

Single-Molecule Real-Time Sequencing for Identifying Sexual-Dimorphism-Related Transcriptomes and Genes in the Chinese Soft-Shelled Turtle (Pelodiscus sinensis)
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Materials
2.2. RNA Extraction
2.3. PacBio Library Construction and Sequencing
2.4. PacBio Data Processing and Error Correction
2.5. Gene and Isoform Identification
2.6. Fusion Transcript Identification
2.7. APA Site Detection
2.8. LncRNA Identification
2.9. RNA-Seq Library Construction and Sequencing
2.10. RNA-Seq Data Analysis
2.11. DEG Validation through RT-qPCR
3. Results
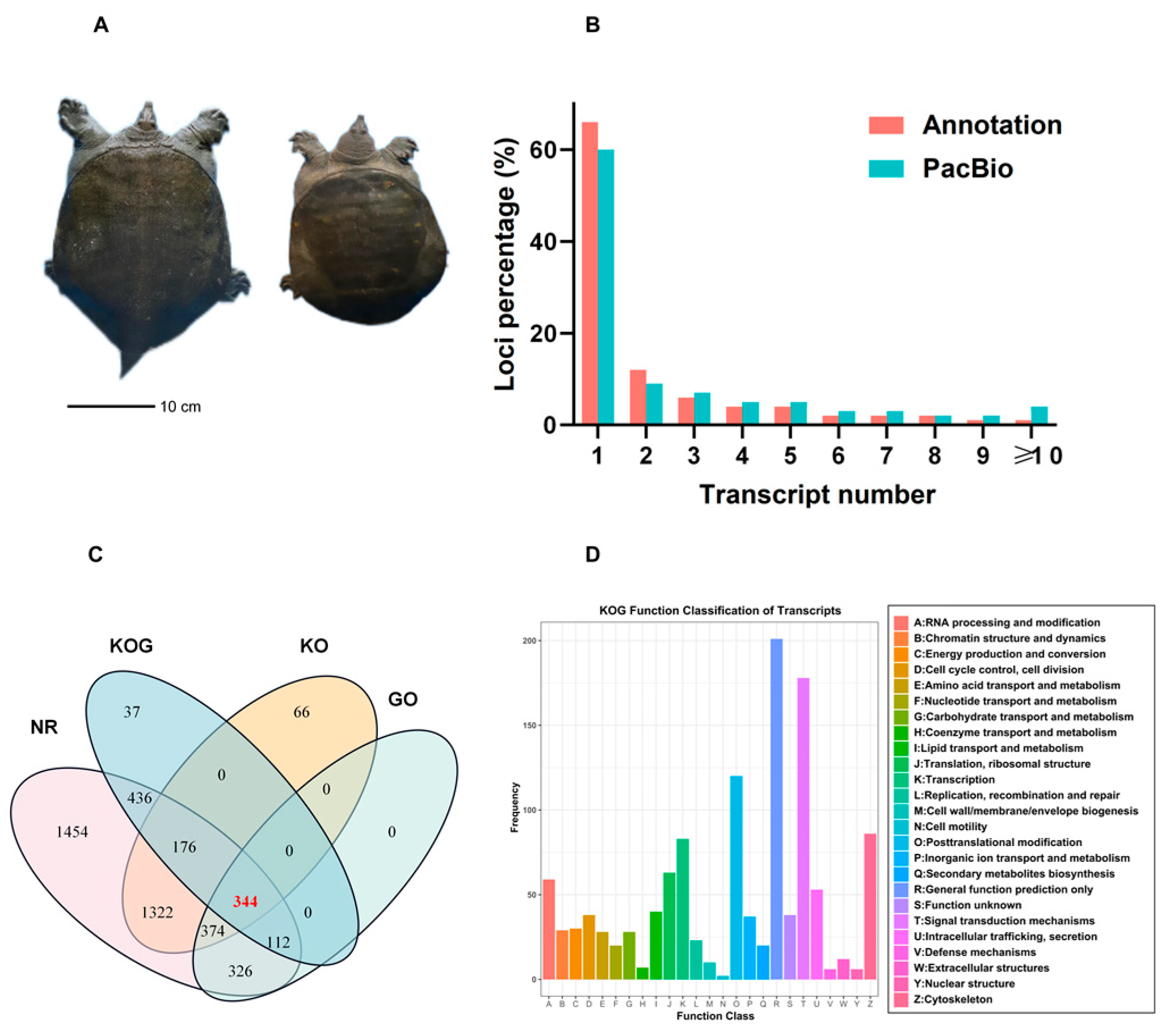
3.1. SMRT Sequence Analysis
3.2. Functional Annotation of Transcripts
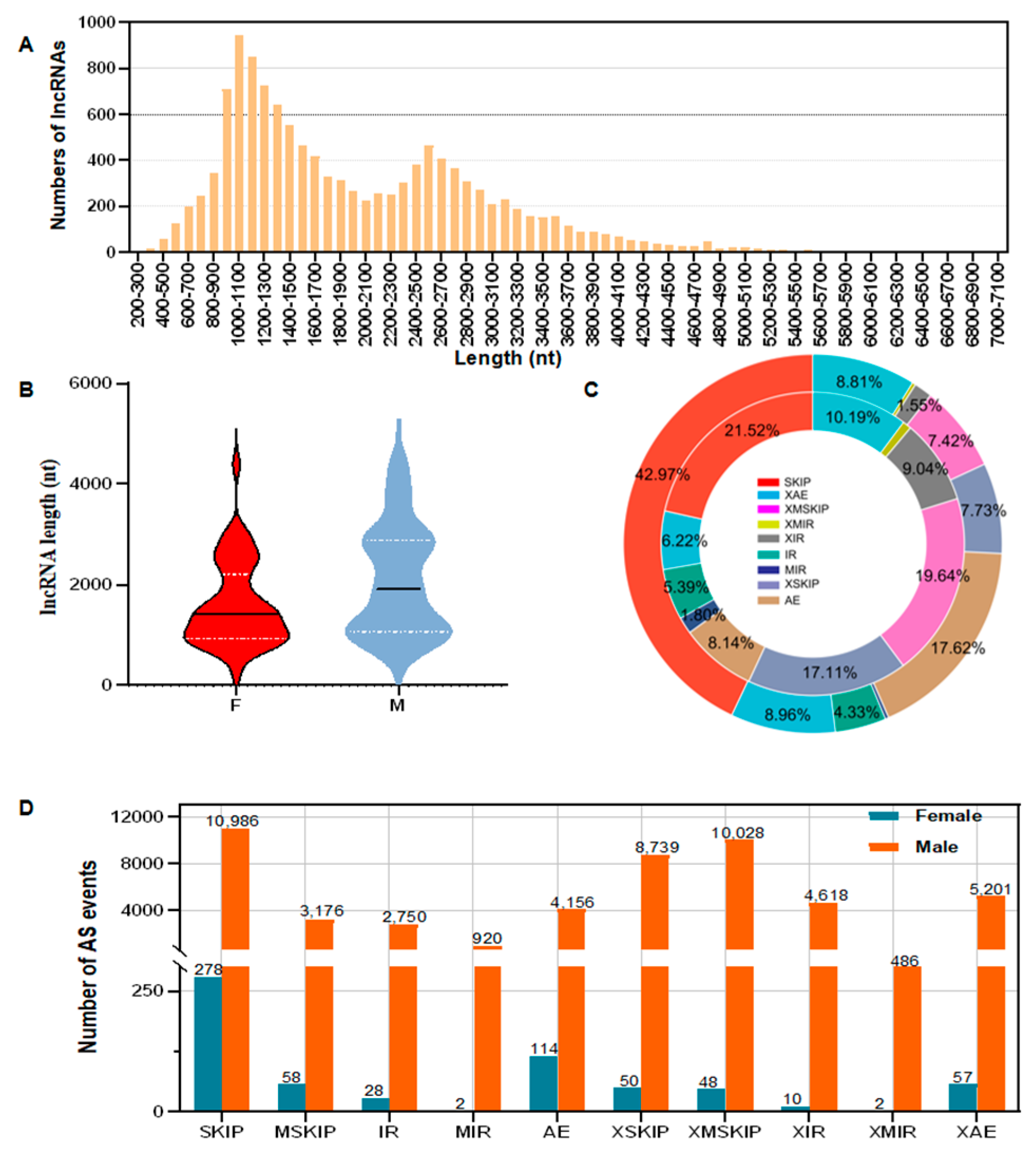
3.3. Analysis of LncRNAs and AS Events
3.4. APA Analysis
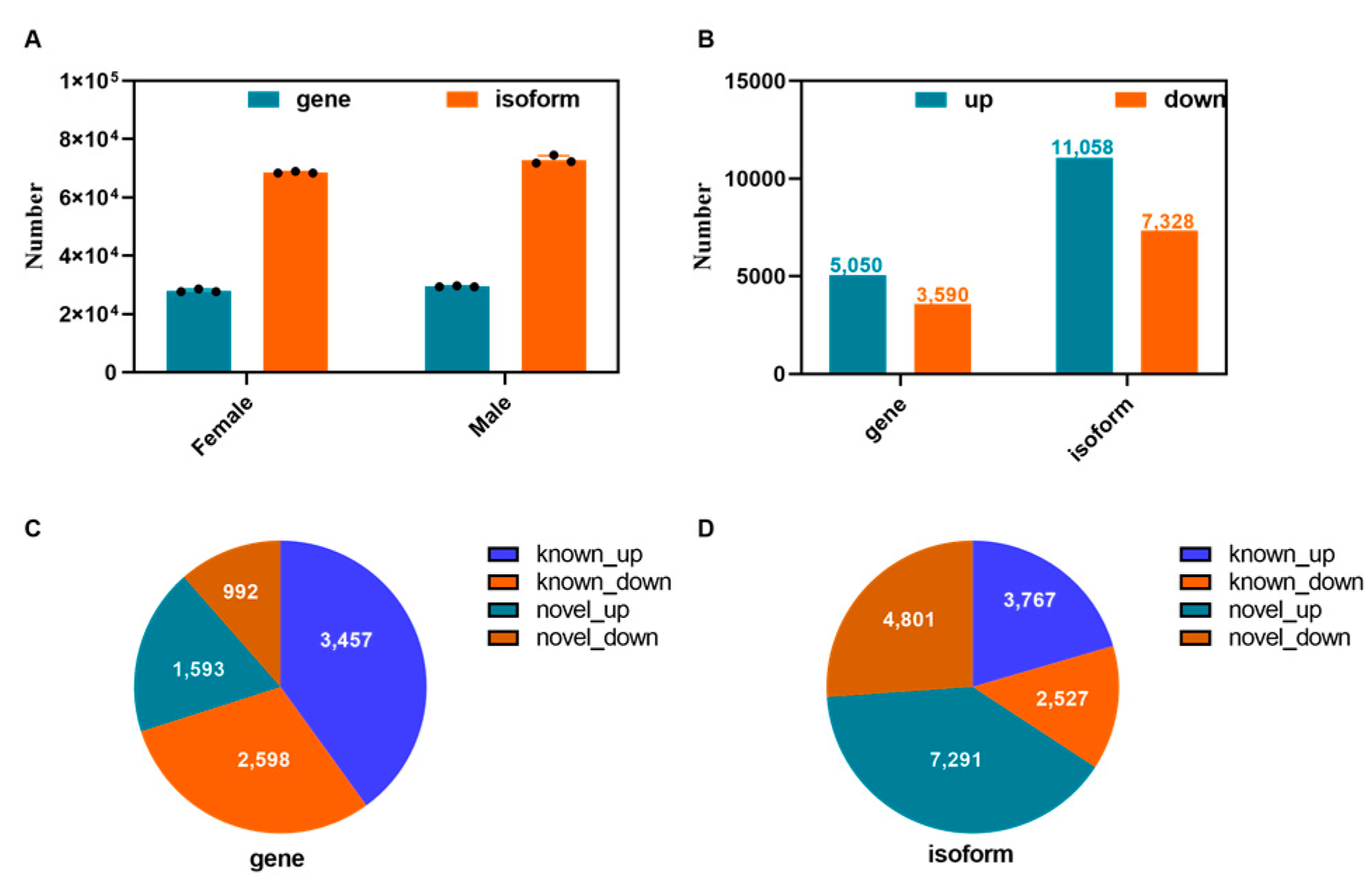
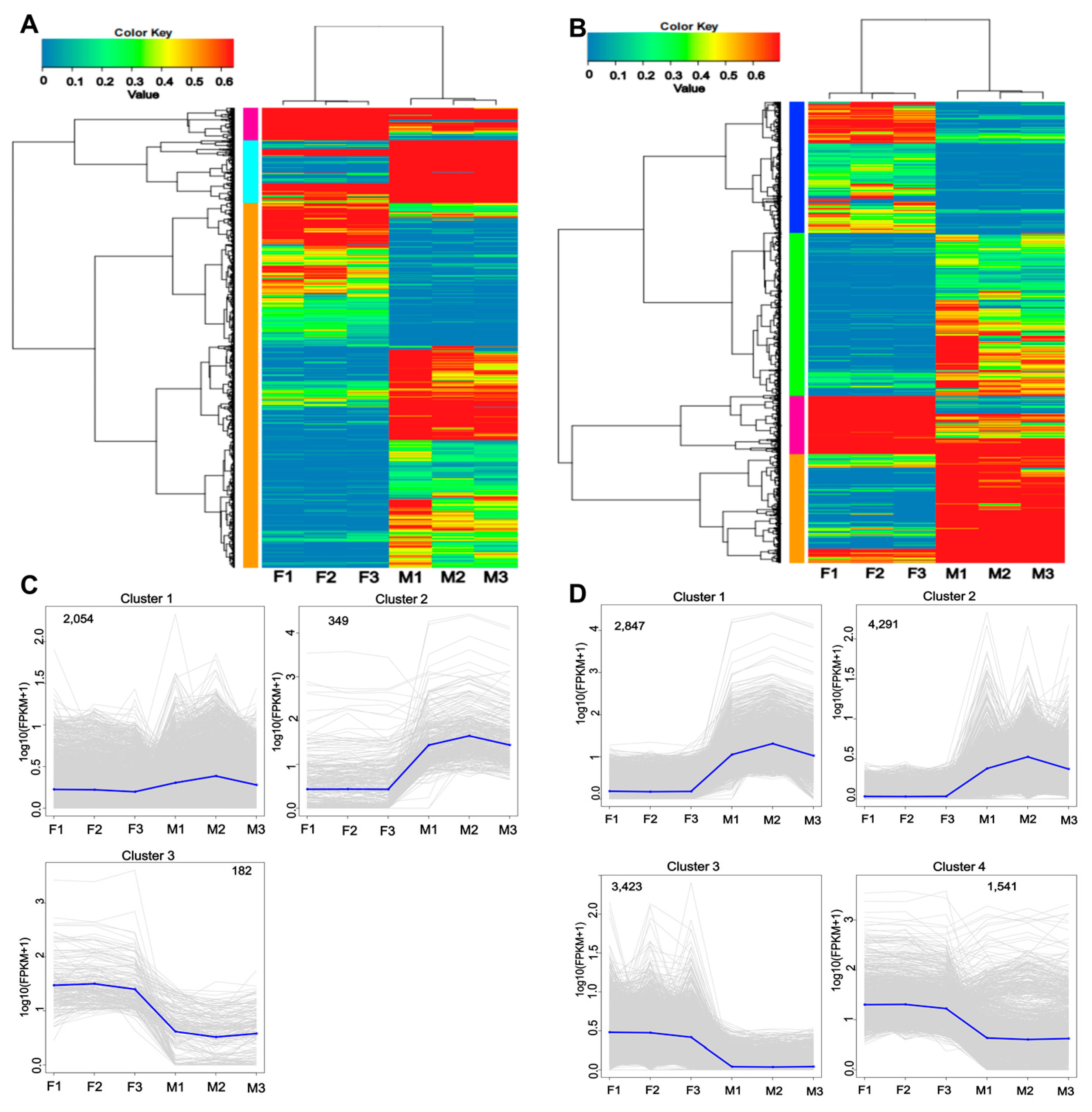
3.5. Analysis of Novel Genes and Isoforms in Males and Females
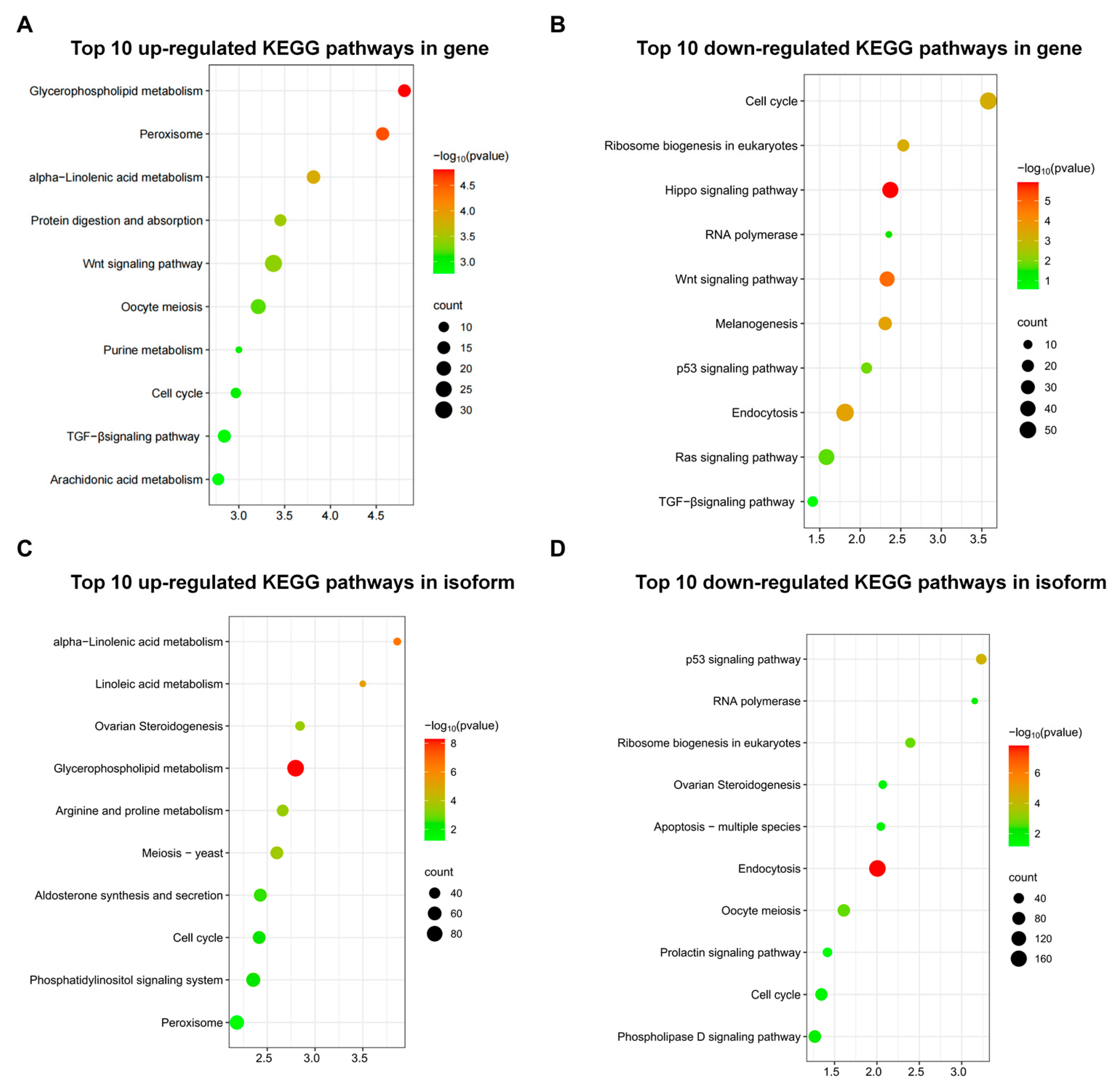
3.6. KEGG Enrichment Analysis of Genes and Isoforms
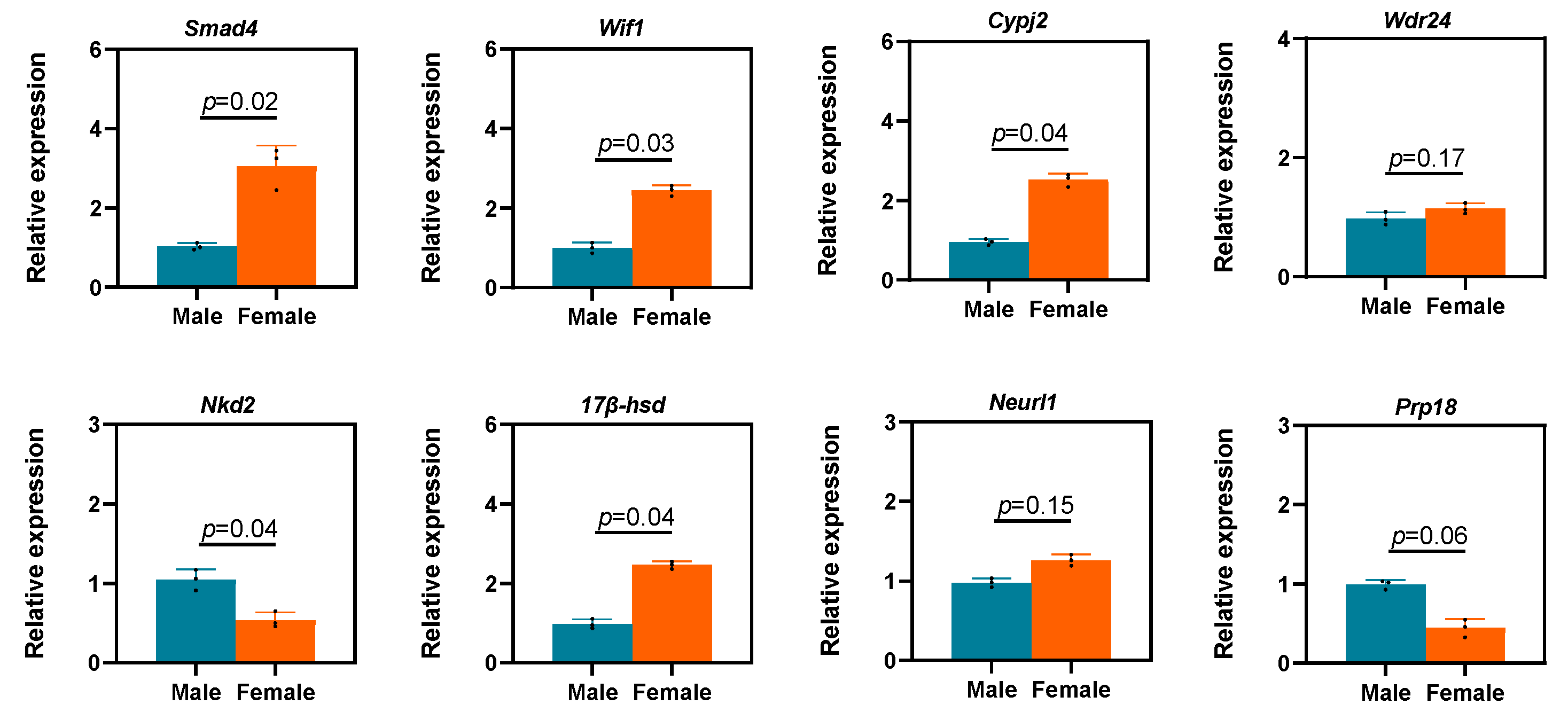
3.7. Validation of DEGs through Quantitative Real-Time Polymerase Chain Reaction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gong, S.; Vamberger, M.; Auer, M.; Praschag, P.; Fritz, U. Millennium-old farm breeding of Chinese softshell turtles (Pelodiscus sinensis) results in massive erosion of biodiversity. Sci. Nat. 2018, 105, 34. [Google Scholar] [CrossRef] [PubMed]
- Bonduriansky, R.; Chenoweth, S.F. Intralocus sexual conflict. Trends Ecol. Evol. 2009, 24, 280–288. [Google Scholar] [CrossRef]
- Liang, H.; Wang, L.; Sha, H.; Zou, G. Development and validation of sex-specific markers in Pelodiscus snensis using restriction site-associated DNA sequencing. Genes 2019, 10, 302. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, Y.; Chakraborty, T.; Paul-Prasanth, B.; Ohta, K.; Nakamura, M. Sex determination, gonadal sex differentiation, and plasticity in vertebrate species. Physiol. Rev. 2021, 101, 1237–1308. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.; Ye, J.; Weber, C.; Sun, W.; Zhang, H.; Zhou, Y.; Cai, C.; Qian, G.; Capel, B. The histone demethylase KDM6B regulates temperature-dependent sex determination in a turtle species. Science 2018, 360, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Y.; Yuan, J.; Liu, F.; Hong, X.; Yu, L.; Chen, C.; Li, W.; Ni, W.; Liu, H.; et al. Chromosome-level genome assembly of Asian yellow pond turtle (Mauremys mutica) with temperature-dependent sex determination system. Sci. Rep. 2022, 12, 7905. [Google Scholar] [CrossRef]
- Liu, X.; Zhu, Y.; Zhao, Y.; Wang, Y.; Li, W.; Hong, X.; Yu, L.; Chen, C.; Xu, H.; Zhu, X. Vasa expression is associated with sex differentiation in the Asian yellow pond turtle, Mauremys mutica. J. Exp. Zool. B Mol. Dev. Evol. 2021, 336, 431–442. [Google Scholar] [CrossRef]
- Stubbs, J.L.; Mitchell, N.J. The Influence of Temperature on Embryonic Respiration, Growth, and Sex Determination in a Western Australian Population of Green Turtles (Chelonia mydas). Physiol. Biochem. Zool. 2018, 91, 1102–1114. [Google Scholar] [CrossRef]
- Williams, T.M.; Carroll, S.B. Genetic and molecular insights into the development and evolution of sexual dimorphism. Nat. Rev. Genet. 2009, 10, 797–804. [Google Scholar] [CrossRef]
- Badenhorst, D.; Stanyon, R.; Engstrom, T.; Valenzuela, N. A ZZ/ZW microchromosome system in the spiny softshell turtle, Apalone spinifera, reveals an intriguing sex chromosome conservation in Trionychidae. Chromosome Res. 2013, 21, 137–147. [Google Scholar] [CrossRef]
- Sun, W.; Cai, H.; Zhang, G.; Zhang, H.; Bao, H.; Wang, L.; Ye, J.; Qian, G.; Ge, C. Dmrt1 is required for primary male sexual differentiation in Chinese soft-shelled turtle Pelodiscus sinensis. Sci. Rep. 2017, 7, 4433. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, N.; Studer, T.; Dufresnes, C.; Ma, W.J.; Veltsos, P.; Perrin, N. Dmrt1 polymorphism and sex-chromosome differentiation in Rana temporaria. Mol. Ecol. 2017, 26, 4897–4905. [Google Scholar] [CrossRef]
