
Transcriptome Profiling of the Liver in Nellore Cattle Phenotypically Divergent for RFI in Two Genetic Groups
 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling Method
2.2. Transcriptome Analyses
2.3. Gene Set Enrichment Analysis
2.4. Non-Coding RNAs Analysis
2.5. Prediction of Footprint Gene Profile
3. Results
3.1. Alignment Statistics
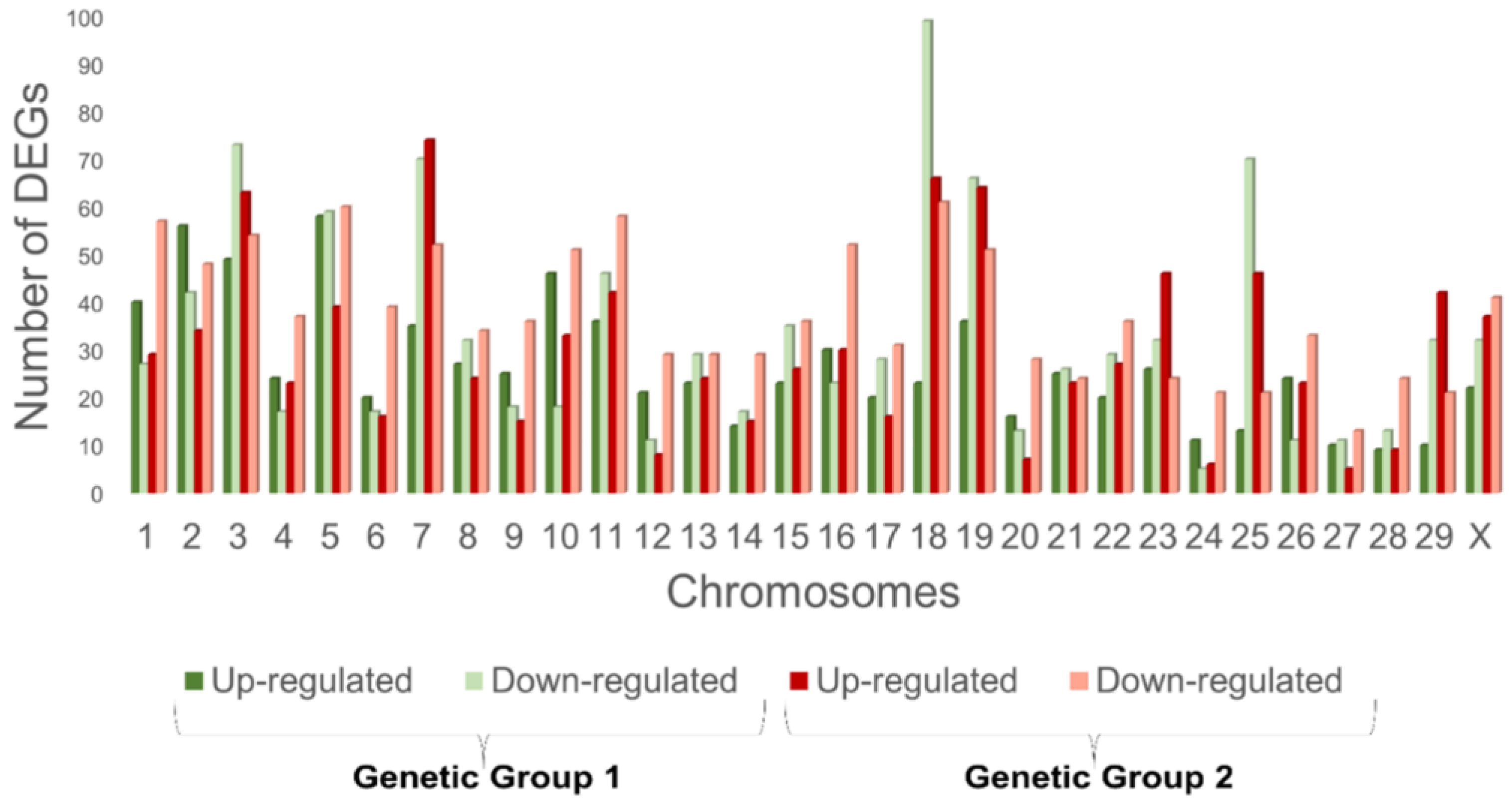
3.2. Differential Expression Analysis of RNA Seq Data
3.3. Biomarkers
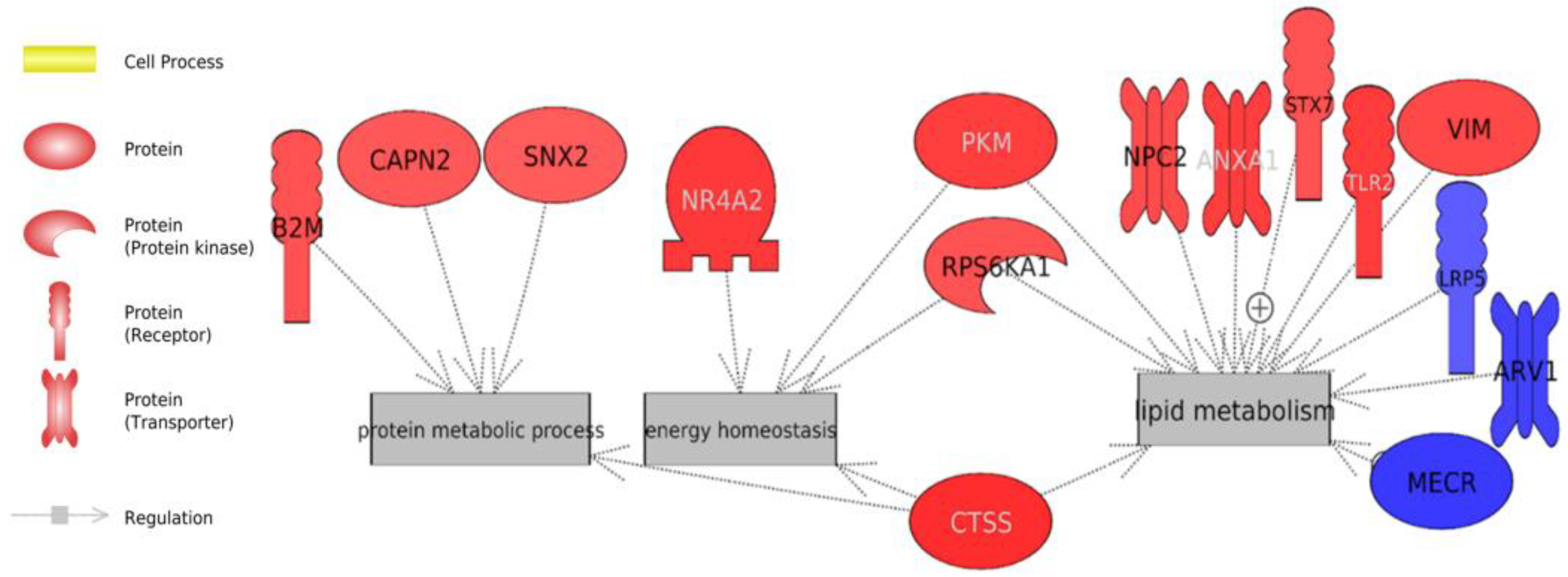
3.4. Gene Set Enrichment Analysis
3.4.1. Biological Analysis of the Whole Transcriptome Data
Energy Metabolism
Protein Turnover
