
Comparative Transcriptome Analysis Reveals Sexually Dimorphic Gene Expression in the Gonads of Brachymystax tsinlingensis Li
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Total RNA Extraction, cDNA Library Construction and Illumine Sequencing
2.2. De Novo Assembly and Functional Annotation
2.3. Identification of DEGs and Functional Enrichment Analysis
2.4. Quantitative Real-Time Polymerase Chain Reaction (qPCR) Verification
2.5. Statistics Analysis
3. Results
3.1. Sex Identification and Histological Characteristics of Juvenile B. tsinlingensis Li Gonads
3.2. Transcriptome Sequencing and Assembly
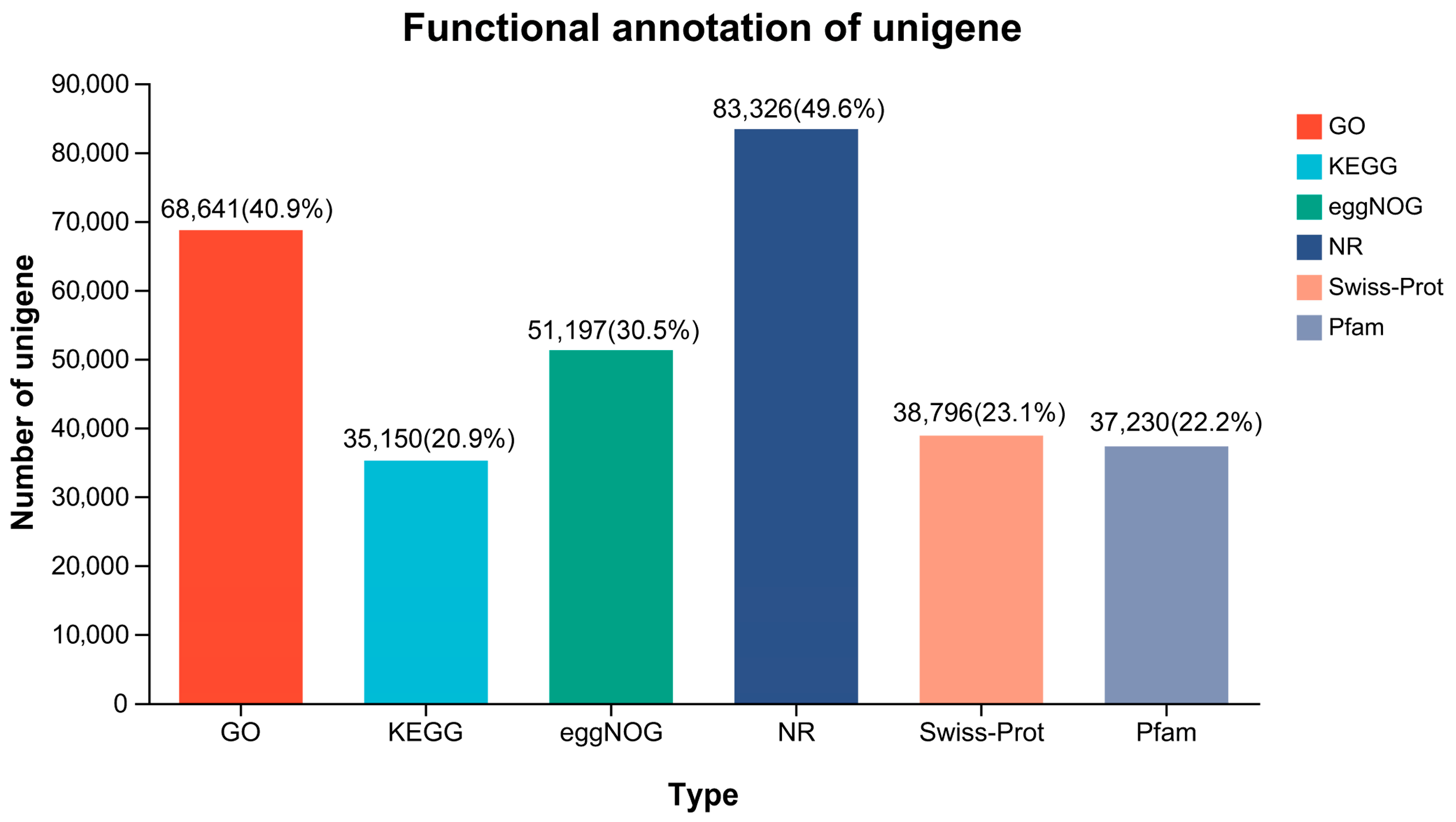
3.3. Functional Annotation
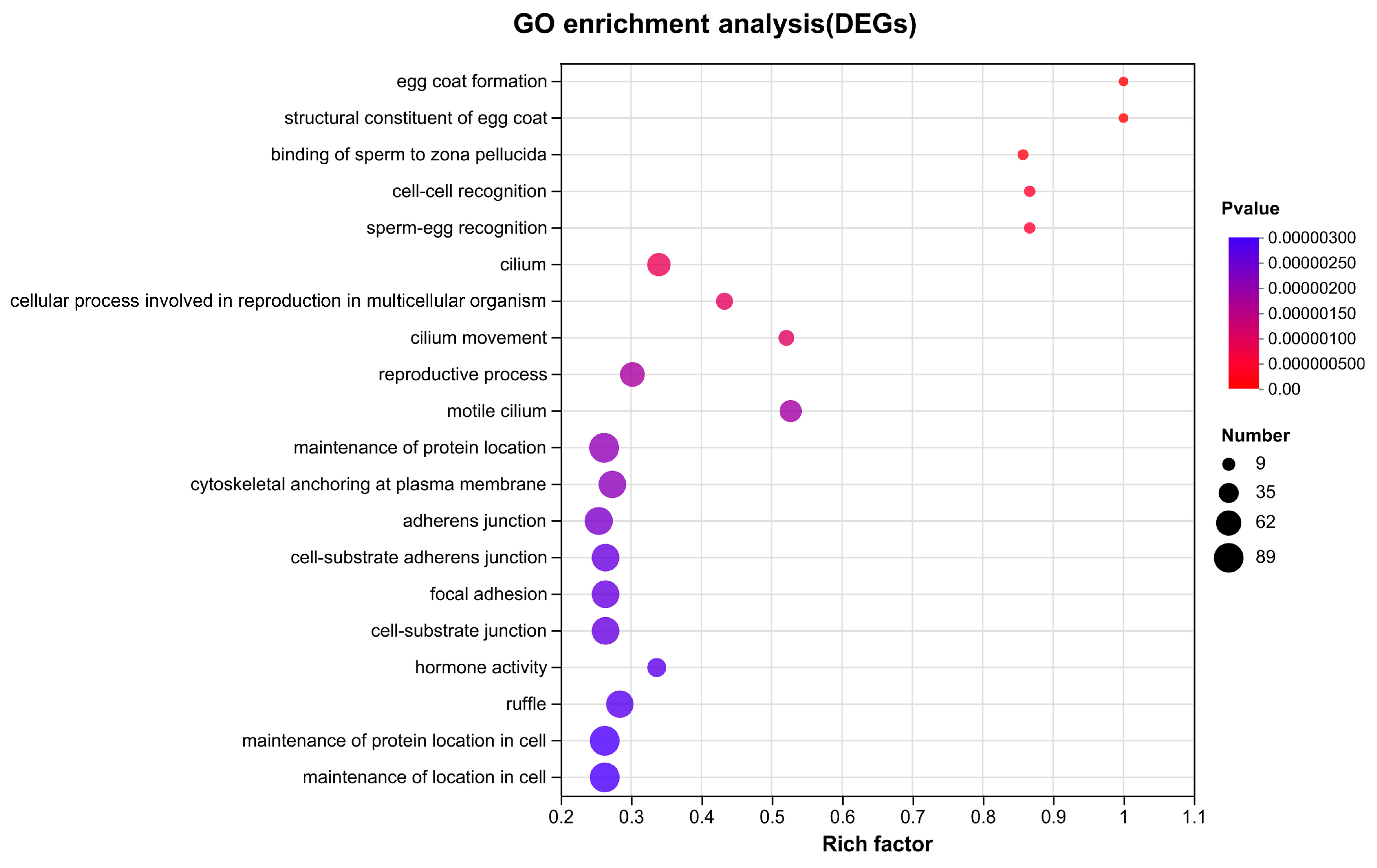
3.4. DEGs Identification and Enrichment Analysis
3.5. Validation of DEGs by qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xiong, D.; Meng, Y.; Zhang, X.; Wang, J.; Feng, G.; Shao, J.; Wang, L.; Meng, Y.; Zhang, X.; Wang, J.; et al. The validity of species of Brachymystax tsinlingensis Li based on mitochondria control region and microsatellite. Acta Hydrobiol. Sin. 2023, 47, 809–818, (In Chinese with English abstract). [Google Scholar]
- Xia, J.; Ma, Y.; Fu, C.; Fu, S.; Cooke, S.J. Effects of temperature acclimation on the critical thermal limits and swimming performance of Brachymystax lenok tsinlingensis: A threatened fish in Qinling Mountain region of China. Ecol. Res. 2017, 32, 61–70. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, C. Threatened fishes of the world: Brachymystax lenok tsinlingensis Li, 1966 (Salmonidae). Environ. Biol. Fishes. 2009, 86, 11–12. [Google Scholar] [CrossRef]
- Li, P.; Liu, Q.; Li, J.; Wang, F.; Wen, S.; Li, N. Transcriptomic responses to heat stress in gill and liver of endangered Brachymystax lenok tsinlingensis. Comp. Biochem. Physiol. Part. D Genom. Proteom. 2021, 38, 100791. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Li, P.; Wang, F.; Li, J.; Liu, H.; Li, N. De novo assembly and microsatellite marker development of the transcriptome of the endangered Brachymystax lenok tsinlingensis. Genes. Genom. 2020, 42, 727–734. [Google Scholar] [CrossRef]
