
Whole-Genome Resequencing Reveals Selection Signal Related to Sheep Wool Fineness
 , , ,
, , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection and Sequencing
2.3. Alignments and Quality Control
2.4. Population Structure Analysis
2.5. Selection Signal Analyses
2.6. Detection and Annotation of Candidate Genes
2.7. Candidate Gene Enrichment Analysis
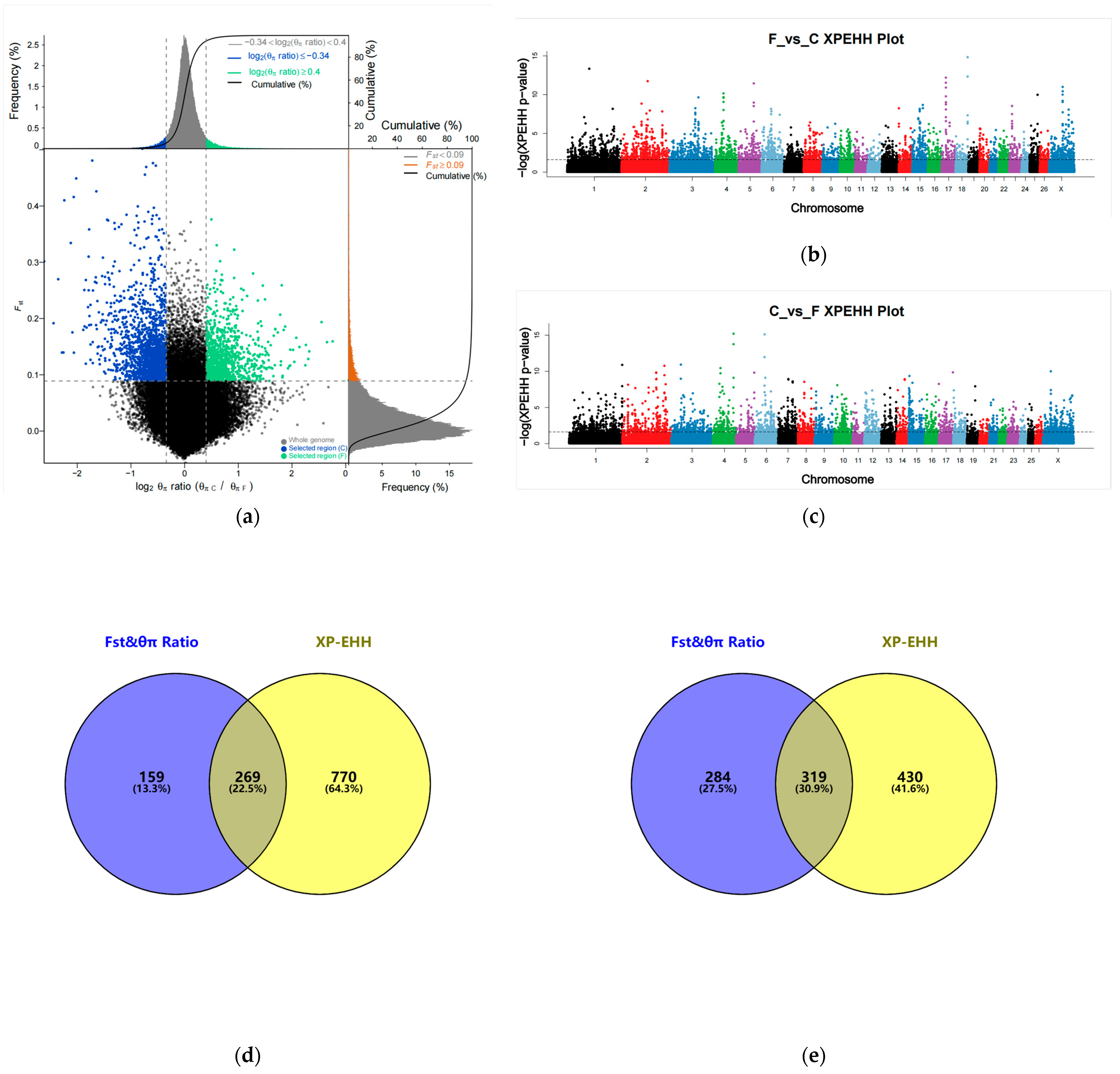
3. Results
3.1. Genetic Variation and Population Genetic Analysis
3.2. Selection Signaling Analysis
3.3. Enrichment Analysis
4. Discussion
4.1. Genetic Variation and Population Genetic Analysis
4.2. Annotation of Candidate Genes
4.3. Analysis of Enrichment Results
4.4. Exploration of Factors Affecting Fineness
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kalds, P.; Zhou, S.; Gao, Y.; Cai, B.; Huang, S.; Chen, Y.; Wang, X. Genetics of the phenotypic evolution in sheep: A molecular look at diversity-driving genes. Genet. Sel. Evol. 2022, 54, 61. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, H.; Yang, H.; Wang, S.; Rong, E.; Pei, W.; Li, H.; Wang, N. Genome-wide association study for wool production traits in a chinese merino sheep population. PLoS ONE 2014, 9, e107101. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, W.; Zheng, X.; Liu, Z.; Yang, G.; Hu, X.; Mou, C. Comparative investigation of coarse and fine wool sheep skin indicates the early regulators for skin and wool diversity. Gene 2020, 758, 144968. [Google Scholar] [CrossRef]
- Zhao, B.; Luo, H.; Huang, X.; Wei, C.; Di, J.; Tian, Y.; Fu, X.; Li, B.; Liu, G.E.; Fang, L.; et al. Integration of a single-step genome-wide association study with a multi-tissue transcriptome analysis provides novel insights into the genetic basis of wool and weight traits in sheep. Genet. Sel. Evol. 2021, 53, 56. [Google Scholar] [CrossRef] [PubMed]
- Langbein, L.; Rogers, M.A.; Praetzel, S.; Aoki, N.; Winter, H.; Schweizer, J. A novel epithelial keratin, hk6irs1, is expressed differentially in all layers of the inner root sheath, including specialized huxley cells (flügelzellen) of the human hair follicle. J. Investig. Dermatol. 2002, 118, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Langbein, L.; Rogers, M.A.; Praetzel, S.; Winter, H.; Schweizer, J. K6irs1, k6irs2, k6irs3, and k6irs4 represent the inner-root-sheath-specific type ii epithelial keratins of the human hair follicle. J. Investig. Dermatol. 2003, 120, 512–522. [Google Scholar]
- Langbein, L.; Rogers, M.A.; Praetzel-Wunder, S.; Helmke, B.; Schirmacher, P.; Schweizer, J. K25 (k25irs1), k26 (k25irs2), k27 (k25irs3), and k28 (k25irs4) represent the type i inner root sheath keratins of the human hair follicle. J. Investig. Dermatol. 2006, 126, 2377–2386. [Google Scholar] [CrossRef]