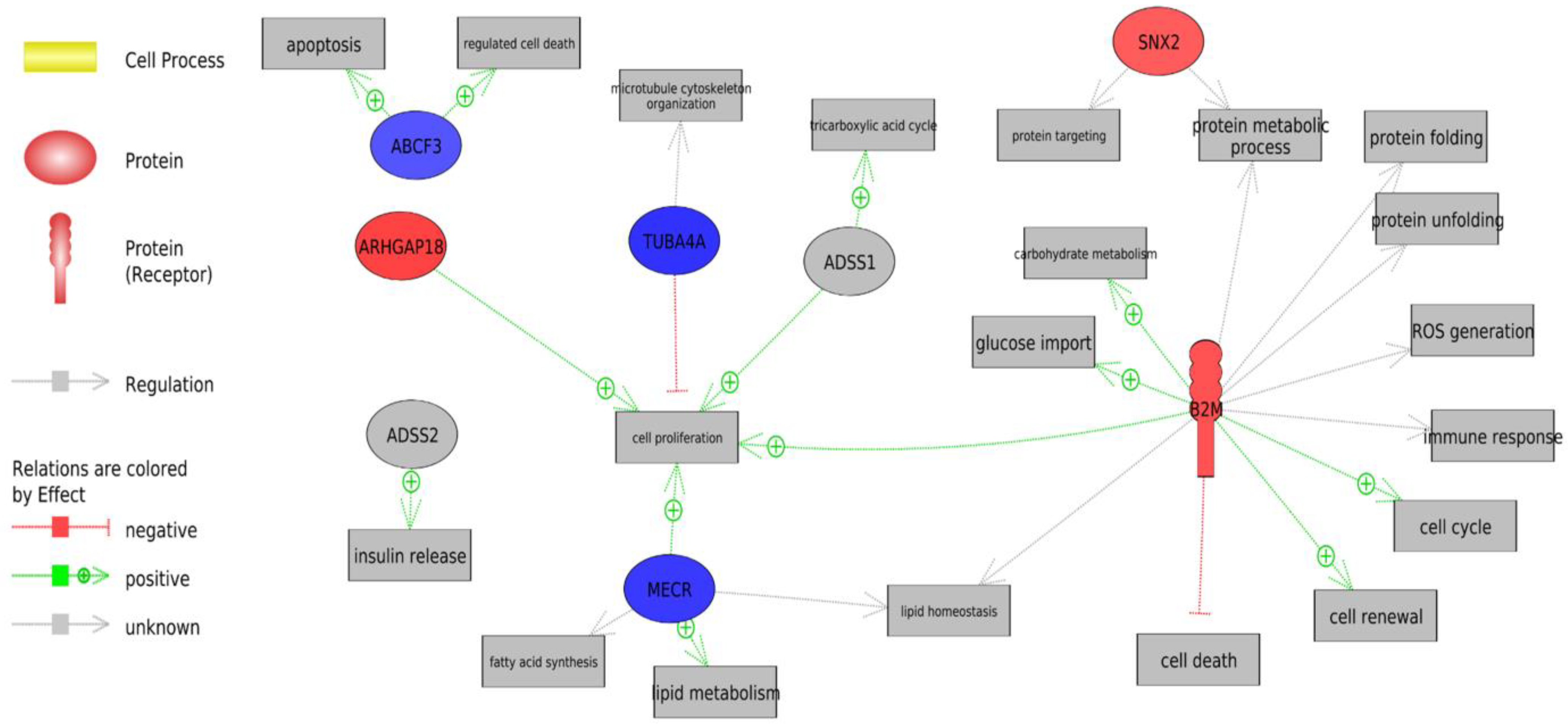
Redox Homeostasis
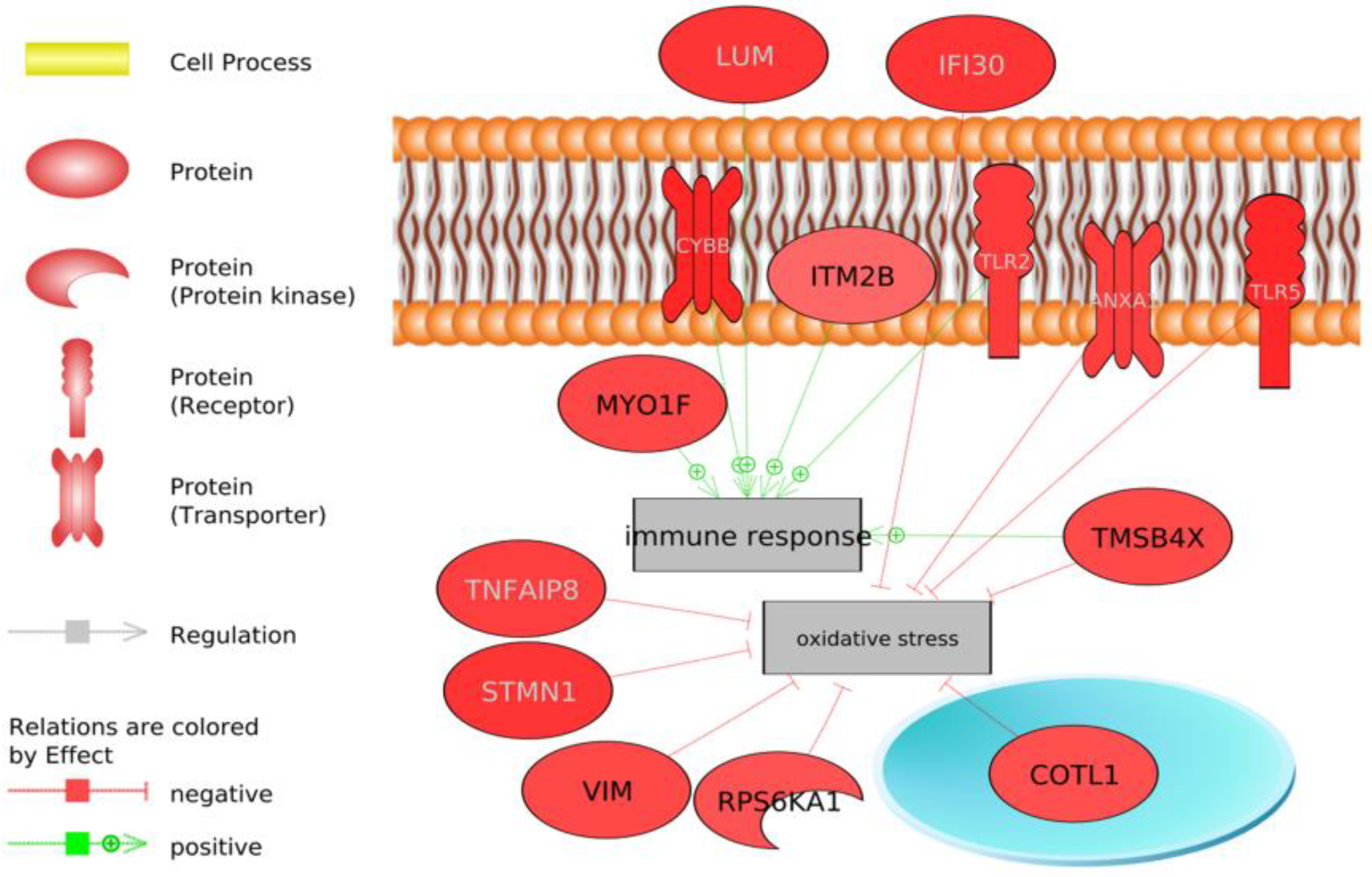
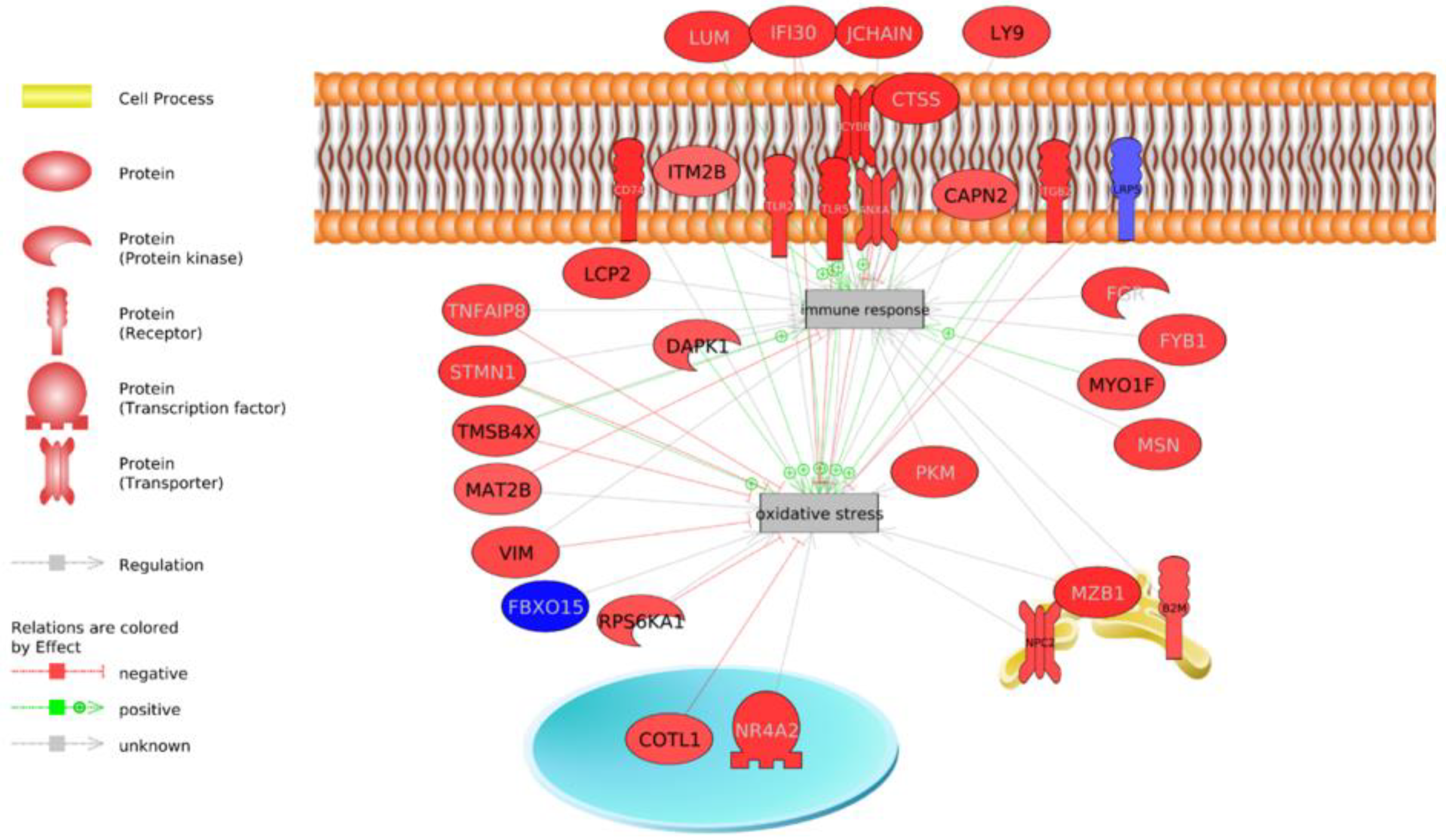
Immune System
3.4.2. Biological Analysis of Differentially-Expressed Genes
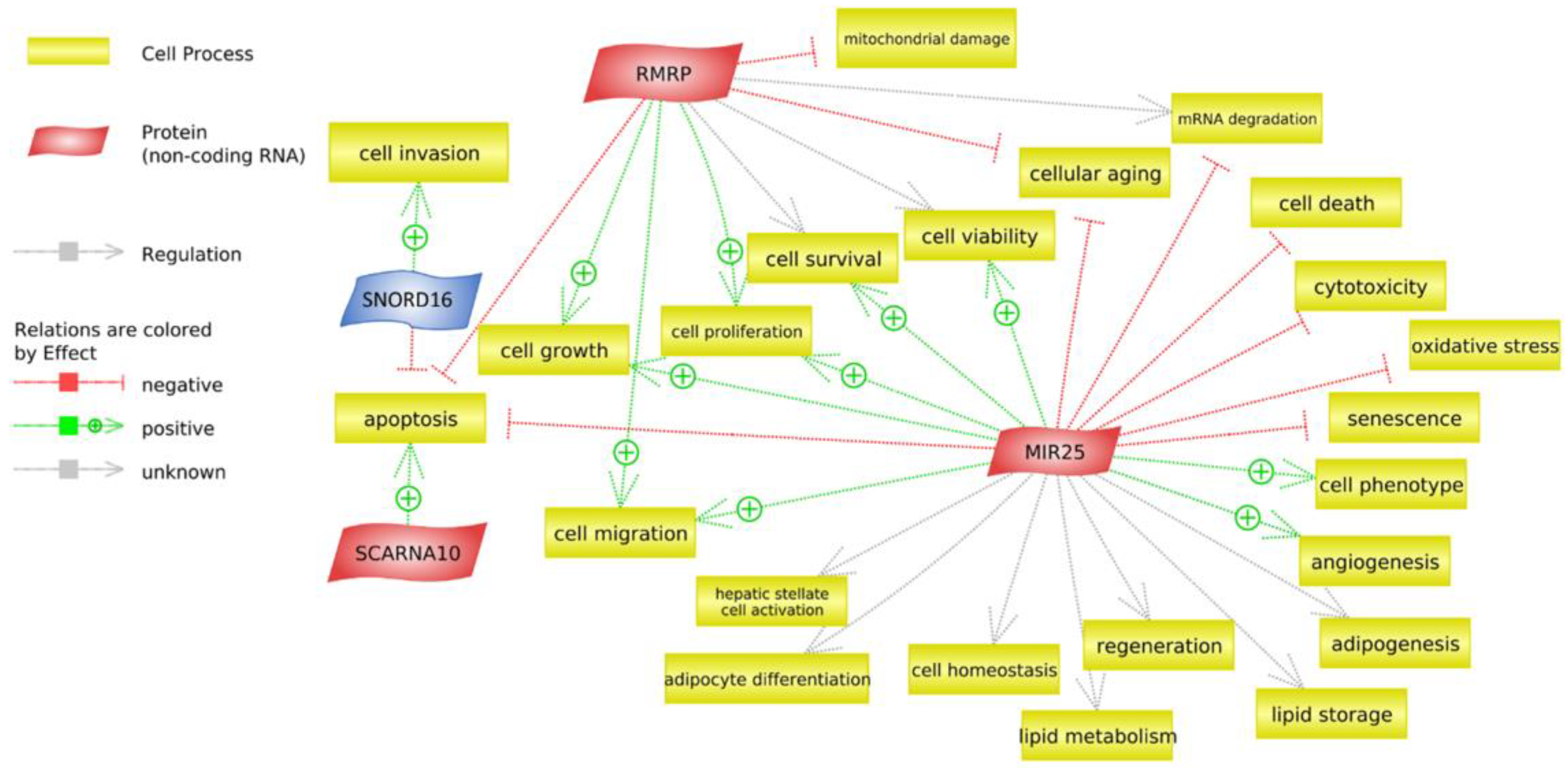
3.5. Analysis of Non-Coding RNAs
4. Discussion
4.1. Energy Metabolism
4.2. Protein Turnover
4.3. Redox Homeostasis
4.4. Immune Response
4.5. Non-Coding RNA
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Martin, P. Climate change, complexity, agriculture and challenged governance. In Research Handbook on Climate Change and Agricultural Law; Edward Elgar Publishing: Cheltenham, UK, 2017; pp. 74–102. [Google Scholar]
- Poore, J.; Nemecek, T. Reducing food’s environmental impacts through producers and consumers. Science 2018, 360, 987–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meat|OECD-FAO Agricultural Outlook 2020–2029|OECD iLibrary. Available online: https://www.oecd-ilibrary.org/sites/29248f46-en/index.html?itemId=/content/component/29248f46-en (accessed on 16 January 2022).
- ABIEC. Perfil da pecuária no Brasil. BeefREPORT 2019; 49. Available online: http://www.abiec.com.br/controle/uploads/arquivos/sumario2019portugues.pdf (accessed on 3 January 2019).