- Ma, F.; Ma, R.; Zou, Y.; Zhao, L. Effect of astaxanthin on the antioxidant capacity and intestinal microbiota of tsinling lenok trout (Brachymystax lenok tsinlingensis). Mar. Biotechnol. 2022, 24, 1125–1137. [Google Scholar] [CrossRef]
- Guo, W.; Shao, J.; Li, P.; Wu, J.; Wei, Q. Morphology and ultrastructure of Brachymystax lenok tsinlingensis spermatozoa by scanning and transmission electron microscopy. Tissue Cell. 2016, 48, 321–327. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, L.; Guan, W.; Zhou, C.; Tang, B.; Cheng, Y.; Huang, J.; Xuan, F. De novo transcriptome sequencing and analysis of male and female swimming crab (Portunus trituberculatus) reproductive systems during mating embrace (stage II). BMC Genet. 2018, 19, 3. [Google Scholar] [CrossRef]
- Lin, X.; Zhou, D.; Zhang, X.; Li, G.; Zhang, Y.; Huang, C.; Zhang, Z.; Tian, C. A first insight into the gonad transcriptome of hong kong catfish (Clarias fuscus). Animals 2021, 11, 1131. [Google Scholar] [CrossRef]
- Guan, W.Z.; Jiang, K.; Lai, X.L.; Dong, Y.T.; Qiu, G.F. Comprehensive transcriptome analysis of gonadal and somatic tissues for identification of sex-related genes in the largemouth bass Micropterus salmoides. Mar. Biotechnol. 2022, 24, 588–598. [Google Scholar] [CrossRef]
- Gou, P.; Wang, Z.; Yang, J.; Wang, X.; Qiu, X. Comparative transcriptome analysis of differentially expressed genes in the testis and ovary of sea urchin (Strongylocentrotus intermedius). Fishes 2022, 7, 152. [Google Scholar] [CrossRef]
- Du, C.; Davis, J.S.; Chen, C.; Li, Z.; Cao, Y.; Sun, H.; Shao, B.S.; Lin, Y.X.; Wang, Y.S.; Yang, L.G.; et al. FGF2/FGRG signaling promotes cumulus-oocyte complex maturation in vitro. Reproduction 2021, 161, 205–214. [Google Scholar] [CrossRef]
- Cheng, J.; Liu, Y. Knockout of cyclin B1 in granulosa cells causes female subfertility. Cell Cycle 2022, 21, 1867–1878. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.L.; Zhou, C.X.; Zhang, X.L.; Liu, P.; Jin, Z.; Zhu, G.Y.; Ma, Y.; Li, J.; Yang, Z.X.; Zhang, D. ZP3 is required for germinal vesicle breakdown in mouse oocyte meiosis. Sci. Rep. 2017, 7, 41272. [Google Scholar] [CrossRef]
