRef] [PubMed]
- Ketmaier, V.; Bianco, P. Understanding and Conserving Genetic Diversity in a World Dominated by Alien Introductions and Native Transfers: The Case Study of Primary and Peripheral Freshwater Fishes in Southern Europe. In Conservation of Freshwater Fishes; Closs, P., Krkosek, M., Olden, J.D., Eds.; Cambridge University Press: Cambridge, UK, 2015; pp. 506–534. [Google Scholar]
- Carosi, A.; Ghetti, L.; Padula, R.; Lorenzoni, M. Population Status and Ecology of the Salmo Trutta Complex in an Italian River Basin under Multiple Anthropogenic Pressures. Ecol. Evol. 2020, 10, 7320–7333. [Google Scholar] [CrossRef] [PubMed]
- Bianco, P.G.; Delmastro, G.B. Recenti Novità Tassonomiche Riguardanti i Pesci d’acqua Dolce Autoctoni in Italia e Descrizione di Una Nuova Specie di Luccio; IGF Publishing: Warsaw, Poland, 2011. [Google Scholar]
- Lucentini, L.; Puletti, M.E.; Ricciolini, C.; Gigliarelli, L.; Fontaneto, D.; Lanfaloni, L.; Bilò, F.; Natali, M.; Panara, F. Molecular and Phenotypic Evidence of a New Species of Genus Esox (Esocidae, Esociformes, Actinopterygii): The Southern Pike, Esox Flaviae. PLoS ONE 2011, 6, e25218. [Google Scholar] [CrossRef]
- Lucentini, L.; Palomba, A.; Lancioni, H.; Gigliarelli, L.; Natali, M.; Panara, F. Microsatellite Polymorphism in Italian Populations of Northern Pike (Esox Lucius L.). Fish. Res. 2006, 80, 251–262. [Google Scholar] [CrossRef]
- Lucentini, L.; Palomba, A.; Gigliarelli, L.; Sgaravizzi, G.; Lancioni, H.; Lanfaloni, L.; Natali, M.; Panara, F. Temporal Changes and Effective Population Size of an Italian Isolated and Supportive-Breeding Managed Northern Pike (Esox Lucius) Population. Fish. Res. 2009, 96, 139–147. [Google Scholar] [CrossRef]
- Gandolfi, A.; Ferrari, C.; Crestanello, B.; Girardi, M.; Lucentini, L.; Meraner, A. Population Genetics of Pike, Genus Esox (Actinopterygii, Esocidae), in Northern Italy: Evidence for Mosaic Distribution of Native, Exotic and Introgressed Populations. Hydrobiologia 2017, 794, 73–92. [Google Scholar] [CrossRef]
- Bekkevold, D.; Jacobsen, L.; Hemmer-Hansen, J.; Berg, S.; Skov, C. From Regionally Predictable to Locally Complex Population Structure in a Freshwater Top Predator: River Systems Are Not Always the Unit of Connectivity in Northern Pike Esox Lucius. Ecol. Freshw. Fish. 2015, 24, 305–316. [Google Scholar] [CrossRef]
- Nicod, J.-C.; Wang, Y.Z.; Excoffier, L.; Largiader, C.R. Low Levels of Mitochondrial DNA Variation among Central and Southern European Esox Lucius Populations. J. Fish Biol. 2004, 64, 1442–1449. [Google Scholar] [CrossRef]
- Eschbach, E.; Nolte, A.W.; Kohlmann, K.; Alós, J.; Schöning, S.; Arlinghaus, R. Genetic Population Structure of a Top Predatory Fish (Northern Pike, Esox Lucius) Covaries with Anthropogenic Alteration of Freshwater Ecosystems. Freshw. Biol. 2021, 66, 884–901. [Google Scholar] [CrossRef]
- La Porta, G.; Angeli, V.; Bicchi, A.; Carosi, A.; Pedicillo, G.; Viali, P.; Lorenzoni, M. Variations in the Fish Community in Lake Piediluco (Italy) Caused by Changes in the Lake’s Trophic Status and the Introduction of Alien Species: Fish Community in Lake Piediluco. J. Appl. Ichthyol. 2010, 26, 53–59. [Google Scholar] [CrossRef]
- Quilodrán, C.S.; Austerlitz, F.; Currat, M.; Montoya-Burgos, J.I. Cryptic Biological Invasions: A General Model of Hybridization. Sci. Rep. 2018, 8, 2414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandolfi, A.; Fontaneto, D.; Natali, M.; Lucentini, L. Mitochondrial genome of Esox flaviae (Southern pike): Announcement and comparison with other Esocidae. Mitochondrial DNA Part A 2015, 27, 3037–3038. [Google Scholar] [CrossRef] [PubMed]
- Mann, R.H.K. Pike: Biology and exploitation. In Fish and Fisheries; Craig, J.F., Ed.; Chapman & Hall: London, UK, 1996; pp. 219–241. [Google Scholar]
- Bianco, P.G. An Update on the Status of Native and Exotic Freshwater Fishes of Italy. J. Appl. Ichthyol. 2014, 30, 62–77. [Google Scholar] [CrossRef]