- Silva de Oliveira, J.; de Moura Zanine, A.; Mauro Santos, E.; Fisiologia, E.M. Fisiologia, manejo e alimentação de bezerros de corte. Arq. Ciênc. Vet. Zool. UNIPAR 2007, 10, 39–48. [Google Scholar]
- Del Claro, A.C.; Mercadante, M.E.Z.; Vasconcelos Silva, J.A. Meta-análise de parâmetros genéticos relacionados ao consumo alimentar residual e a suas características componentes em bovinos. Pesqui Agropecu. Bras. 2012, 47, 302–310. [Google Scholar] [CrossRef]
- Moore, S.S.; Mujibi, F.D.; Sherman, E.L. Molecular basis for residual feed intake in beef cattle. J. Anim. Sci. 2009, 87, E41–E47. [Google Scholar] [CrossRef] [Green Version]
- Koch, R.M.; Swiger, L.A.; Chambers, D.; Gregory, K.E. Efficiency of Feed Use in Beef Cattle. J. Anim. Sci. 1963, 22, 486–494. [Google Scholar] [CrossRef]
- Herd, R.M.; Archer, J.A.; Arthur, P.F. Reducing the cost of beef production through genetic improvement in residual feed intake: Opportunity and challenges to application. J. Anim. Sci. 2003, 81, E9–E17. [Google Scholar]
- Crews, D.H. Genetics of efficient feed utilization and national cattle evaluation: A review. Genet. Mol. Res. 2005, 4, 152–165. [Google Scholar]
- Nkrumah, J.D.; Basarab, J.A.; Wang, Z.; Li, C.; Price, M.A.; Okine, E.K.; Crews, D.H., Jr.; Moore, S.S. Genetic and phenotypic relationships of feed intake and measures of efficiency with growth and carcass merit of beef cattle. J. Anim. Sci. 2007, 85, 2711–2720. [Google Scholar] [CrossRef]
- de Oliveira, P.S.N.; Cesar, A.S.M.; do Nascimento, M.L.; Chaves, A.S.; Tizioto, P.C.; Tullio, R.R.; Lanna, D.P.; Rosa, A.N.; Sonstegard, T.S.; Mourao, G.B.; et al. Identification of genomic regions associated with feed efficiency in Nelore cattle. BMC Genet. 2014, 15, 100. [Google Scholar] [CrossRef] [Green Version]
- Archer, J.A.; Richardson, E.C.; Herd, R.M.; Arthur, P.F. Potential for selection to improve efficiency of feed use in beef cattle: A review. Aust. J. Agric. Res. 1999, 50, 147–162. [Google Scholar] [CrossRef]
- Yang, C.; Zhu, Y.; Ding, Y.; Huang, Z.; Dan, X.; Shi, Y.; Kang, X. Identifying the key genes and functional enrichment pathways associated with feed efficiency in cattle. Gene 2022, 807, 145934. [Google Scholar] [CrossRef] [PubMed]
- Basarab, J.A.; Price, M.A.; Aalhus, J.L.; Okine, E.K.; Snelling, W.M.; Lyle, K.L. Residual feed intake and body composition in young growing cattle. Can. J. Anim. Sci. 2003, 83, 189–204. [Google Scholar] [CrossRef]
- Zhao, L.; Ding, Y.; Yang, C.; Wang, P.; Zhao, Z.; Ma, Y.; Shi, Y.; Kang, X. Identification and characterization of hypothalamic circular RNAs associated with bovine residual feed intake. Gene 2023, 851, 147017. [Google Scholar] [CrossRef]
- Fonseca, L.F.S.; Gimenez, D.F.J.; Mercadante, M.E.Z.; Bonilha, S.F.M.; Ferro, J.A.; Baldi, F.; De Souza, F.R.P.; De Albuquerque, L.G. Expression of genes related to mitochondrial function in Nellore cattle divergently ranked on residual feed intake. Mol. Biol. Rep. 2015, 42, 559–565. [Google Scholar] [CrossRef]
- Tizioto, P.C.; Coutinho, L.L.; Decker, J.E.; Schnabel, R.D.; Rosa, K.O.; Oliveira, P.S.N.; Souza, M.M.; Mourão, G.B.; Tullio, R.R.; Chaves, A.S.; et al. Global liver gene expression differences in Nelore steers with divergent residual feed intake phenotypes. BMC Genom. 2015, 16, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, D.F.; de Albuquerque, L.G.; Reimer, C.; Qanbari, S.; Erbe, M.; do Nascimento, A.V.; Venturini, G.C.; Scalez, D.C.B.; Baldi, F.; de Camargo, G.M.F.; et al. Genome-wide scan reveals population stratification and footprints of recent selection in Nelore cattle. Genet. Sel. Evol. 2018, 50, 22. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Alexandre, P.A.; Ribeiro, G.; Fukumasu, H.; Sun, W.; Reverter, A.; Li, Y. Identification of Predictor Genes for Feed Efficiency in Beef Cattle by Applying Machine Learning Methods to Multi-Tissue Transcriptome Data. Front. Genet. 2021, 12, 619857. [Google Scholar] [CrossRef]
- Du, L.; Chang, T.; An, B.; Liang, M.; Duan, X.; Cai, W.; Zhu, B.; Gao, X.; Chen, Y.; Xu, L.; et al. Transcriptome profiling analysis of muscle tissue reveals potential candidate genes affecting water holding capacity in Chinese Simmental beef cattle. Sci. Rep. 2021, 11, 11897. [Google Scholar] [CrossRef]
- Martin, P.; Ducrocq, V.; Faverdin, P.; Friggens, N.C. Invited review: Disentangling residual feed intake—Insights and approaches to make it more fit for purpose in the modern context. J. Dairy Sci. 2021, 104, 6329–6342. [Google Scholar] [CrossRef]
- Kenny, D.A.; Fitzsimons, C.; Waters, S.M.; McGee, M. Improving feed efficiency of beef cattle—The current state of the art and future challenges. Animal 2018, 12, 1815–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef] [PubMed]
- Ferrell, C.L.; Jenkins, T.G. Cow Type and the Nutritional Environment: Nutritional Aspects. J. Anim. Sci. 1985, 61, 725–741. [Google Scholar] [CrossRef] [PubMed]
- Di Agostino, S.; Vahabi, M.; Turco, C.; Fontemaggi, G. Secreted Non-Coding RNAs: Functional Impact on the Tumor Microenvironment and Clinical Relevance in Triple-Negative Breast Cancer. Non-Coding RNA 2022, 8, 5. [Google Scholar] [CrossRef]
- Winkle, M.; El-Daly, S.M.; Fabbri, M.; Calin, G.A. Noncoding RNA therapeutics—Challenges and potential solutions. Nat. Rev. Drug Discov. 2021, 20, 629–651. [Google Scholar] [CrossRef]
- Rönnau, C.G.H.; Verhaegh, G.W.; Luna-Velez, M.V.; Schalken, J.A. Noncoding RNAs as Novel Biomarkers in Prostate Cancer. Biomed. Res. Int. 2014, 2014, 591703. [Google Scholar] [CrossRef]
- Velonas, V.M.; Woo, H.H.; dos Remedios, C.G.; Assinder, S.J. Current status of biomarkers for prostate cancer. Int. J. Mol. Sci. 2013, 14, 11034–11060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Z.; Wu, H.; Xiong, Y.; Wei, D.; Wang, X.; Luoreng, Z.; Cai, X.; Ma, Y. ncRNAs regulate bovine adipose tissue deposition. Mol. Cell. Biochem. 2021, 476, 2837–2845. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Li, S.; Lai, Z.; Zhou, Z.; Wu, F.; Huang, Y.; Lan, X.; Lei, C.; Chen, H.; Dang, R. Analysis of Long Non-Coding RNA and mRNA Expression Profiling in Immature and Mature Bovine (Bos taurus) Testes. Front. Genet. 2019, 10, 646. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Koganti, P.P.; Yao, J. Systematic identification of long intergenic non-coding RNAs expressed in bovine oocytes. Reprod. Biol. Endocrinol. 2020, 18, 13. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, D.P.; Lourenco, D.A.L.; Tsuruta, S.; Misztal, I.; Santos, D.J.A.; de Araújo Neto, F.R.; Aspilcueta-Borquis, R.R.; Baldi, F.; Carvalheiro, R.; De Camargo, G.M.F.; et al. Reaction norm for yearling weight in beef cattle using single-step genomic evaluation1. J. Anim. Sci. 2018, 96, 27–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandre, P.A.; Reverter, A.; Berezin, R.B.; Porto-Neto, L.R.; Ribeiro, G.; Santana, M.H.A.; Ferraz, J.; Fukumasu, H. Exploring the Regulatory Potential of Long Non-Coding RNA in Feed Efficiency of Indicine Cattle. Genes 2020, 11, 997. [Google Scholar] [CrossRef] [PubMed]
- Al-Husseini, W.; Chen, Y.; Gondro, C.; Herd, R.M.; Gibson, J.P.; Arthur, P.F. Characterization and Profiling of Liver microRNAs by RNA-sequencing in Cattle Divergently Selected for Residual Feed Intake. Asian-Australas. J. Anim. Sci. 2016, 29, 1371–1382. [Google Scholar] [CrossRef] [PubMed]