- Shi, R.; Li, S.; Liu, P.; Zhang, S.; Wu, Z.; Wu, T.; Gong, S.; Wan, Y. Identification of key genes and signaling pathways related to hetian sheep wool density by rna-seq technology. PLoS ONE 2022, 17, e0265989. [Google Scholar] [CrossRef]
- Liang, B.; Bai, T.; Zhao, Y.; Han, J.; He, X.; Pu, Y.; Wang, C.; Liu, W.; Ma, Q.; Tian, K.; et al. Two mutations at krt74 and edar synergistically drive the fine-wool production in chinese sheep. J. Adv. Res. 2023; Epub ahead of print. [Google Scholar] [CrossRef]
- Kimura, R.; Watanabe, C.; Kawaguchi, A.; Kim, Y.-I.; Park, S.-B.; Maki, K.; Ishida, H.; Yamaguchi, T. Common polymorphisms in wnt10a affect tooth morphology as well as hair shape. Hum. Mol. Genet. 2015, 24, 2673–2680. [Google Scholar] [CrossRef]
- Cai, W.; Xu, Y.; Bai, Z.; Lin, G.; Wang, L.; Dou, X.; Han, D.; Wang, Z.; Wang, J.; Zhang, X.; et al. Association analysis for snps of baat and col1a1 genes with cashmere production performance and other production traits in liaoning cashmere goats. Anim. Biotechnol. 2022. [Google Scholar] [CrossRef]
- Zhao, B.; Luo, H.; He, J.; Huang, X.; Chen, S.; Fu, X.; Zeng, W.; Tian, Y.; Liu, S.; Li, C.-J.; et al. Comprehensive transcriptome and methylome analysis delineates the biological basis of hair follicle development and wool-related traits in merino sheep. BMC Biol. 2021, 19, 197. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Guo, T.; Lu, Z.; Liu, J.; Zhu, S.; Qiao, G.; Han, M.; Yuan, C.; Wang, T.; Li, F.; et al. Genome-wide association studies detects candidate genes for wool traits by re-sequencing in chinese fine-wool sheep. BMC Genom. 2021, 22, 127. [Google Scholar] [CrossRef] [PubMed]
- Thulabandu, V.; Nehila, T.; Ferguson, J.W.; Atit, R.P. Dermal ezh2 orchestrates dermal differentiation and epidermal proliferation during murine skin development. Dev. Biol. 2021, 478, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Du, K.-T.; Deng, J.-Q.; He, X.-G.; Liu, Z.-P.; Peng, C.; Zhang, M.-S. Mir-214 regulates the human hair follicle stem cell proliferation and differentiation by targeting ezh2 and wnt/β-catenin signaling way in vitro. Tissue Eng. Regen. Med. 2018, 15, 341–350. [Google Scholar] [CrossRef]
- Ren, X.; Xia, W.; Xu, P.; Shen, H.; Dai, X.; Liu, M.; Shi, Y.; Ye, X.; Dang, Y. Lgr4 deletion delays the hair cycle and inhibits the activation of hair follicle stem cells. J. Investig. Dermatol. 2020, 140, 1706–1712.e4. [Google Scholar] [CrossRef]