- Su, Y.Q.; Sugiura, K.; Wigglesworth, K.; O’Brien, M.J.; Affourtit, J.P.; Pangas, S.A.; Matzuk, M.M.; Eppig, J.J. Oocyte regulation of metabolic cooperativity between mouse cumulus cells and oocytes: BMP15 and GDF9 control cholesterol biosynthesis in cumulus cells. Development 2008, 135, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Dranow, D.B.; Hu, K.; Bird, A.M.; Lawry, S.T.; Adams, M.T.; Sanchez, A.; Amatruda, J.F.; Draper, B.W. Bmp15 is an oocyte-produced signal required for maintenance of the adult female sexual phenotype in zebrafish. PLoS Genet. 2016, 12, e1006323. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, D.; Li, M.; Mustapha, U.F.; Tian, C.; Chen, H.; Huang, Y.; Deng, S.; Wu, T.; Zhu, C.; et al. Genome survey of male and female spotted scat (Scatophagus argus). Animals 2019, 9, 1117. [Google Scholar] [CrossRef]
- Nanda, I.; Kondo, M.; Hornung, U.; Asakawa, S.; Winkler, C.; Shimizu, A.; Shan, Z.; Haaf, T.; Shimizu, N.; Shima, A.; et al. A duplicated copy of DMRT1 in the sex-determining region of the Y chromosome of the medaka, Oryzias latipes. Proc. Natl. Acad. Sci. USA 2002, 99, 11778–11783. [Google Scholar] [CrossRef]
- Lei, N.; Hornbaker, K.I.; Rice, D.A.; Karpova, T.; Agbor, V.A.; Heckert, L.L. Sex-specific differences in mouse DMRT1 expression are both cell type- and stage-dependent during gonad development. Biol. Reprod. 2007, 77, 466–475. [Google Scholar] [CrossRef]
- Raymond, C.S.; Kettlewell, J.R.; Hirsch, B.; Bardwell, V.J.; Zarkower, D. Expression of Dmrt1 in the genital ridge of mouse and chicken embryos suggests a role in vertebrate sexual development. Dev. Biol. 1999, 215, 208–220. [Google Scholar] [CrossRef]
- Huang, S.; Ye, L.; Chen, H. Sex determination and maintenance: The role of DMRT1 and FOXL2. Asian J. Androl. 2017, 19, 619–624. [Google Scholar] [PubMed]
- Wei, Y.D.; Du, X.M.; Yang, D.H.; Ma, F.L.; Yu, X.W.; Zhang, M.F.; Li, N.; Peng, S.; Liao, M.Z.; Li, G.P.; et al. Dmrt1 regulates the immune response by repressing the TLR4 signaling pathway in goat male germline stem cells. Zool. Res. 2021, 42, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Kajiura-Kobayashi, H.; Guan, G.; Nagahama, Y. Sexual dimorphic expression of DMRT1 and Sox9a during gonadal differentiation and hormone-induced sex reversal in