- Bratuś, A.; Słota, E. DMRT1/Dmrt1, the sex determining or sex differentiating gene in Vertebrata. Folia Biol. 2006, 54, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Pannetier, M.; Chassot, A.A.; Chaboissier, M.C.; Pailhoux, E. Involvement of FOXL2 and RSPO1 in Ovarian Determination, Development, and Maintenance in Mammals. Sex. Dev. 2016, 10, 167–184. [Google Scholar] [CrossRef] [PubMed]
- Parma, P.; Radi, O.; Vidal, V.; Chaboissier, M.C.; Dellambra, E.; Valentini, S.; Guerra, L.; Schedl, A.; Camerino, G. R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat. Genet. 2006, 38, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Sun, W.; Bao, H.; Liang, X.; Li, P.; Shi, S.; Wang, Z.; Qian, G.; Ge, C. The forkhead factor Foxl2 participates in the ovarian differentiation of Chinese soft-shelled turtle Pelodiscus sinensis. Dev. Biol. 2022, 492, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wu, Q.; Wang, G.; Zhang, S.; Ma, W.; Shi, X.; Liu, H.; Wu, L.; Tian, X.; Li, X.; et al. Histomorphic analysis and expression of mRNA and miRNA in embryonic gonadal differentiation in Chinese soft-shelled turtle (Pelodiscus sinensis). Gene 2023, 893, 147913. [Google Scholar] [CrossRef]
- Liang, H.; Meng, Y.; Cao, L.; Li, X.; Zou, G. Effect of exogenous hormones on R-spondin 1 (RSPO1) gene expression and embryo development in Pelodiscus sinensis. Reprod. Fertil. Dev. 2019, 31, 1425–1433. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, X.; Qu, C.; Xu, T.; Zou, G.; Liang, H. The Important Role of Sex-Related Sox Family Genes in the Sex Reversal of the Chinese Soft-Shelled Turtle (Pelodiscus sinensis). Biology 2022, 11, 83. [Google Scholar] [CrossRef]
- Hu, Q.; Xiao, H.; Tian, H.; Meng, Y. Characterization and expression of cyp19a gene in the Chinese giant salamander Andrias davidianus. Comp. Biochem. Phys. B 2016, 192, 21–29. [Google Scholar] [CrossRef]
- Liang, H.W.; Meng, Y.; Cao, L.H.; Li, X.; Zou, G.W. Expression and characterization of the cyp19a gene and its responses to estradiol/letrozole exposure in Chinese soft-shelled turtle (Pelodiscus sinensis). Mol. Reprod. Dev. 2019, 86, 480–490. [Google Scholar] [CrossRef]