- Casu, M.; Scarpa, F.; Cossu, P.; Lai, T.; Curini-Galletti, M.; Varcasia, A.; Sanna, D. First Record of Esox Cisalpinus (Teleostea: Esocidae) in Sardinia with Insight on Its Mitochondrial DNA Genetic Variability. Ital. J. Zool. 2016, 83, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Orrù, F.; Deiana, A.M.; Cau, A. Introduction and Distribution of Alien Freshwater Fishes on the Island of Sardinia (Italy): An Assessment on the Basis of Existing Data Sources: Alien Freshwater Fishes in Sardinia. J. Appl. Ichthyol. 2010, 26, 46–52. [Google Scholar] [CrossRef]
- Orrù, F.; Niffoi, A.; Sanna, D.; Varcasia, A.; Buscarinu, P.; Casu, M. Prima segnalazione di Leucos aula (Teleostei: Cyprinidae) in Sardegna basata su analisi morfologiche e DNA barcoding. Italy J. Freshw. Ichthyol. 2019, 5, 166–172. [Google Scholar]
- Azzena, I.; Scarpa, F.; Locci, C.; Cossu, P.; Niffoi, A.; Orrù, F.; Bovero, S.; Sotgiu, G.; Sanna, D.; Casu, M. Mitochondrial DNA of Sardinian and North-West Italian Populations Revealed a New Piece in the Mosaic of Phylogeography and Phylogeny of Salariopsis Fluviatilis (Blenniidae). Animals 2022, 12, 3403. [Google Scholar] [CrossRef]
- Cottiglia, M. La Distribuzione Dell’ittiofauna Dulciacquicola in Sardegna. Riv. Idrobiol. 1968, 7, 63–116. [Google Scholar]
- Toonen, R.J.; Hughes, S. Increased Throughput for Fragment Analysis on an ABI PRISM 377 Automated Sequencer Using a Membrane Comb and STRand Software. BioTechniques 2001, 31, 1320–1324. [Google Scholar]
- Waples, R.S. Testing for Hardy–Weinberg Proportions: Have We Lost the Plot? J. Hered. 2015, 106, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Rousset, F. Genepop’007: A Complete Re-Implementation of the Genepop Software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Yekutieli, D. The Control of the False Discovery Rate in Multiple Testing under Dependency. Ann. Stat. 2001, 29, 25. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Cossu, P.; Scarpa, F.; Sanna, D.; Lai, T.; Dedola, G.L.; Curini-Galletti, M.; Mura, L.; Fois, N.; Casu, M. Influence of Genetic Drift on Patterns of Genetic Variation: The Footprint of Aquaculture Practices in Sparus Aurata (Teleostei: Sparidae). Mol. Ecol. 2019, 28, 3012–3024. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Schroeder, K.B.; Rosenberg, N.A. A Maximum-Likelihood Method to Correct for Allelic Dropout in Microsatellite Data with No Replicate Genotypes. Genetics 2012, 192, 651–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foll, M.; Gaggiotti, O. A Genome-Scan Method to Identify Selected Loci Appropriate for Both Dominant and Codominant Markers: A Bayesian Perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Excoffier, L.; Lischer, H.E.L. Arlequin Suite Ver 3.5: A New Series of Programs to Perform Population Genetics Analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, M.A.; Nichols, R.A. Evaluating Loci for Use in the Genetic Analysis of Population Structure. Proceedings of the Royal Society of London. Proc. R. Soc. B Biol. Sci. 1996, 263, 1619–1626. [Google Scholar] [CrossRef]
- Paris, M.; Boyer, S.; Bonin, A.; Collado, A.; David, J.-P.; Despres, L. Genome Scan in the Mosquito Aedes Rusticus: Population Structure and Detection of Positive Selection after Insecticide Treatment: Population Structure at a Regional Scale. Mol. Ecol. 2010, 19, 325–337. [Google Scholar] [CrossRef]
- Keenan, K.; McGinnity, P.; Cross, T.F.; Crozier, W.W.; Prodöhl, P.A. DiveRsity: An R Package for the Estimation and Exploration of Population Genetics Parameters and Their Associated Errors. Methods Ecol. Evol. 2013, 4, 782–788. [Google Scholar] [CrossRef] [Green Version]
- Hill, W.G. Estimation of Effective Population Size from Data on Linkage Disequilibrium. Genet. Res. 1981, 38, 209–216. [Google Scholar] [CrossRef] [Green Version]