- Nolte, W.; Weikard, R.; Brunner, R.M.; Albrecht, E.; Hammon, H.M.; Reverter, A.; Küehn, C. Identification and Annotation of Potential Function of Regulatory Antisense Long Non-Coding RNAs Related to Feed Efficiency in Bos taurus Bulls. Int. J. Mol. Sci. 2020, 21, 3292. [Google Scholar] [CrossRef] [PubMed]
- Zorzi, K.; Bonilha, S.F.; Queiroz, A.C.; Branco, R.H.; Sobrinho, T.L.; Duarte, M.S. Meat quality of young Nellore bulls with low and high residual feed intake. Meat Sci. 2013, 93, 593–599. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Law, C.W.; Alhamdoosh, M.; Su, S.; Smyth, G.K.; Ritchie, M.E. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Research 2016, 5, 1408. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Hoo, Z.H.; Candlish, J.; Teare, D. What is an ROC curve? Emerg. Med. J. 2017, 34, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, P.S.P.; Folger, J.K.; Rajput, S.K.; Lv, L.; Yao, J.; Ireland, J.J.; Smith, G.W. Regulation and Regulatory Role of WNT Signaling in Potentiating FSH Action during Bovine Dominant Follicle Selection. PLoS ONE 2014, 9, e100201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakab, L. The liver and the immune system. Orv. Hetil. 2015, 156, 1203–1213. [Google Scholar] [CrossRef]
- Hegde, V.R.; Vogel, R.; Feany, M.B. Glia are critical for the neuropathology of complex I deficiency in Drosophila. Hum. Mol. Genet. 2014, 23, 4686–4692. [Google Scholar] [CrossRef] [Green Version]
- Grummer, R.R. Etiology of lipid-related metabolic disorders in periparturient dairy cows. J. Dairy Sci. 1993, 76, 3882–3896. [Google Scholar] [CrossRef] [PubMed]
- Connor, E.E.; Kahl, S.; Elsasser, T.H.; Parker, J.S.; Li, R.W.; Tassell, C.P.; Baldwin, R.L.; Barao, S.M. Enhanced mitochondrial complex gene function and reduced liver size may mediate improved feed efficiency of beef cattle during compensatory growth. Funct. Integr. Genom. 2009, 10, 39–51. [Google Scholar] [CrossRef]
- Bottje, W.; Tang, Z.X.; Iqbal, M.; Cawthon, D.; Okimoto, R.; Wing, T.; Cooper, M. Association of mitochondrial function with feed efficiency within a single genetic line of male broilers. Poult. Sci. 2002, 81, 546–555. [Google Scholar] [CrossRef]
- Herd, R.M.; Arthur, P.F. Physiological basis for residual feed intake. J. Anim. Sci. 2009, 87, E64–E71. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.K.; McGee, M.; Crews, D.H.; Fahey, A.G.; Wylie, A.R.; Kenny, D.A. Effect of divergence in residual feed intake on feeding behavior, blood metabolic variables, and body composition traits in growing beef heifers. J. Anim. Sci. 2010, 88, 109–123. [Google Scholar] [CrossRef] [Green Version]
- Fitzsimons, C.; Kenny, D.A.; McGee, M. Visceral organ weights, digestion and carcass characteristics of beef bulls differing in residual feed intake offered a high concentrate diet. Animal 2014, 8, 949–959. [Google Scholar] [CrossRef] [Green Version]
- Chaffey, N.; Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular biology of the cell. 4th edn. Ann. Bot. 2003, 91, 401. [Google Scholar] [CrossRef] [Green Version]
- Assifi, M.M.; Suchankova, G.; Constant, S.; Prentki, M.; Saha, A.K.; Ruderman, N.B. AMP-activated protein kinase and the coordination of hepatic fatty acid metabolism of starved/carbohydrate-refed rats. Am. J. Physiol.-Endocrinol. Metab. 2005, 289, E794–E800. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, I.; Kahn, A.; Doiron, B. The 5′-AMP-activated protein kinase inhibits the transcriptional stimulation by glucose in liver cells, acting through the glucose response complex. FEBS Lett. 1998, 431, 180–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viollet, B.; Foretz, M.; Guigas, B.; Horman, S.; Dentin, R.; Bertrand, L.; Hue, L.; Andreelli, F. Activation of AMP-activated protein kinase in the liver: A new strategy for the management of metabolic hepatic disorders. J. Physiol. 2006, 574, 41–53. [Google Scholar] [CrossRef]
- Carrasco-Chaumel, E.; Roselló-Catafau, J.; Bartrons, R.; Franco-Gou, R.; Xaus, C.; Casillas, A.; Gelpí, E.; Rodés, J.; Peralta, C. Adenosine monophosphate-activated protein kinase and nitric oxide in rat steatotic liver transplantation. J. Hepatol. 2005, 43, 997–1006. [Google Scholar] [CrossRef]
- Goceri, E.; Shah, Z.K.; Layman, R.; Jiang, X.; Gurcan, M.N. Quantification of liver fat: A comprehensive review. Comput. Biol. Med. 2016, 71, 174–189. [Google Scholar] [CrossRef]
- Drackley, J.K.; Dann, H.M.; Douglas, G.N.; Janovick Guretzky, N.A.; Litherland, N.B.; Underwood, J.P.; Loor, J.J.; Douglas, G.N. Physiological and pathological adaptations in dairy cows that may increase susceptibility to periparturient diseases and disorders. Ital. J. Anim. Sci. 2016, 4, 323–344. [Google Scholar] [CrossRef] [Green Version]
- Lagomarsino, A. Regeneración hepática en enfermedad de hígado graso no-alcohólica. Medwave 2012, 12, e5559. [Google Scholar] [CrossRef]
- Alexandre, P.A.; Kogelman, L.J.A.; Santana, M.H.A.; Passarelli, D.; Pulz, L.H.; Fantinato-Neto, P.; Silva, P.L.; Leme, P.R.; Strefezzi, R.F.; Coutinho, L.L.; et al. Liver transcriptomic networks reveal main biological processes associated with feed efficiency in beef cattle. BMC Genom. 2015, 16, 1073. [Google Scholar] [CrossRef] [Green Version]
- Tizioto, P.C.; Coutinho, L.L.; Oliveira, P.S.N.; Cesar, A.S.M.; Diniz, W.J.S.; Lima, A.O.; Rocha, M.I.; Decker, J.E.; Schnabel, R.D.; Mourão, G.B.; et al. Gene expression differences in Longissimus muscle of Nelore steers genetically divergent for residual feed intake. Sci. Rep. 2016, 6, 39493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lkhagvadorj, S.; Qu, L.; Cai, W.; Couture, O.P.; Barb, C.R.; Hausman, G.J.; Nettleton, D.; Anderson, L.L.; Dekkers, J.C.M.; Tuggle, C.K. Gene expression profiling of the short-term adaptive response to acute caloric restriction in liver and adipose tissues of pigs differing in feed efficiency. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R494–R507. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gondro, C.; Quinn, K.; Herd, R.M.; Parnell, P.F.; Vanselow, B. Global gene expression profiling reveals genes expressed differentially in cattle with high and low residual feed intake. Anim. Genet. 2011, 42, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Santana, M.H.A.; Freua, M.C.; Do, D.N.; Ventura, R.V.; Kadarmideen, H.N.; Ferraz, J.B.S. Systems genetics and genome-wide association approaches for analysis of feed intake, feed efficiency, and performance in beef cattle. Genet. Mol. Res. 2016, 15, 10.4238. [Google Scholar] [CrossRef]
- Zanou, N.; Gailly, P. Skeletal muscle hypertrophy and regeneration: Interplay between the myogenic regulatory factors (MRFs) and insulin-like growth factors (IGFs) pathways. Cell. Mol. Life Sci. 2013, 70, 4117–4130. [Google Scholar] [CrossRef]
- Casal, A.; Garcia-Roche, M.; Navajas, E.A.; Cassina, A.; Carriquiry, M. Differential hepatic oxidative status in steers with divergent residual feed intake phenotype. Animal 2020, 14, 78–85. [Google Scholar] [CrossRef]
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxidative Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef] [Green Version]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef]