- Wang, J.; Xiang, H.; Lu, Y.; Wu, T. Role and clinical significance of TGF-beta 1 and TGF-beta R1 in malignant tumors (Review). Int. J. Biol. Sci. 2021, 47, 55. [Google Scholar]
- Wang, T.; Zhang, D.; Song, T.; Sun, M.; Zhang, J. Advances in research of TGF-B1 in human testis. Food Sci. Technol. Int. 2022, 42, 7–8. [Google Scholar]
- Steijger, T.; Abril, J.F.; Engstrom, P.G.; Kokocinski, F.; Hubbard, T.J.; Guigo, R.; Harrow, J.; Bertone, P.; Consortium, R. Assessment of transcript reconstruction methods for RNA-seq. Nat. Methods 2013, 10, 1177–1184. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Li, Y.; Fang, C.; Fu, Y.; Hu, A.; Li, C.; Zou, C.; Li, X.; Zhao, S.; Zhang, C.; Li, C. A survey of transcriptome complexity in Sus scrofa using single-molecule long-read sequencing. DNA Res. 2018, 25, 421–437. [Google Scholar] [CrossRef]
- Vembar, S.S.; Seetin, M.; Lambert, C.; Nattestad, M.; Schatz, M.C.; Baybayan, P.; Scherf, A.; Smith, M.L. Complete telomere-to-telomere de novo assembly of the Plasmodium falciparum genome through long-read (>11 kb), single molecule, real-time sequencing. DNA Res. 2016, 23, 339–351. [Google Scholar] [CrossRef]
- Roberts, R.J.; Carneiro, M.O.; Schatz, M.C. The advantages of SMRT sequencing. Genome Biol. 2013, 14, 405. [Google Scholar] [CrossRef]
- Sharon, D.; Tilgner, H.; Grubert, F.; Snyder, M. A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 2014, 31, 1009–1014. [Google Scholar] [CrossRef]
- Kuo, R.I.; Cheng, Y.; Zhang, R.; Brown, J.W.S.; Smith, J.; Archibald, A.L.; Burt, D.W. Illuminating the dark side of the human transcriptome with long read transcript sequencing. BMC Genom. 2020, 21, 751. [Google Scholar] [CrossRef]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S.N. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef]
- Zeng, D.; Chen, X.; Peng, J.; Yang, C.; Peng, M.; Zhu, W.; Xie, D.; He, P.; Wei, P.; Lin, Y.; et al. Single-molecule long-read sequencing facilitates shrimp transcriptome research. Sci. Rep. 2018, 8, 16920. [Google Scholar] [CrossRef]