- Waples, R.S. A Bias Correction for Estimates of Effective Population Size Based on Linkage Disequilibrium at Unlinked Gene Loci*. Conserv. Genet 2006, 7, 167–184. [Google Scholar] [CrossRef] [Green Version]
- Waples, R.S.; Do, C. Linkage Disequilibrium Estimates of Contemporary Ne Using Highly Variable Genetic Markers: A Largely Untapped Resource for Applied Conservation and Evolution. Evol. Appl. 2010, 3, 244–262. [Google Scholar] [CrossRef] [PubMed]
- Do, C.; Waples, R.S.; Peel, D.; Macbeth, G.M.; Tillett, B.J.; Ovenden, J.R. NeEstimatorv2: Re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 2013, 14, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Jost, L. GST and Its Relatives Do Not Measure Differentiation. Mol. Ecol. 2008, 17, 4015–4026. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of Population Structure Using Multilocus Genotype Data: Linked Loci and Correlated Allele Frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the Number of Clusters of Individuals Using the Software STRUCTURE: A Simulation Study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Puechmaille, S.J. The Program Structure Does Not Reliably Recover the Correct Population Structure When Sampling Is Uneven: Subsampling and New Estimators Alleviate the Problem. Mol. Ecol. Resour. 2016, 16, 608–627. [Google Scholar] [CrossRef]
- Li, Y.-L.; Liu, J.-X. StructureSelector: A Web-Based Software to Select and Visualize the Optimal Number of Clusters Using Multiple Methods. Mol. Ecol. Resour. 2018, 18, 176–177. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A Program for Identifying Clustering Modes and Packaging Population Structure Inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, R.M. Pophelper: An R Package and Web App to Analyse and Visualize Population Structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant Analysis of Principal Components: A New Method for the Analysis of Genetically Structured Populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T. Adegenet: A R Package for the Multivariate Analysis of Genetic Markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, E.C.; Thompson, E.A. A Model-Based Method for Identifying Species Hybrids Using Multilocus Genetic Data. Genetics 2002, 160, 1217–1229. [Google Scholar] [CrossRef] [PubMed]
- Beugin, M.-P.; Gayet, T.; Pontier, D.; Devillard, S.; Jombart, T. A Fast Likelihood Solution to the Genetic Clustering Problem. Methods Ecol. Evol. 2018, 9, 1006–1016. [Google Scholar] [CrossRef] [Green Version]
- Cordonnier, M.; Gayet, T.; Escarguel, G.; Kaufmann, B. From Hybridization to Introgression between Two Closely Related Sympatric Ant Species. J. Zool. Syst. Evol. Res. 2019, 57, 778–788. [Google Scholar] [CrossRef]
- Balloux, F. EASYPOP (Version 1.7): A Computer Program for Population Genetics Simulations. J. Hered. 2001, 92, 301–302. [Google Scholar] [CrossRef] [Green Version]
- Miller, L.M.; Kapuscinski, A.R. Historical analysis of genetic variation reveals low effective population size in a northern pike (Esox lucius) population. Genetics 1997, 147, 1249–1258. [Google Scholar] [CrossRef]
- Wang, J.; Santiago, E.; Caballero, A. Prediction and Estimation of Effective Population Size. Heredity 2016, 117, 193–206. [Google Scholar] [CrossRef] [Green Version]
- Russell, T.; Cullingham, C.; Ball, M.; Pybus, M.; Coltman, D. Extent and Direction of Introgressive Hybridization of Mule and White-tailed Deer in Western Canada. Evol. Appl. 2021, 14, 1914–1925. [Google Scholar] [CrossRef] [PubMed]
- Sanz, N.; Araguas, R.M.; Fernández, R.; Vera, M.; García-Marín, J.-L. Efficiency of Markers and Methods for Detecting Hybrids and Introgression in Stocked Populations. Conserv. Genet. 2009, 10, 225–236. [Google Scholar] [CrossRef]
- Vähä, J.-P.; Primmer, C.R. Efficiency of Model-Based Bayesian Methods for Detecting Hybrid Individuals under Different Hybridization Scenarios and with Different Numbers of Loci. Mol. Ecol. 2006, 15, 63–72. [Google Scholar] [CrossRef]