the teleost fish nile tilapia (Oreochromis niloticus). Dev. Dyn. 2008, 237, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Masuyama, H.; Yamada, M.; Kamei, Y.; Fujiwara-Ishikawa, T.; Todo, T.; Nagahama, Y.; Matsuda, M. Dmrt1 mutation causes a male-to-female sex reversal after the sex determination by Dmy in the medaka. Chromosom. Res. 2012, 20, 163–176. [Google Scholar] [CrossRef]
- Webster, K.A.; Schach, U.; Ordaz, A.; Steinfeld, J.S.; Draper, B.W.; Siegfried, K.R. Dmrt1 is necessary for male sexual development in zebrafish. Dev. Biol. 2017, 422, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Koh, E.; Sakugawa, N.; Sato, H.; Hayashi, H.; Namiki, M.; Sengoku, K. Two single nucleotide polymorphisms in PRDM9 (MEISETZ) gene may be a genetic risk factor for japanese patients with azoospermia by meiotic arrest. J. Assist. Reprod. Genet. 2008, 25, 553–557. [Google Scholar] [CrossRef]
- Zangen, D.; Kaufman, Y.; Zeligson, S.; Perlberg, S.; Fridman, H.; Kanaan, M.; Abdulhadi-Atwan, M.; Abu Libdeh, A.; Gussow, A.; Kisslov, I.; et al. XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that abolishes coactivation of estrogen-driven transcription. Am. J. Hum. Genet. 2011, 89, 572–579. [Google Scholar] [CrossRef]
- Karahalil, B.; Hogue, B.A.; de Souza-Pinto, N.C.; Bohr, V.A. Base excision repair capacity in mitochondria and nuclei: Tissue-specific variations. FASEB J. 2002, 16, 1895–1902. [Google Scholar] [CrossRef]
- Lindahl, T.; Karran, P.; Wood, R.D. DNA excision repair pathways. Curr. Opin. Genet. Dev. 1997, 7, 158–169. [Google Scholar] [CrossRef]
- Walter, C.A.; Trolian, D.A.; McFarland, M.B.; Street, K.A.; Gurram, G.R.; McCarrey, J.R. Xrcc-1 expression during male meiosis in the mouse. Biol. Reprod. 1996, 55, 630–635. [Google Scholar] [CrossRef]
- Walter, C.A.; Lu, J.; Bhakta, M.; Zhou, Z.Q.; Thompson, L.H.; McCarrey, J.R. Testis and somatic Xrcc-1 DNA repair gene expression. Somat. Cell Mol. Genet. 