- Lim, D.; Cho, Y.-M.; Lee, K.-T.; Kang, Y.; Sung, S.; Nam, J.; Park, E.-W.; Oh, S.-J.; Im, S.-K.; Kim, H. The Pig Genome Database (PiGenome): An integrated database for pig genome research. Mamm. Genome 2009, 20, 60–66. [Google Scholar] [CrossRef]
- Luo, H.; Liu, H.; Zhang, J.; Hu, B.; Zhou, C.; Xiang, M.; Yang, Y.; Zhou, M.; Jing, T.; Li, Z.; et al. Full-length transcript sequencing accelerates the transcriptome research of Gymnocypris namensis, an iconic fish of the Tibetan Plateau. Sci. Rep. 2020, 10, 9668. [Google Scholar] [CrossRef]
- Zhang, Y.; Lou, F.; Chen, J.; Han, Z.; Yang, T.; Gao, T.; Song, N. Single-molecule Real-time (SMRT) Sequencing Facilitates Transcriptome Research and Genome Annotation of the Fish Sillago sinica. Mar. Biotechnol. 2022, 24, 1002–1013. [Google Scholar] [CrossRef]
- Tilgner, H.; Grubert, F.; Sharon, D.; Snyder, M.P. Defining a personal, allele-specific, and single-molecule long-read transcriptome. Proc. Natl. Acad. Sci. USA 2014, 111, 9869–9874. [Google Scholar] [CrossRef]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucleic Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef]
- Zimin, A.V.; Puiu, D.; Luo, M.-C.; Zhu, T.; Koren, S.; Marcais, G.; Yorke, J.A.; Dvorak, J.; Salzberg, S.L. Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res. 2017, 27, 787–792. [Google Scholar] [CrossRef]
- Redwan, R.M.; Saidin, A.; Kumar, S.V. The draft genome of MD-2 pineapple using hybrid error correction of long reads. DNA Res. 2016, 23, 427–439. [Google Scholar] [CrossRef]
- Stevant, I.; Nef, S. Genetic Control of Gonadal Sex Determination and Development. Trends Genet. 2019, 35, 346–358. [Google Scholar] [CrossRef]
- Fernandino, J.I.; Hattori, R.S. Sex determination in Neotropical fish: Implications ranging from aquaculture technology to ecological assessment. Gen. Comp. Endocrinol. 2019, 273, 172–183. [Google Scholar] [CrossRef]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef]
- Gordon, S.P.; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V.; Figueroa, M.; et al. Widespread Polycistronic Transcripts in Fungi Revealed by Single-Molecule mRNA Sequencing. PLoS ONE 2015, 10, e0132628. [Google Scholar] [CrossRef]