- Rueda, E.C.; Mullaney, K.A.; Conte-Grand, C.; Habit, E.M.; Cussac, V.; Ortí, G. Displacement of Native Patagonian Freshwater Silverside Populations (Odontesthes Hatcheri, Atherinopsidae) by Introgressive Hybridization with Introduced O. Bonariensis. Biol. Invasions 2017, 19, 971–988. [Google Scholar] [CrossRef]
- Skog, A.; Vøllestad, L.A.; Stenseth, N.C.; Kasumyan, A.; Jakobsen, K.S. Circumpolar Phylogeography of the Northern Pike (Esox Lucius) and Its Relationship to the Amur Pike (E. reichertii). Front. Zool. 2014, 11, 67. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.D.; Moyer, G.R. Linkage Disequilibrium and Effective Population Size When Generations Overlap. Evol. Appl. 2013, 6, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Waples, R.S.; Antao, T.; Luikart, G. Effects of Overlapping Generations on Linkage Disequilibrium Estimates of Effective Population Size. Genetics 2014, 197, 769–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waples, R.S.; Hindar, K.; Karlsson, S.; Hard, J.J. Evaluating the Ryman–Laikre Effect for Marine Stock Enhancement and Aquaculture. Curr. Zool. 2016, 62, 617–627. [Google Scholar] [CrossRef]
- Sanna, D.; Merella, P.; Lai, T.; Farjallah, S.; Francalacci, P.; Curini-Galletti, M.; Pais, A.; Casu, M. Combined analysis of four mitochondrial regions allowed the detection of several matrilineal lineages of the lessepsian fish Fistularia commersonii in the Mediterranean Sea. J. Mar. Biolog. Assoc. 2011, 91, 1289–1293. [Google Scholar] [CrossRef]
- Sanna, D.; Scarpa, F.; Lai, T.; Cossu, P.; Falautano, M.; Castriota, L.; Andaloro, F.; Francalacci, P.; Curini-Galletti, M.; Casu, M. Fistularia commersonii: Walking through the Lessepsian paradox of mitochondrial DNA. Ital. J. Zool. 2015, 82, 499–512. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | NA ± SD | AR ± SD | HE ± SD | HO ± SD | FIS (95% CI) | |
|---|---|---|---|---|---|---|
| FLU | 58 | 4.73 ± 1.68 | 3.91 ± 1.18 | 0.580 ± 0.187 | 0.605 ± 0.194 | −0.042 (−0.086, 0.002) |
| TRA | 20 | 3.30 ± 2.36 | 2.90 ± 1.79 | 0.385 ± 0.245 | 0.478 ± 0.311 | −0.217 (−0.346, −0.084) |
| CAR | 28 | 4.18 ± 2.18 | 3.59 ± 1.64 | 0.417 ± 0.205 | 0.435 ± 0.227 | −0.043 (−0.131, 0.034) |
| DRA | 22 | 5.54 ± 3.56 | 4.86 ± 3.04 | 0.481 ± 0.318 | 0.465 ± 0.319 | 0.036 (−0.024, 0.089) |
| Ne˄ LD | |||
|---|---|---|---|
| Harmonic Mean | Lower 95% CI | Upper 95% CI | |
| FLU | 66 (12) | 36 (4) | 189 (48) |
| TRA | 6 | 2 | 23 |
| CAR | 13 (12) | 3 (2) | 56 (∞) |
| DRA | 14 (13) | 6 (3) | 46 (105) |
| FLU | TRA | CAR | DRA | |
|---|---|---|---|---|
| FLU | — | (0.203, 0.254) | (0.210, 0.276) | (0.293, 0.348) |
| TRA | 0.227 | — | (0.322, 0.409) | (0.421, 0.485) |
| CAR | 0.243 | 0.365 | — | (0.440, 0.519) |
| DRA | 0.320 | 0.451 | 0.478 | — |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casu, M.; Azzena, I.; Scarpa, F.; Locci, C.; Niffoi, A.; Delmastro, G.B.; Lo Conte, P.; Varcasia, A.; Bovero, S.; Candiotto, A.; et al. Appraising the Genetic Makeup of an Allochthonous Southern Pike Population: An Opportunity to Predict the Evolution of Introgressive Hybridization in Isolated Populations? Animals 2023, 13, 380. https://doi.org/10.3390/ani13030380
Casu M, Azzena I, Scarpa F, Locci C, Niffoi A, Delmastro GB, Lo Conte P, Varcasia A, Bovero S, Candiotto A, et al. Appraising the Genetic Makeup of an Allochthonous Southern Pike Population: An Opportunity to Predict the Evolution of Introgressive Hybridization in Isolated Populations? Animals. 2023; 13(3):380. https://doi.org/10.3390/ani13030380
Chicago/Turabian StyleCasu, Marco, Ilenia Azzena, Fabio Scarpa, Chiara Locci, Alessio Niffoi, Giovanni Battista Delmastro, Paolo Lo Conte, Antonio Varcasia, Stefano Bovero, Alessandro Candiotto, and et al. 2023. "Appraising the Genetic Makeup of an Allochthonous Southern Pike Population: An Opportunity to Predict the Evolution of Introgressive Hybridization in Isolated Populations?" Animals 13, no. 3: 380. https://doi.org/10.3390/ani13030380