- Hekimi, S.; Wang, Y.; Noë, A. Mitochondrial ROS and the effectors of the intrinsic apoptotic pathway in aging cells: The discerning killers! Front. Genet. 2016, 7, 161. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Sagua, R.; Parra, V.; López-Crisosto, C.; Díaz, P.; Quest, A.F.G.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. Compr. Physiol. 2017, 7, 623–634. [Google Scholar]
- Annesley, S.J.; Fisher, P.R. Mitochondria in Health and Disease. Cells 2019, 8, 680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moehle, E.A.; Shen, K.; Dillin, A. Mitochondrial proteostasis in the context of cellular and organismal health and aging. J. Biol. Chem. 2019, 294, 5396–5407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Wang, X.; He, T.; Xiong, F.; Chen, X.; Chen, X.; Jin, S.; Geng, Z. Association of residual feed intake with growth performance, carcass traits, meat quality, and blood variables in native chickens. J. Anim. Sci. 2020, 98, skaa121. [Google Scholar] [CrossRef] [PubMed]
- Ojano-Dirain, C.; Iqbal, M.; Wing, T.; Cooper, M.; Bottje, W. Glutathione and respiratory chain complex activity in duodenal mitochondria of broilers with low and high feed efficiency. Poult. Sci. 2005, 84, 782–788. [Google Scholar] [CrossRef]
- Ferguson, M.; Mockett, R.J.; Shen, Y.; Orr, W.C.; Sohal, R.S. Age-associated decline in mitochondrial respiration and electron transport in Drosophila melanogaster. Biochem. J. 2005, 390, 501–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, B.W.; Lassiter, K.; Piekarski-Welsher, A.; Dridi, S.; Reverter-Gomez, A.; Hudson, N.J.; Bottje, W.G. Proteomics of Breast Muscle Tissue Associated with the Phenotypic Expression of Feed Efficiency within a Pedigree Male Broiler Line: I. Highlight on Mitochondria. PLoS ONE 2016, 11, e0155679. [Google Scholar] [CrossRef] [Green Version]
- Acetoze, G.; Weber, K.L.; Ramsey, J.J.; Rossow, H.A. Relationship between Liver Mitochondrial Respiration and Proton Leak in Low and High RFI Steers from Two Lineages of RFI Angus Bulls. Int. Sch. Res. Not. 2015, 2015, 194014. [Google Scholar] [CrossRef] [Green Version]
- Zuelke, K.A.; Jeffay, S.C.; Zucker, R.M.; Perreault, S.D. Glutathione (GSH) concentrations vary with the cell cycle in maturing hamster oocytes, zygotes, and pre-implantation stage embryos. Mol. Reprod. Dev. 2003, 64, 106–112. [Google Scholar] [CrossRef]
- Barendse, W.; Reverter, A.; Bunch, R.J.; Harrison, B.E.; Barris, W.; Thomas, M.B. A validated whole-genome association study of efficient food conversion in cattle. Genetics 2007, 176, 1893–1905. [Google Scholar] [CrossRef] [Green Version]
- Lindholm-Perry, A.K.; Butler, A.R.; Kern, R.J.; Hill, R.; Kuehn, L.A.; Wells, J.E.; Oliver, W.T.; Hales, K.E.; Foote, A.P.; Freetly, H.C. Differential gene expression in the duodenum, jejunum and ileum among crossbred beef steers with divergent gain and feed intake phenotypes. Anim. Genet. 2016, 47, 408–427. [Google Scholar] [CrossRef]
- Kong, B.W.; Hudson, N.; Seo, D.; Lee, S.; Khatri, B.; Lassiter, K.; Cook, D.; Piekarski, A.; Dridi, S.; Anthony, N.; et al. RNA sequencing for global gene expression associated with muscle growth in a single male modern broiler line compared to a foundational Barred Plymouth Rock chicken line. BMC Genom. 2017, 18, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafizadeh, M.; Ahmadi, Z.; Farkhondeh, T.; Samarghandian, S. Resveratrol targeting the Wnt signaling pathway: A focus on therapeutic activities. J. Cell. Physiol. 2020, 235, 4135–4145. [Google Scholar] [CrossRef] [PubMed]
- Segditsas, S.; Tomlinson, I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 2006, 25, 7531–7537. [Google Scholar] [CrossRef] [Green Version]
- Horodyska, J.; Oster, M.; Wimmers, K.; Mullen, A.M.; Lawlor, P.G.; Hamill, R.M. P3024 Transcriptome analysis of longissimus thoracis et lumborum from pigs divergent in residual feed intake. J. Anim. Sci. 2016, 94, 63–64. [Google Scholar] [CrossRef]
- Huang, W.C.; Zhau, H.E.; Chung, L.W.K. Androgen receptor survival signaling is blocked by anti-β2- microglobulin monoclonal antibody via a MAPK/lipogenic pathway in human prostate cancer cells. J. Biol. Chem. 2010, 285, 7947–7956. [Google Scholar] [CrossRef] [Green Version]
- Good, S.C.; Dewison, K.M.; Radford, S.E.; van Oosten-Hawle, P. Global proteotoxicity caused by human β2 microglobulin variants impairs the unfolded protein response in c. elegans. Int. J. Mol. Sci. 2021, 22, 10752. [Google Scholar] [CrossRef]
- Taurines, R.; Dudley, E.; Grassl, J.; Warnke, A.; Gerlach, M.; Coogan, A.N.; Thome, J. Review: Proteomic research in psychiatry. J. Psychopharmacol. 2011, 25, 151–196. [Google Scholar] [CrossRef] [PubMed]
- León-Letelier, R.A.; Bonifaz, L.C.; Fuentes-Pananá, E.M. OMIC signatures to understand cancer immunosurveillance and immunoediting: Melanoma and immune cells interplay in immunotherapy. J. Leukoc. Biol. 2019, 105, 915–933. [Google Scholar] [CrossRef]
- Chevaliez, S.; Balanant, J.; Maillard, P.; Lone, Y.C.; Lemonnier, F.A.; Delpeyroux, F. Role of class I human leukocyte antigen molecules in early steps of echovirus infection of rhabdomyosarcoma cells. Virology 2008, 381, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Kollnberger, S. The Role of HLA-Class I Heavy-Chain Interactions with Killer-Cell Immunoglobulin-Like Receptors in Immune Regulation. Crit. Rev. Immunol. 2016, 36, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Prizment, A.E.; Linabery, A.M.; Lutsey, P.L.; Selvin, E.; Nelson, H.H.; Folsom, A.R.; Church, T.R.; Drake, C.G.; Platz, E.A.; Joshu, C. Circulating Beta-2 Microglobulin and Risk of Cancer: The Atherosclerosis Risk in Communities Study (ARIC). Cancer Epidemiol. Biomark. Prev. 2016, 25, 657–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.J.; Luo, J. The PIN-FORMED Auxin Efflux Carriers in Plants. Int. J. Mol. Sci. 2018, 19, 2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar]
- Montelli, N.L.L.L.; Alvarenga, T.I.R.C.; Almeida, A.K.; Alvarenga, F.A.P.; Furusho-Garcia, I.F.; Greenwood, P.L.; Pereira, I.G. Associations of feed efficiency with circulating IGF-1 and leptin, carcass traits and meat quality of lambs. Meat Sci. 2021, 173, 108379. [Google Scholar] [CrossRef]
- Prieto, M.; Rivas, J.V.; López Novoa, J.M.; Pérez Barriocanal, F. El TGF-beta síntesis y mecanismo de acción. Nefrologia 2002, 22, 135–143. [Google Scholar]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta—Mol. Cell Res. 2007, 1773, 1213–1226. [Google Scholar] [CrossRef]
- Kontny, E.; Kurowska, M.; Szczepanska, K.; Maslinski, W. Rottlerin, a PKC isozyme-selective inhibitor, affects signaling events and cytokine production in human monocytes. J. Leukoc. Biol. 2000, 67, 249–258. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-κB Family of Transcription Factors and Its Regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Ramayo-Caldas, Y.; Ballester, M.; Sánchez, J.P.; González-Rodríguez, O.; Revilla, M.; Reyer, H.; Wimmers, K.; Torrallardona, D.; Quintanilla, R. Integrative approach using liver and duodenum RNA-Seq data identifies candidate genes and pathways associated with feed efficiency in pigs. Sci. Rep. 2018, 8, 558. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Wisse, E.; Tian, Z. Liver natural killer cells: Subsets and roles in liver immunity. Cell. Mol. Immunol. 2015, 13, 328–336. [Google Scholar] [CrossRef] [Green Version]
- Racanelli, V.; Rehermann, B. The liver as an immunological organ. Hepatology 2006, 43, S54–S62. [Google Scholar] [CrossRef]