- Diegoli, T.M.; Farr, M.; Cromartie, C.; Coble, M.D.; Bille, T.W. An optimized protocol for forensic application of the PreCR (TM) Repair Mix to multiplex STR amplification of UV-damaged DNA. Forensic Sci. Int.-Gen. 2012, 6, 498–503. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Fryxell, D.C.; Weiler, D.E.; Kinnison, M.T.; Palkovacs, E.P. Eco-Evolutionary Dynamics of Sexual Dimorphism. Trends Ecol. Evol. 2019, 34, 591–594. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Song, D.; Yu, X.; Li, Y.; Wang, X.; Cui, X.; Si, T.; Zou, X.; Wang, Y.; Wang, M.; Zhang, X. Full-Length Transcriptome Analysis of Cultivated and Wild Tetraploid Peanut. Phyton-Int. J. Exp. Bot. 2023, 92, 439–453. [Google Scholar] [CrossRef]
- Shih, J.-W.; Kung, H.-J. Long non-coding RNA and tumor hypoxia: New players ushered toward an old arena. J. Biomed. Sci. 2017, 24, 53. [Google Scholar] [CrossRef]
- Lekka, E.; Hall, J. Noncoding RNAs in disease. FEBS Lett. 2018, 592, 2884–2900. [Google Scholar] [CrossRef]
- Chang, M.X.; Zhang, J. Alternative Pre-mRNA Splicing in Mammals and Teleost Fish: A Effective Strategy for the Regulation of Immune Responses Against Pathogen Infection. Int. J. Mol. Sci. 2017, 18, 1530. [Google Scholar] [CrossRef]
- Song, H.; Wang, L.; Chen, D.; Li, F. The Function of Pre-mRNA Alternative Splicing in Mammal Spermatogenesis. Int. J. Biol. Sci. 2020, 16, 38–48. [Google Scholar] [CrossRef]
- Zhang, X.-O.; Dong, R.; Zhang, Y.; Zhang, J.-L.; Luo, Z.; Zhang, J.; Chen, L.-L.; Yang, L. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 2016, 26, 1277–1287. [Google Scholar] [CrossRef]
- Kalsotra, A.; Cooper, T.A. Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 2011, 12, 715–729. [Google Scholar] [CrossRef]
- Tian, B.; Manley, J.L. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 2017, 18, 18–30. [Google Scholar] [CrossRef]
- Guo, S.; Lin, S. mRNA alternative polyadenylation (APA) in regulation of gene expression and diseases. Genes. Dis. 2023, 10, 165–174. [Google Scholar] [CrossRef]