- Liu, Y.; Guerrero-Juarez, C.F.; Xiao, F.; Shettigar, N.U.; Ramos, R.; Kuan, C.-H.; Lin, Y.-C.; de Jesus Martinez Lomeli, L.; Park, J.M.; Oh, J.W.; et al. Hedgehog signaling reprograms hair follicle niche fibroblasts to a hyper-activated state. Dev. Cell 2022, 57, 1758–1775.e7. [Google Scholar] [CrossRef]
- Krugluger, W.; Stiefsohn, K.; Moser, K.; Moser, C.; Laciak, K. Evaluation of gene expression patterns in micrografts demonstrate induction of catagen-like processes during storage. Dermatol. Surg. 2017, 43, 275–280. [Google Scholar] [CrossRef]
- Zhang, Y.; Tomann, P.; Andl, T.; Gallant, N.M.; Huelsken, J.; Jerchow, B.; Birchmeier, W.; Paus, R.; Piccolo, S.; Mikkola, M.L.; et al. Reciprocal requirements for eda/edar/nf-kappab and wnt/beta-catenin signaling pathways in hair follicle induction. Dev. Cell 2009, 17, 49–61. [Google Scholar] [CrossRef]
- Zhang, Y.; Andl, T.; Yang, S.H.; Teta, M.; Liu, F.; Seykora, J.T.; Tobias, J.W.; Piccolo, S.; Schmidt-Ullrich, R.; Nagy, A.; et al. Activation of beta-catenin signaling programs embryonic epidermis to hair follicle fate. Development 2008, 135, 2161–2172. [Google Scholar] [CrossRef]
- Hochfeld, L.M.; Bertolini, M.; Broadley, D.; Botchkareva, N.V.; Betz, R.C.; Schoch, S.; Nöthen, M.M.; Heilmann-Heimbach, S. Evidence for a functional interaction of wnt10a and ebf1 in male-pattern baldness. PLoS ONE 2021, 16, e0256846. [Google Scholar] [CrossRef]
- Melnik, B.C. The role of transcription factor foxo1 in the pathogenesis of acne vulgaris and the mode of isotretinoin action. G. Ital. Dermatol. Venereol. 2010, 145, 559–571. [Google Scholar] [PubMed]
- Wang, S.; Wu, T.; Sun, J.; Li, Y.; Yuan, Z.; Sun, W. Single-cell transcriptomics reveals the molecular anatomy of sheep hair follicle heterogeneity and wool curvature. Front. Cell Dev. Biol. 2021, 9, 800157. [Google Scholar] [CrossRef] [PubMed]
- Nissimov, J.N.; Das Chaudhuri, A.B. Hair curvature: A natural dialectic and review. Biol. Rev. Camb. Philos. Soc. 2014, 89, 723–766. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Yanase, T.; Morinaga, H.; Okabe, T.; Nomura, M.; Daitoku, H.; Fukamizu, A.; Kato, S.; Takayanagi, R.; Nawata, H. Insulin-like growth factor 1/insulin signaling activates androgen signaling through direct interactions of foxo1 with androgen receptor. J. Biol. Chem. 2007, 282, 7329–7338. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C. Foxo1—The key for the pathogenesis and therapy of acne? J. Dtsch. Dermatol. Ges. 2010, 8, 105–114. [Google Scholar] [CrossRef]
- Bulbach, S. The importance of wool. Orient. Rug Rev. 1988, 8. Available online: http://www.bulbach.com/library/orientalRugReview.pdf (accessed on 11 September 2023).