1994, 20, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Mackey, Z.B.; Ramos, W.; Levin, D.S.; Walter, C.A.; McCarrey, J.R.; Tomkinson, A.E. An alternative splicing event which occurs in mouse pachytene spermatocytes generates a form of DNA ligase III with distinct biochemical properties that may function in meiotic recombination. Mol. Cell. Biol. 1997, 17, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Alcivar, A.A.; Hake, L.E.; Hecht, N.B. DNA polymerase-beta and poly (ADP) ribose polymerase mRNAs are differentially expressed during the development of male germinal cells. Biol. Reprod. 1992, 46, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Intano, G.W.; McMahan, C.A.; McCarrey, J.R.; Walter, R.B.; McKenna, A.E.; Matsumoto, Y.; MacInnes, M.A.; Chen, D.J.; Walter, C.A. Base excision repair is limited by different proteins in male germ cell nuclear extracts prepared from young and old mice. Mol. Cell. Biol. 2002, 22, 2410–2418. [Google Scholar] [CrossRef] [PubMed]
- Garcia, T.X.; DeFalco, T.; Capel, B.; Hofmann, M.C. Constitutive activation of NOTCH1 signaling in Sertoli cells causes gonocyte exit from quiescence. Dev. Biol. 2013, 377, 188–201. [Google Scholar] [CrossRef]
- Garcia, T.X.; Parekh, P.; Gandhi, P.; Sinha, K.; Hofmann, M.C. The NOTCH ligand JAG1 regulates GDNF expression in Sertoli cells. Stem Cells Dev. 2017, 26, 585–598. [Google Scholar] [CrossRef]
- Kamińska, A.; Marek, S.; Pardyak, L.; Brzoskwinia, M.; Bilinska, B.; Hejmej, A. Crosstalk between androgen-ZIP9 signaling and notch pathway in rodent sertoli cells. Int. J. Mol. Sci. 2020, 21, 8275. [Google Scholar] [CrossRef]
- Wang, X.; Guo, X.; He, X.; Di, R.; Zhang, X.; Zhang, J.; Chu, M. Integrated proteotranscriptomics of the hypothalamus reveals altered regulation associated with the FecB mutation in the BMPR1B gene that affects prolificacy in small tail han sheep. Biology 2022, 12, 72. [Google Scholar] [CrossRef]
- Chi, M.L.; Ni, M.; Li, J.F.; He, F.; Qian, K.; Zhang, P.; Chai, S.H.; Wen, H.S. Molecular cloning and characterization of gonadotropin subunits (GTHα, FSHβ and LHβ) and their regulation by hCG and GnRHa in Japanese sea bass (Lateolabrax japonicas) in vivo. Fish Physiol. Biochem. 2015, 41, 587–601. [Google Scholar] [CrossRef]
- Mu, R.; Yu, Y.Y.; Gegen, T.; Wen, D.; Wang, F.; Chen, Z.; Xu, W.B. Transcriptome analysis of ovary tissues from low- and high-yielding Changshun green-shell laying hens. BMC Genom. 2021, 22, 349. [Google Scholar] [CrossRef]