- Malouf, G.G.; Camparo, P.; Oudard, S.; Schleiermacher, G.; Theodore, C.; Rustine, A.; Dutcher, J.; Billemont, B.; Rixe, O.; Bompas, E.; et al. Targeted agents in metastatic Xp11 translocation/TFE3 gene fusion renal cell carcinoma (RCC): A report from the Juvenile RCC Network. Eur. J. Cancer Suppl. 2010, 7, 1834–1838. [Google Scholar] [CrossRef]
- Yamada, Y.; Rothenberg, M.E.; Lee, A.W.; Akei, H.S.; Brandt, E.B.; Williams, D.A.; Cancelas, J.A. The FIP1L1-PDGFRA fusion gene cooperates with IL-5 to induce murine hypereosinophilic syndrome (HES)/chronic eosinophilic leukemia (CEL)-like disease. Blood 2006, 107, 4071–4079. [Google Scholar] [CrossRef]
- Zhao, M.; Mishra, L.; Deng, C.X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef]
- Du, X.; Li, Q.; Yang, L.; Liu, L.; Cao, Q.; Li, Q. SMAD4 activates Wnt signaling pathway to inhibit granulosa cell apoptosis. Cell Death Dis. 2020, 11, 373. [Google Scholar] [CrossRef]
- Poggi, L.; Casarosa, S.; Carl, M. An Eye on the Wnt Inhibitory Factor Wif1. Front. Cell Dev. Biol. 2018, 6, 167. [Google Scholar] [CrossRef]
- Cai, W.; Wei, Y.; Jarnik, M.; Reich, J.; Lilly, M.A. The GATOR2 Component Wdr24 Regulates TORC1 Activity and Lysosome Function. PLoS Genet. 2016, 12, e1006036. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, W.J.; Ratamess, N.A.; Hymer, W.C.; Nindl, B.C.; Fragala, M.S. Growth Hormone(s), Testosterone, Insulin-Like Growth Factors, and Cortisol: Roles and Integration for Cellular Development and Growth with Exercise. Front. Endocrinol. 2020, 11, 33. [Google Scholar] [CrossRef]
- Börjesson, A.E.; Lagerquist, M.K.; Liu, C.; Shao, R.; Windahl, S.H.; Karlsson, C.; Sjögren, K.; Movérare-Skrtic, S.; Antal, M.C.; Krust, A.; et al. The role of estrogen receptor α in growth plate cartilage for longitudinal bone growth. J. Bone Miner. Res. 2010, 25, 2690–2700. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Library | Total Bases (bp) | ROIs | Mean Length (bp) | Read N50 |
|---|---|---|---|---|
| F 0–2 k | 12,461,189,105 | 661,271 | 18,844 | 42,250 |
| F 2 k+ | 10,011,411,220 | 686,536 | 14,582 | 33,750 |