- Yang, J.; Li, W.-R.; Lv, F.-H.; He, S.-G.; Tian, S.-L.; Peng, W.-F.; Sun, Y.-W.; Zhao, Y.-X.; Tu, X.-L.; Zhang, M.; et al. Whole-genome sequencing of native sheep provides insights into rapid adaptations to extreme environments. Mol. Biol. Evol. 2016, 33, 2576–2592. [Google Scholar] [CrossRef]
- Porto-Neto, L.R.; Lee, S.H.; Lee, H.K.; Gondro, C. Detection of signatures of selection using fst. Methods Mol. Biol. 2013, 1019, 423–436. [Google Scholar]
- Weir, B.S.; Cockerham, C.C. Estimating f-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Lu, J.; Fei, X.; Lu, Z.; Quan, K.; Liu, Y.; Chu, M.; Di, R.; Wei, C.; Wang, H. Selection signatures analysis reveals genes associated with high-altitude adaptation in tibetan goats from nagqu, tibet. Animals 2020, 10, 1599. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. Plink: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Wei, C.; Wang, H.; Liu, G.; Zhao, F.; Kijas, J.W.; Ma, Y.; Lu, J.; Zhang, L.; Cao, J.; Wu, M.; et al. Genome-wide analysis reveals adaptation to high altitudes in tibetan sheep. Sci. Rep. 2016, 6, 26770. [Google Scholar] [CrossRef]
- Bruno, W.J.; Socci, N.D.; Halpern, A.L. Weighted neighbor joining: A likelihood-based approach to distance-based phylogeny reconstruction. Mol. Biol. Evol. 2000, 17, 189–197. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. Mega6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (itol) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Sabeti, P.C.; Schaffner, S.F.; Fry, B.; Lohmueller, J.; Varilly, P.; Shamovsky, O.; Palma, A.; Mikkelsen, T.S.; Altshuler, D.; Lander, E.S. Positive natural selection in the human lineage. Science 2006, 312, 1614–1620. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and vcftools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Scheet, P.; Stephens, M. A fast and flexible statistical model for large-scale population genotype data: Applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 2006, 78, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. Genetical structure of populations. Nature 1950, 166, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-C.; Na, Y.-P.; Lee, J.-B. Expression of peroxiredoxin ii in vascular tumors of the skin: A novel vascular marker of endothelial cells. J. Am. Acad. Dermatol. 2003, 49, 487–491. [Google Scholar] [CrossRef]
- Li, X.; Su, R.; Wan, W.; Zhang, W.; Jiang, H.; Qiao, X.; Fan, Y.; Zhang, Y.; Wang, R.; Liu, Z. Identification of selection signals by large-scale whole-genome resequencing of cashmere goats. Sci. Rep. 2017, 7, 15142. [Google Scholar] [CrossRef]
- Ye, R.; Tian, Y.; Huang, Y.; Zhang, Y.; Wang, J.; Sun, X.; Zhou, H.; Zhang, D.; Pan, W. Genome-wide analysis of genetic diversity in plasmodium falciparum isolates from china–myanmar border. Front. Genet. 2019, 10, 1065. [Google Scholar] [CrossRef]
- Valipour, S.; Karimi, K.; Do, D.N.; Barrett, D.; Sargolzaei, M.; Plastow, G.; Wang, Z.; Miar, Y. Genome-wide detection of selection signatures for pelt quality traits and coat color using whole-genome sequencing data in american mink. Genes 2022, 13, 1939. [Google Scholar] [CrossRef]