- Johnson, K.A.; Michal, J.J.; Carstens, G.E.; Hafla, A.N.; Forbes, T.D.A. Differential expression of innate immune system genes in liver of beef cattle with divergent phenotypes for RFI. In Energy and Protein Metabolism and Nutrition in Sustainable Animal Production; Wageningen Academic Publishers: Wageningen, The Netherlands, 2013; pp. 371–372. [Google Scholar]
- Verhagen, J.M.F. Energy Metabolism and Immune Function. In Energy Metabolism in Farm Animals; Springer: Dordrecht, The Netherlands, 1987; pp. 291–303. [Google Scholar]
- Paradis, F.; Yue, S.; Grant, J.R.; Stothard, P.; Basarab, J.A.; Fitzsimmons, C. Transcriptomic analysis by RNA sequencing reveals that hepatic interferon-induced genes may be associated with feed efficiency in beef heifers. J. Anim. Sci. 2015, 93, 3331–3341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarek, C.M.; Lindholm-Perry, A.K.; Kuehn, L.A.; Freetly, H.C. Differential expression of genes related to gain and intake in the liver of beef cattle. BMC Res. Notes 2017, 10, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallard, B.A.; Emam, M.; Paibomesai, M.; Thompson-Crispi, K.; Wagter-Lesperance, L. Genetic selection of cattle for improved immunity and health. Jpn. J. Vet. Res. 2015, 63 (Suppl. S1), S37–S44. [Google Scholar] [PubMed]
- Gondret, F.; Vincent, A.; Houée-Bigot, M.; Siegel, A.; Lagarrigue, S.; Causeur, D.; Gilbert, H.; Louveau, I. A transcriptome multi-tissue analysis identifies biological pathways and genes associated with variations in feed efficiency of growing pigs. BMC Genom. 2017, 18, 244. [Google Scholar] [CrossRef] [Green Version]
- Patience, J.F.; Rossoni-Serão, M.C.; Gutiérrez, N.A. A review of feed efficiency in swine: Biology and application. J. Anim. Sci. Biotechnol. 2015, 6, 33. [Google Scholar] [CrossRef]
- Rakhshandeh, A.; Dekkers, J.C.M.; Kerr, B.J.; Weber, T.E.; English, J.; Gabler, N.K. Effect of immune system stimulation and divergent selection for residual feed intake on digestive capacity of the small intestine in growing pigs. J. Anim. Sci. 2012, 90 (Suppl. S4), 233–235. [Google Scholar] [CrossRef] [Green Version]
- Klasing, K.C.; Austic, R.E. Changes in protein synthesis due to an inflammatory challenge. Proc. Soc. Exp. Biol. Med. 1984, 176, 285–291. [Google Scholar] [CrossRef]
- Mashaly, M.M.; Heetkamp, M.J.W.; Parmentier, H.K.; Schrama, J.W. Influence of genetic selection for antibody production against sheep blood cells on energy metabolism in laying hens. Poult. Sci. 2000, 79, 519–524. [Google Scholar] [CrossRef]
- Saw, P.E.; Xu, X.; Chen, J.; Song, E.W. Non-coding RNAs: The new central dogma of cancer biology. Sci. China Life Sci. 2020, 64, 22–50. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Dozmorov, M.G.; Giles, C.B.; Koelsch, K.A.; Wren, J.D. Systematic classification of non-coding RNAs by epigenomic similarity. BMC Bioinform. 2013, 14, S2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sárközy, M.; Kahán, Z.; Csont, T. A myriad of roles of miR-25 in health and disease. Oncotarget 2018, 9, 21580. [Google Scholar] [CrossRef] [Green Version]
- Varga, Z.V.; Kupai, K.; Szucs, G.; Gáspár, R.; Pálóczi, J.; Faragó, N.; Zvara, Á.; Puskás, L.G.; Rázga, Z.; Tiszlavicz, L.; et al. MicroRNA-25-dependent up-regulation of NADPH oxidase 4 (NOX4) mediates hypercholesterolemia-induced oxidative/nitrative stress and subsequent dysfunction in the heart. J. Mol. Cell. Cardiol. 2013, 62, 111–121. [Google Scholar] [CrossRef]
- Pan, L.; Huang, B.J.; Ma, X.E.; Wang, S.Y.; Feng, J.; Lv, F.; Liu, Y.; Liu, Y.; Li, C.-M.; Liang, D.-D.; et al. MiR-25 protects cardiomyocytes against oxidative damage by targeting the mitochondrial calcium uniporter. Int. J. Mol. Sci. 2015, 16, 5420–5433. [Google Scholar] [CrossRef] [Green Version]
- Magenta, A.; Dellambra, E.; Ciarapica, R.; Capogrossi, M.C. Oxidative stress, microRNAs and cytosolic calcium homeostasis. Cell Calcium 2016, 60, 207–217. [Google Scholar] [CrossRef]
- Yao, Y.; Sun, F.; Lei, M. miR-25 inhibits sepsis-induced cardiomyocyte apoptosis by targetting PTEN. Biosci. Rep. 2018, 38, BSR20171511. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Thorne, R.F.; Zhang, X.D.; Wu, M.; Liu, L. Non-coding RNAs, guardians of the p53 galaxy. Semin. Cancer Biol. 2021, 75, 72–83. [Google Scholar] [CrossRef]
- Genz, B.; Coleman, M.A.; Irvine, K.M.; Kutasovic, J.R.; Miranda, M.; Gratte, F.D.; Tirnitz-Parker, J.E.E.; Olynyk, J.K.; Calvopina, D.A.; Weis, A.; et al. Overexpression of miRNA-25-3p inhibits Notch1 signaling and TGF-β-induced collagen expression in hepatic stellate cells. Sci. Rep. 2019, 9, 8541. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, E.E.; Oltjen, J.W.; Sainz, R.D. Mitochondrial abundance and function in muscle from beef steers with divergent residual feed intakes. Animal 2020, 14, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Kropp, J.; Salih, S.M.; Khatib, H. Expression of microRNAs in bovine and human pre-implantation embryo culture media. Front. Genet. 2014, 5, 91. [Google Scholar] [CrossRef]
- Pohler, K.G.; Green, J.A.; Moley, L.A.; Gunewardena, S.; Hung, W.T.; Payton, R.R.; Hong, X.; Christenson, L.K.; Geary, T.W.; Smith, M.F. Circulating microRNA as candidates for early embryonic viability in cattle. Mol. Reprod. Dev. 2017, 84, 731–743. [Google Scholar] [CrossRef]
- He, J.-Y.; Liu, X.; Qi, Z.-H.; Wang, Q.; Lu, W.-Q.; Zhang, Q.-T.; He, S.-Y.; Wang, Z.D. Small Nucleolar RNA, C/D Box 16 (SNORD16) Acts as a Potential Prognostic Biomarker in Colon Cancer. Dose Response 2020, 18, 1559325820917829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirisola, V.; Mora, R.; Esposito, A.I.; Guastini, L.; Tabacchiera, F.; Paleari, L.; Amaro, A.; Angelini, G.; Dellepiane, M.; Pfeffer, U.; et al. A prognostic multigene classifier for squamous cell carcinomas of the larynx. Cancer Lett. 2011, 307, 37–46. [Google Scholar] [CrossRef] [PubMed]
- López-Corral, L.; Mateos, M.V.; Corchete, L.A.; Sarasquete, M.E.; de la Rubia, J.; de Arriba, F.; Lahuerta, J.-J.; Garcia-Sanz, R.; Miguel, J.S.; Gutierrez, N.; et al. Genomic analysis of high-risk smoldering multiple myeloma. Haematologica 2012, 97, 1439–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weikard, R.; Demasius, W.; Kuehn, C. Mining long noncoding RNA in livestock. Anim. Genet. 2017, 48, 3–18. [Google Scholar] [CrossRef]
- Deniz, E.; Erman, B. Long noncoding RNA (lincRNA), a new paradigm in gene expression control. Funct. Integr. Genom. 2017, 17, 135–143. [Google Scholar] [CrossRef]
- Nolte, W.; Weikard, R.; Brunner, R.M.; Albrecht, E.; Hammon, H.M.; Reverter, A.; Kühn, C. Biological Network Approach for the Identification of Regulatory Long Non-Coding RNAs Associated With Metabolic Efficiency in Cattle. Front. Genet. 2019, 10, 1130. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Cui, Q.; Zhang, X.; Luo, X.; Liu, Y.; Zuo, J.; Peng, Y. Long non-coding RNAs regulation in adipogenesis and lipid metabolism: Emerging insights in obesity. Cell. Signal. 2018, 51, 47–58. [Google Scholar] [CrossRef]
- Serna-García, M. Determinação do perfil transcriptômico associados à eficiência alimentar de bovinos Nelore (BOS TAURUS INDICUS). Doctoral Thesis, Universidade Estadual Paulista (UNESP), Jaboticabal, Brazil, Grant number 88887.529047/2020-00. 2022. Available online: http://hdl.handle.net/11449/234839 (accessed on 20 November 2022).