- Wei, S.; Kang, X.; Yang, C.; Wang, F.; Dai, T.; Guo, X.; Ma, Z.; Li, C.; Zhao, H.; Dan, X. Analysis of reproduction-related transcriptomes on pineal-hypothalamic-pituitary-ovarian tissues during estrus and anestrus in Tan sheep. Front. Vet. Sci. 2022, 9, 1068882. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Q.; Hu, S.; Wang, G.; Hu, J.; Zhang, J.; Li, L.; Hu, B.; He, H.; Liu, H.; Xia, L.; et al. Comparative transcriptome analysis suggests key roles for 5-hydroxytryptamlne receptors in control of goose egg production. Genes 2020, 11, 455. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.M. Regulation of the ovarian follicular vasculature. Reprod. Biol. Endocrinol. 2006, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.M.; Duncan, W.C. SRB reproduction, fertility and development award lecture 2008. Regulation and manipulation of angiogenesis in the ovary and endometrium. Reprod. Fertil. Dev. 2009, 21, 377–392. [Google Scholar] [CrossRef]
- Baerwald, A.R.; Adams, G.P.; Pierson, R.A. Ovarian antral folliculogenesis during the human menstrual cycle: A review. Hum. Reprod. Update 2012, 18, 73–91. [Google Scholar] [CrossRef]
- Asiamah, C.A.; Liu, Y.; Ye, R.; Pan, Y.; Lu, L.L.; Zou, K.; Zhao, Z.; Jiang, P.; Su, Y. Polymorphism analysis and expression profile of the estrogen receptor 2 gene in leizhou black duck. Poult. Sci. 2022, 101, 101630. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Bases | Clean Bases | Raw Reads | Clean Reads | Mapped Reads(Ratio) | Error Rate (%) | Q30(%) | GC Content (%) |
|---|---|---|---|---|---|---|---|---|
| F_1 | 13,429,748,532 | 12,982,936,212 | 88,938,732 | 87,804,560 | 69,352,106 (78.98%) | 0.025 | 94.2 | 49.59 |
| F_2 | 11,051,384,074 | 10,653,775,488 | 73,187,974 | 72,275,082 | 57,395,498 (79.41%) | 0.025 | 94.26 | 49.87 |
| F_3 | 14,737,855,492 | 14,309,137,436 | 97,601,692 | 96,375,314 | 76,066,826 (78.93%) | 0.0251 | 94.18 | 49.47 |
| F_4 | 14,257,279,268 | 13,826,078,968 | 94,419,068 | 93,274,920 | 73,624,950 (78.93%) | 0.0249 | 94.3 | 49.54 |
| F_5 | 11,127,009,404 | 10,761,812,681 | 73,688,804 | 72,655,432 | 57,117,684 (78.61%) | 0.0251 | 94.13 | 49.58 |