- Wang, F.; Zha, Z.; He, Y.; Li, J.; Zhong, Z.; Xiao, Q.; Tan, Z. Genome-wide re-sequencing data reveals the population structure and selection signature analysis of tunchang pigs in china. Animals 2023, 13, 1835. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. Kobas-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- Utsunomiya, Y.T.; Pérez O’Brien, A.M.; Sonstegard, T.S.; Van Tassell, C.P.; do Carmo, A.S.; Meszaros, G.; Sölkner, J.; Garcia, J.F. Detecting loci under recent positive selection in dairy and beef cattle by combining different genome-wide scan methods. PLoS ONE 2013, 8, e64280. [Google Scholar] [CrossRef] [PubMed]
- Foll, M.; Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A bayesian perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef] [PubMed]
- Voight, B.F.; Kudaravalli, S.; Wen, X.; Pritchard, J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006, 4, e72. [Google Scholar]
- Arzik, Y.; Kizilaslan, M. Genome-wide scan of wool production traits in akkaraman sheep. Genes 2023, 14, 713. [Google Scholar] [CrossRef]
- Wu, C.; Ma, S.; Zhao, B.; Qin, C.; Wu, Y.; Di, J.; Suo, L.; Fu, X. Drivers of plateau adaptability in cashmere goats revealed by genomic and transcriptomic analyses. BMC Genom. 2023, 24, 428. [Google Scholar] [CrossRef]
- Mohri, Y.; Kato, S.; Umezawa, A.; Okuyama, R.; Nishimori, K. Impaired hair placode formation with reduced expression of hair follicle-related genes in mice lacking lgr4. Dev. Dyn. 2008, 237, 2235–2242. [Google Scholar] [CrossRef]
- Tian, Y.Z.; Usman, T.; Tian, K.C.; Di, J.; Huang, X.X.; Xu, X.M.; Tulafu, H.; Wu, W.W.; Fu, X.F.; Bai, Y.; et al. Comparative study of 13 candidate genes applying multi-reference normalization to detect the expression of different fineness in skin tissues of wool sheep. Genet. Mol. Res. 2017, 16, gmr16018905. [Google Scholar] [CrossRef]
- Zhong, H.-B.; Chu, Q.-S.; Xiang, J.J.; Zhang, A.L.; Chen, E.Q.; Shen, X.-R.; Liao, X.-H. Spatial association of sema3c with nerve endings/terminal schwann cells in hair follicle isthmus region. Int. J. Dev. Neurosci. 2020, 80, 737–741. [Google Scholar] [CrossRef]
- Wang, F.H.; Zhang, L.; Gong, G.; Yan, X.C.; Zhang, L.T.; Zhang, F.T.; Liu, H.F.; Lv, Q.; Wang, Z.Y.; Wang, R.J.; et al. Genome-wide association study of fleece traits in inner mongolia cashmere goats. Anim. Genet. 2021, 52, 375–379. [Google Scholar] [CrossRef]
- Yu, M.; Bell, R.H.; Ho, M.M.; Leung, G.; Haegert, A.; Carr, N.; Shapiro, J.; McElwee, K.J. Deficiency in nucleotide excision repair family gene activity, especially ercc3, is associated with non-pigmented hair fiber growth. PLoS ONE 2012, 7, e34185. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, M.; Shen, S.; He, L.; Zhang, X.; Zuo, X.; Yang, S. Variants in sell, mrps36p2, tp63, ddb2, cacna1h, adam19, gnai1, cdh13 and gabrg2 interact to confer risk of acne in chinese population. J. Dermatol. 2015, 42, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-W.; Su, C.-H.; Chen, H.-H.; Huang, C.-W.; Tsao, L.-P.; Tsao, Y.-P.; Chen, S.-L. Ddb2 is a novel ar interacting protein and mediates ar ubiquitination/degradation. Int. J. Biochem. Cell Biol. 2012, 44, 1952–1961. [Google Scholar] [CrossRef] [PubMed]
- Russell, L.E.; Harrison, W.J.; Bahta, A.W.; Zouboulis, C.C.; Burrin, J.M.; Philpott, M.P. Characterization of liver x receptor expression and function in human skin and the pilosebaceous unit. Exp. Dermatol. 2007, 16, 844–852. [Google Scholar] [CrossRef]