| M 0–2 k | 10,292,266,219 | 680,809 | 15,118 | 36,750 |
| M 2 k+ | 10,677,216,778 | 752,012 | 14,198 | 32,750 |
| Total | 43,442,083,322 | 2,780,628 | - | - |
| Library | CCS | 5′ Primer | 3′ Primer | Poly(A) | FL | Of FLNC | Mean FLNC Length (bp) |
|---|---|---|---|---|---|---|---|
| F 0–2 k | 427,494 | 386,518 | 387,826 | 367,398 | 347,788 | 327,713 | 1675 |
| F 2 k+ | 337,154 | 286,996 | 288,005 | 269,671 | 244,810 | 239,600 | 3124 |
| M 0–2 k | 400,518 | 352,792 | 356,931 | 333,916 | 312,660 | 296,777 | 1572 |
| M 2 k+ | 371,683 | 304,514 | 310,103 | 288,588 | 259,044 | 257,442 | 2856 |
| - | 1,536,849 | 1,330,820 | 1,342,865 | 1,259,573 | 1,164,302 | 1,121,532 | - |
| Gene | Description | Log2 (Fold Change) (F vs. M) |
|---|---|---|
| Smad4 | Smad family 4 | 2.33 |
| Wif1 | Wnt inhibitory factor 1 | 2.73 |
| Nkd2 | Naked cuticle 2 | −3.25 |
| 17β-hsd | 17-β-Hydroxysteroid dehydrogenase | −6.22 |
| Cypj2 | Cytochrome P450, subfamily J, family 2 | 2.11 |
| Neurl1 | Neuralized E3 ubiquitin protein ligase 1 | −3.25 |
| Prp18 | Proline-rich protein 18-like | −2.46 |
| Wdr24 | Wd repeat domain 24 | 2.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, T.; Chen, G.; Cao, J.; Wang, J.; Zou, G.; Liang, H. Single-Molecule Real-Time Sequencing for Identifying Sexual-Dimorphism-Related Transcriptomes and Genes in the Chinese Soft-Shelled Turtle (Pelodiscus sinensis). Animals 2023, 13, 3704. https://doi.org/10.3390/ani13233704
Zhou T, Chen G, Cao J, Wang J, Zou G, Liang H. Single-Molecule Real-Time Sequencing for Identifying Sexual-Dimorphism-Related Transcriptomes and Genes in the Chinese Soft-Shelled Turtle (Pelodiscus sinensis). Animals. 2023; 13(23):3704. https://doi.org/10.3390/ani13233704
Chicago/Turabian StyleZhou, Tong, Guobin Chen, Jizeng Cao, Jiahui Wang, Guiwei Zou, and Hongwei Liang. 2023. "Single-Molecule Real-Time Sequencing for Identifying Sexual-Dimorphism-Related Transcriptomes and Genes in the Chinese Soft-Shelled Turtle (Pelodiscus sinensis)" Animals 13, no. 23: 3704. https://doi.org/10.3390/ani13233704