| M_1 | 12,286,990,498 | 11,868,538,929 | 81,370,798 | 80,223,798 | 63,205,096 (78.79%) | 0.0247 | 94.44 | 49.32 |
| M_2 | 12,421,889,066 | 11,962,639,831 | 82,264,166 | 81,048,350 | 62,965,986 (77.69%) | 0.025 | 94.17 | 50.2 |
| M_3 | 11,909,431,910 | 11,467,120,995 | 78,870,410 | 77,846,102 | 61,366,132 (78.83%) | 0.0245 | 94.69 | 50.05 |
| M_4 | 12,657,506,144 | 12,189,596,284 | 83,824,544 | 82,737,530 | 64,825,426 (78.35%) | 0.0245 | 94.68 | 49.86 |
| M_5 | 12,159,281,644 | 11,741,956,029 | 80,525,044 | 79,461,734 | 61,365,664 (77.23%) | 0.0247 | 94.53 | 50.09 |
| Type | Unigene | Transcript |
|---|---|---|
| 200~500 (bp) | 95,750 (57%) | 123,592 (51%) |
| 501~1000 (bp) | 38,003 (23%) | 53,103 (22%) |
| 1001~2000 (bp) | 18,088 (11%) | 31,700 (13%) |
| 2001~3000 (bp) | 7510 (5%) | 15,145 (7%) |
| 3001~4000 (bp) | 4002 (2%) | 8236 (3%) |
| >4001 (bp) | 4551 (3%) | 9338 (4%) |
| Total number | 167,904 | 241,114 |
| Total base | 140,382,062 | 240,137,939 |
| Largest length (bp) | 26,461 | 26,461 |
| Smallest length (bp) | 201 | 201 |
| Average length (bp) | 836.09 | 995.95 |
| N50 length (bp) | 1452 | 1943 |
| E90N50 length (bp) | 3227 | 3100 |
| Fragment mapped percent (%) | 60.856 | 77.793 |
| GC percent (%) | 45.79 | 46.68 |
| TransRate score | 0.27696 | 0.35206 |
| BUSCO score | C:93.6% [S:78.8%; D:14.8%] | C:93.6% [S:78.8%; D:14.8%] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, L.; Ye, H.; Yue, H.; Leng, X.; Ruan, R.; Du, H.; Li, C.; Wu, J. Comparative Transcriptome Analysis Reveals Sexually Dimorphic Gene Expression in the Gonads of Brachymystax tsinlingensis Li. Animals 2023, 13, 3690. https://doi.org/10.3390/ani13233690
Huang L, Ye H, Yue H, Leng X, Ruan R, Du H, Li C, Wu J. Comparative Transcriptome Analysis Reveals Sexually Dimorphic Gene Expression in the Gonads of Brachymystax tsinlingensis Li. Animals. 2023; 13(23):3690. https://doi.org/10.3390/ani13233690
Chicago/Turabian StyleHuang, Ling, Huan Ye, Huamei Yue, Xiaoqian Leng, Rui Ruan, Hao Du, Chuangju Li, and Jinming Wu. 2023. "Comparative Transcriptome Analysis Reveals Sexually Dimorphic Gene Expression in the Gonads of Brachymystax tsinlingensis Li" Animals 13, no. 23: 3690. https://doi.org/10.3390/ani13233690