- Sun, X.; Jiang, J.; Wang, G.; Zhou, P.; Li, J.; Chen, C.; Liu, L.; Li, N.; Xia, Y.; Ren, H. Genome-wide association analysis of nine reproduction and morphological traits in three goat breeds from southern china. Anim. Biosci. 2023, 36, 191–199. [Google Scholar] [CrossRef]
- Zhao, R.; Li, J.; Liu, N.; Li, H.; Liu, L.; Yang, F.; Li, L.; Wang, Y.; He, J. Transcriptomic analysis reveals the involvement of lncrna-mirna-mrna networks in hair follicle induction in aohan fine wool sheep skin. Front. Genet. 2020, 11, 590. [Google Scholar] [CrossRef]
- Udhane, S.S.; Pandey, A.V.; Hofer, G.; Mullis, P.E.; Flück, C.E. Retinoic acid receptor beta and angiopoietin-like protein 1 are involved in the regulation of human androgen biosynthesis. Sci. Rep. 2015, 5, 10132. [Google Scholar] [CrossRef]
- Wolf, S.S.; Patchev, V.K.; Obendorf, M. A novel variant of the putative demethylase gene, s-jmjd1c, is a coactivator of the ar. Arch. Biochem. Biophys. 2007, 460, 56–66. [Google Scholar] [CrossRef]
- Jin, G.; Sun, J.; Kim, S.-T.; Feng, J.; Wang, Z.; Tao, S.; Chen, Z.; Purcell, L.; Smith, S.; Isaacs, W.B.; et al. Genome-wide association study identifies a new locus jmjd1c at 10q21 that may influence serum androgen levels in men. Hum. Mol. Genet. 2012, 21, 5222–5228. [Google Scholar] [CrossRef]
- Wu, H.; Stanley, J.R.; Cotsarelis, G. Desmoglein isotype expression in the hair follicle and its cysts correlates with type of keratinization and degree of differentiation. J. Investig. Dermatol. 2003, 120, 1052–1057. [Google Scholar] [CrossRef]
- Brennan-Crispi, D.M.; Hossain, C.; Sahu, J.; Brady, M.; Riobo, N.A.; Mahoney, M.G. Crosstalk between desmoglein 2 and patched 1 accelerates chemical-induced skin tumorigenesis. Oncotarget 2015, 6, 8593–8605. [Google Scholar] [CrossRef] [PubMed]
- Adam, R.C.; Yang, H.; Ge, Y.; Infarinato, N.R.; Gur-Cohen, S.; Miao, Y.; Wang, P.; Zhao, Y.; Lu, C.P.; Kim, J.E.; et al. Nfi transcription factors provide chromatin access to maintain stem cell identity while preventing unintended lineage fate choices. Nat. Cell Biol. 2020, 22, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, M.M.; Elliott, A.D.; DeGraff, D.J.; Anderson, P.D.; Anumanthan, G.; Yamashita, H.; Sun, Q.; Friedman, D.B.; Hachey, D.L.; Yu, X.; et al. Nfi transcription factors interact with foxa1 to regulate prostate-specific gene expression. Mol. Endocrinol. 2014, 28, 949–964. [Google Scholar] [CrossRef]
- Song, C.S.; Jung, M.H.; Supakar, P.C.; Chatterjee, B.; Roy, A.K. Negative regulation of the androgen receptor gene promoter by nfi and an adjacently located multiprotein-binding site. Mol. Endocrinol. 1999, 13, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sun, Y.; Huang, C.-P.; Luo, J.; Zhang, L.; Meng, J.; Liang, C.; Chang, C. Targeting the lnc-ophn1-5/androgen receptor/hnrnpa1 complex increases enzalutamide sensitivity to better suppress prostate cancer progression. Cell Death Dis. 2021, 12, 855. [Google Scholar] [CrossRef]
- Cai, B.; Li, M.; Zheng, Y.; Yin, Y.; Jin, F.; Li, X.; Dong, J.; Jiao, X.; Liu, X.; Zhang, K.; et al. Ezh2-mediated inhibition of microrna-22 promotes differentiation of hair follicle stem cells by elevating stk40 expression. Aging 2020, 12, 12726–12739. [Google Scholar] [CrossRef]
- Oliveira, I.O.; Lhullier, C.; Brum, I.S.; Spritzer, P.M. Gene expression of type 2 17 beta hydroxysteroid dehydrogenase in scalp hairs of hirsute women. Steroids 2003, 68, 641–649. [Google Scholar] [CrossRef]