- Zhang, K.; Han, Y.; Hu, Z.; Zhang, Z.; Shao, S.; Yao, Q.; Zheng, L.; Wang, J.; Han, X.; Zhang, Y.; et al. SCARNA10, a nuclear-retained long non-coding RNA, promotes liver fibrosis and serves as a potential biomarker. Theranostics 2019, 9, 3622. [Google Scholar] [CrossRef] [PubMed]
- Hussen, B.M.; Azimi, T.; Hidayat, H.J.; Taheri, M.; Ghafouri-Fard, S. Long Non-coding RNA RMRP in the Pathogenesis of Human Disorders. Front. Cell Dev. Biol. 2021, 9, 1130. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Cai, Y.; Lai, X.; Wang, Z.; Wei, S.; Tan, K.; Xu, M.; Xie, H. lncRNA RMRP Prevents Mitochondrial Dysfunction and Cardiomyocyte Apoptosis via the miR-1-5p/hsp70 Axis in LPS-Induced Sepsis Mice. Inflammation 2020, 43, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Trainor, P.A.; Merrill, A.E. Ribosome biogenesis in skeletal development and the pathogenesis of skeletal disorders. Biochim. Biophys. Acta—Mol. Basis Dis. 2014, 1842, 769–778. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Chen, X.; Zhang, F.; Zhao, M. RMRP inhibition prevents NAFLD progression in rats via regulating miR-206/PTPN1 axis. Mamm. Genome 2022, 33, 480–489. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Go Term | Biological Processes | Genes | p-Value |
|---|---|---|---|
| GO:0071803 | positive regulation of podosome assembly | ARGEF5, LCP1, MSN | 8.0 × 10−4 |
| GO:0045087 | innate immune response | FGR, ANXA1, B2M, CYBB, JCHAIN, LYA, TLR2, TLR5 | 2.4 × 10−3 |
| GO:0051493 | regulation of cytoskeleton organization | ARGEF5, CAPN2, STMN1 | 3.0 × 10−3 |
| GO:0008360 | regulation of cell shape | FGR, ARHGAP18, ANXA1, ITGB2, MSN | 3.1 × 10−3 |
| GO:0001666 | response to hypoxia | CAPN2, NR4A2, PAK1, PKM, TLR2 | 6.4 × 10−3 |
| GO:0035556 | intracellular signal transduction | ARHGEP5, ADRAIA, ASB8, DAPK1, LCP1, ARS6KA1, STMN1 | 7.6 × 10−3 |
| GO:0032366 | intracellular sterol transport | ARV1, NPC2 | 8.6 × 10−3 |
| GO:0007409 | axonogenesis | CNTN4.LUM, PAK, STMN1 | 8.6 × 10−3 |
| GO:0007165 | signal transduction | CD74, ARHGAP30, ADRAIA, APBBIKP, AMXA1, DAPK1, GRN, MRC2, NR4A2, RPS6KA1, STMN1, TLR2 | 1.0 × 10−2 |
| GO:0006915 | apoptotic process | TNFAIP8, ADRAIA, DAPK1, ITGB2, MZB1, PAK1, RPRGKA1, TLR2 | 1.1 × 10−2 |
| GO:0001895 | retina homeostasis | ACTB, B2M, JCHAIN | 1.3 × 10−2 |
| GO:0031532 | actin cytoskeleton reorganization | ANXA1, PAK1, PARVG | 1.7 × 10−2 |
| GO:0006886 | intracellular protein transport | CD74, AP4B1, SNX2, SNX6, STX7 | 1.9 × 10−2 |
| GO:0034123 | positive regulation of toll-like receptor signaling pathway | TLR2, TLR5 | 2.1 × 10−2 |
| GO:0006897 | endocytosis | LRP5, MRC2, SNX2, SNX6 | 2.2 × 10−2 |
| GO:0098609 | cell–cell adhesion | ABI3, LRRFIPI, ARHGAP18, PKM, SNX2 | 2.9 × 10−2 |
| GO:0006955 | immune response | CD74, B2M, CTSS, JCHAIN, LCP2, TLR2 | 3.4 × 10−2 |
| GO:0022617 | extracellular matrix disassembly | CAPN2, CTSS, LCP1 | 4.2 × 10−2 |
| GO:0050707 | regulation of cytokine secretion | TLR2, TLR5 | 4.6 × 10−2 |
| Genetic group1 | |||
| ID | Name | Fold change (log2) * | p-value |
| SNORD16 | Small nucleolar RNA, C/D box 16 | −0.356 | 0.01276 |
| MIR25 | MicroRNA 25 | 0.511 | 0.04741 |
| Genetic group 2 | |||
| ID | Name | Fold-change (log2) * | p-value |
| SCARNA10 | Small Cajal body-specific RNA 10 | 0.636 | 0.01855 |
| RNase_MRP | RNA component of mitochondrial RNA processing endoribonuclease | 0.664 | 0.02635 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serna-García, M.; Fonseca, L.F.S.; Panadero Romero, J.J.; Carretero Asuncion, J.; dos Santos Silva, D.B.; Salatta, B.M.; Frezarim, G.B.; Mercadante, M.E.Z.; Bonilha, S.F.M.; Ferro, J.A.; et al. Transcriptome Profiling of the Liver in Nellore Cattle Phenotypically Divergent for RFI in Two Genetic Groups. Animals 2023, 13, 359. https://doi.org/10.3390/ani13030359
Serna-García M, Fonseca LFS, Panadero Romero JJ, Carretero Asuncion J, dos Santos Silva DB, Salatta BM, Frezarim GB, Mercadante MEZ, Bonilha SFM, Ferro JA, et al. Transcriptome Profiling of the Liver in Nellore Cattle Phenotypically Divergent for RFI in Two Genetic Groups. Animals. 2023; 13(3):359. https://doi.org/10.3390/ani13030359
Chicago/Turabian StyleSerna-García, Marta, Larissa Fernanda Simielli Fonseca, Joaquin Javier Panadero Romero, Julian Carretero Asuncion, Danielly Beraldo dos Santos Silva, Bruna Maria Salatta, Gabriela Bonfá Frezarim, Maria Eugênia Zerlotti Mercadante, Sarah Figueiredo Martins Bonilha, Jesus Aparecido Ferro, and et al. 2023. "Transcriptome Profiling of the Liver in Nellore Cattle Phenotypically Divergent for RFI in Two Genetic Groups" Animals 13, no. 3: 359. https://doi.org/10.3390/ani13030359