- Drzewiecka, H.; Jarmołowska-Jurczyszyn, D.; Kluk, A.; Gałęcki, B.; Dyszkiewicz, W.; Jagodziński, P.P. Altered expression of 17-β-hydroxysteroid dehydrogenase type 2 and its prognostic significance in non-small cell lung cancer. Int. J. Oncol. 2020, 56, 1352–1372. [Google Scholar] [CrossRef]
- Zhongyi, S.; Rantakari, P.; Lamminen, T.; Toppari, J.; Poutanen, M. Transgenic male mice expressing human hydroxysteroid dehydrogenase 2 indicate a role for the enzyme independent of its action on sex steroids. Endocrinology 2007, 148, 3827–3836. [Google Scholar] [CrossRef]
- Gao, X.; Dai, C.; Huang, S.; Tang, J.; Chen, G.; Li, J.; Zhu, Z.; Zhu, X.; Zhou, S.; Gao, Y.; et al. Functional silencing of hsd17b2 in prostate cancer promotes disease progression. Clin. Cancer Res. 2019, 25, 1291–1301. [Google Scholar] [CrossRef]
- Fu, D.; Huang, J.; Li, K.; Chen, Y.; He, Y.; Sun, Y.; Guo, Y.; Du, L.; Qu, Q.; Miao, Y.; et al. Dihydrotestosterone-induced hair regrowth inhibition by activating androgen receptor in c57bl6 mice simulates androgenetic alopecia. Biomed. Pharmacother. 2021, 137, 111247. [Google Scholar] [CrossRef]
- Ceruti, J.M.; Leirós, G.J.; Balañá, M.E. Androgens and androgen receptor action in skin and hair follicles. Mol. Cell Endocrinol. 2018, 465, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Teta, M.; Choi, Y.S.; Okegbe, T.; Wong, G.; Tam, O.H.; Chong, M.M.W.; Seykora, J.T.; Nagy, A.; Littman, D.R.; Andl, T.; et al. Inducible deletion of epidermal dicer and drosha reveals multiple functions for mirnas in postnatal skin. Development 2012, 139, 1405–1416. [Google Scholar] [CrossRef] [PubMed]
- Mill, P.; Mo, R.; Hu, M.C.; Dagnino, L.; Rosenblum, N.D.; Hui, C.-C. Shh controls epithelial proliferation via independent pathways that converge on n-myc. Dev. Cell 2005, 9, 293–303. [Google Scholar] [CrossRef]
- Chiang, C.; Swan, R.Z.; Grachtchouk, M.; Bolinger, M.; Litingtung, Y.; Robertson, E.K.; Cooper, M.K.; Gaffield, W.; Westphal, H.; Beachy, P.A.; et al. Essential role for sonic hedgehog during hair follicle morphogenesis. Dev. Biol. 1999, 205, 1–9. [Google Scholar] [CrossRef]
- Mill, P.; Mo, R.; Fu, H.; Grachtchouk, M.; Kim, P.C.; Dlugosz, A.A.; Hui, C.C. Sonic hedgehog-dependent activation of gli2 is essential for embryonic hair follicle development. Genes Dev. 2003, 17, 282–294. [Google Scholar] [CrossRef]
- St-Jacques, B.; Dassule, H.R.; Karavanova, I.; Botchkarev, V.A.; Li, J.; Danielian, P.S.; McMahon, J.A.; Lewis, P.M.; Paus, R.; McMahon, A.P. Sonic hedgehog signaling is essential for hair development. Curr. Biol. 1998, 8, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- Kasper, M.; Schnidar, H.; Neill, G.W.; Hanneder, M.; Klingler, S.; Blaas, L.; Schmid, C.; Hauser-Kronberger, C.; Regl, G.; Philpott, M.P.; et al. Selective modulation of hedgehog/gli target gene expression by epidermal growth factor signaling in human keratinocytes. Mol. Cell Biol. 2006, 26, 6283–6298. [Google Scholar] [CrossRef] [PubMed]
- Megdiche, S.; Mastrangelo, S.; Ben Hamouda, M.; Lenstra, J.A.; Ciani, E. A combined multi-cohort approach reveals novel and known genome-wide selection signatures for wool traits in merino and merino-derived sheep breeds. Front. Genet. 2019, 10, 1025. [Google Scholar] [CrossRef]
- Hung, R.-J.; Spaeth, C.S.; Yesilyurt, H.G.; Terman, J.R. Selr reverses mical-mediated oxidation of actin to regulate f-actin dynamics. Nat. Cell Biol. 2013, 15, 1445–1454. [Google Scholar] [CrossRef]
- Furumura, M.; Ishikawa, H. Actin bundles in human hair follicles as revealed by confocal laser microscopy. Cell Tissue Res. 1996, 283, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Seroussi, E.; Rosov, A.; Shirak, A.; Lam, A.; Gootwine, E. Unveiling genomic regions that underlie differences between afec-assaf sheep and its parental awassi breed. Genet. Sel. Evol. 2017, 49, 19. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Long, L.; Huang, X.; Tian, K.; Tian, Y.; Wu, C.; Zhao, Z. Transcriptome analysis reveals genes associated with wool fineness in merinos. PeerJ 2023, 11, e15327. [Google Scholar] [CrossRef] [PubMed]
- Ramos, Z.; Garrick, D.J.; Blair, H.T.; De Barbieri, I.; Ciappesoni, G.; Montossi, F.; Kenyon, P.R. Genetic and phenotypic relationships between ewe reproductive performance and wool and growth traits in uruguayan ultrafine merino sheep. J. Anim. Sci. 2023, 101, skad071. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NO. | Breed | Abbreviation | Location | Photo | Size | Use | Wool Type |
|---|---|---|---|---|---|---|---|
| 1 | Australian Merino | AME | Europe |  | 2 | Wool sheep | Fine wool sheep |
| 2 | |||||||
| 3 | German Mutton Merino | GME |  | 3 | Wool sheep | Fine wool sheep | |
| 4 | |||||||
| 5 | |||||||
| 6 | Tong Sheep | TON | Eastern Asia |  | 3 | Wool and meat sheep | Semi-fine wool sheep |
| 7 | |||||||
| 8 | |||||||
| 9 | Hanzhong sheep | HZS |  | 2 | Wool and meat sheep | Semi-fine wool sheep | |
| 10 | |||||||
| 11 | Lop sheep | LOP |  | 5 | Wool and meat sheep | Coarse wool sheep | |
| 12 | |||||||
| 13 | |||||||
| 14 | |||||||
| 15 | |||||||
| 16 | Hu Sheep | HUS |  | 3 | Wool and meat sheep | Coarse wool sheep | |
| 17 | |||||||
| 18 | |||||||
| 19 | Small Tailed Han Sheep | STH |  | 5 | Wool and meat sheep | Coarse wool sheep | |
| 20 | |||||||
| 21 | |||||||
| 22 | |||||||
| 23 | |||||||
| 24 | Ujimqin sheep | UJI |  | 5 | Wool and meat sheep | Coarse wool sheep | |
| 25 | |||||||
| 26 | |||||||
| 27 | |||||||
| 28 |
| Catalogue | SNP Numbers |
|---|---|
| Upstream | 93,281 |
| Stop gain (Exonic) | 734 |
| Stop loss (Exonic) | 188 |
| Synonymous (Exonic) | 68,110 |
| Non-synonymous (Exonic) | 82,379 |
| Intronic | 8,123,336 |
| Splicing | 4227 |
| Downstream | 125,137 |
| Upstream/downstream | 2550 |
| Intergenic | 13,414,800 |
| ts | 14,501,815 |
| tv | 7,631,392 |
| ts/tv | 1.9 |
| Total | 22,133,207 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Jin, M.; Li, T.; Lu, Z.; Wang, H.; Yuan, Z.; Wei, C. Whole-Genome Resequencing Reveals Selection Signal Related to Sheep Wool Fineness. Animals 2023, 13, 2944. https://doi.org/10.3390/ani13182944
Zhang W, Jin M, Li T, Lu Z, Wang H, Yuan Z, Wei C. Whole-Genome Resequencing Reveals Selection Signal Related to Sheep Wool Fineness. Animals. 2023; 13(18):2944. https://doi.org/10.3390/ani13182944
Chicago/Turabian StyleZhang, Wentao, Meilin Jin, Taotao Li, Zengkui Lu, Huihua Wang, Zehu Yuan, and Caihong Wei. 2023. "Whole-Genome Resequencing Reveals Selection Signal Related to Sheep Wool Fineness" Animals 13, no. 18: 2944. https://doi.org/10.3390/ani13182944