
Genotypic Comparison of Pasteurella multocida from Healthy Animals at Entry to the Feedlots with That and from Bovine Respiratory Disease-Affected Animals during the Fattening Period
 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animal Sampling
2.3. P. multocida Isolation and Identification
2.4. Capsular, Lipopolysaccharide, and Pulsed-Field Gel Electrophoresis (PFGE) Typing of P. multocida Isolates
2.5. Whole-Genome Sequencing (WGS), Assembly, and Multilocus Sequence Typing
2.6. Detection of Virulence-Associated Genes, Antimicrobial Resistance Genes, and Mobile Genetic Elements
2.7. Phylogenetic Analysis
2.8. Data Analysis
3. Results
3.1. P. multocida Capsular-LPS Genotypes in Apparently Healthy and Clinically Affected Animals
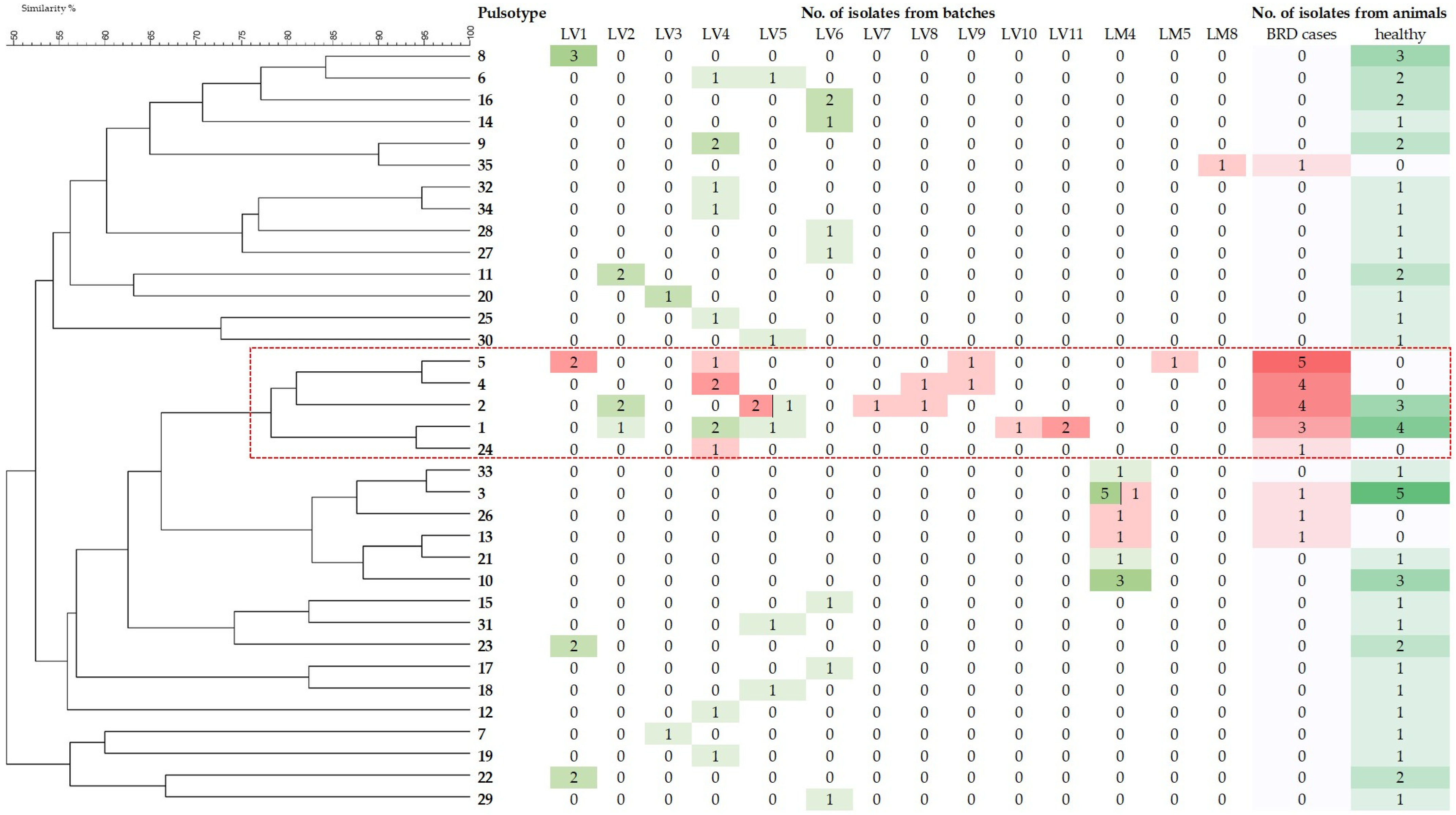
3.2. PFGE Typing
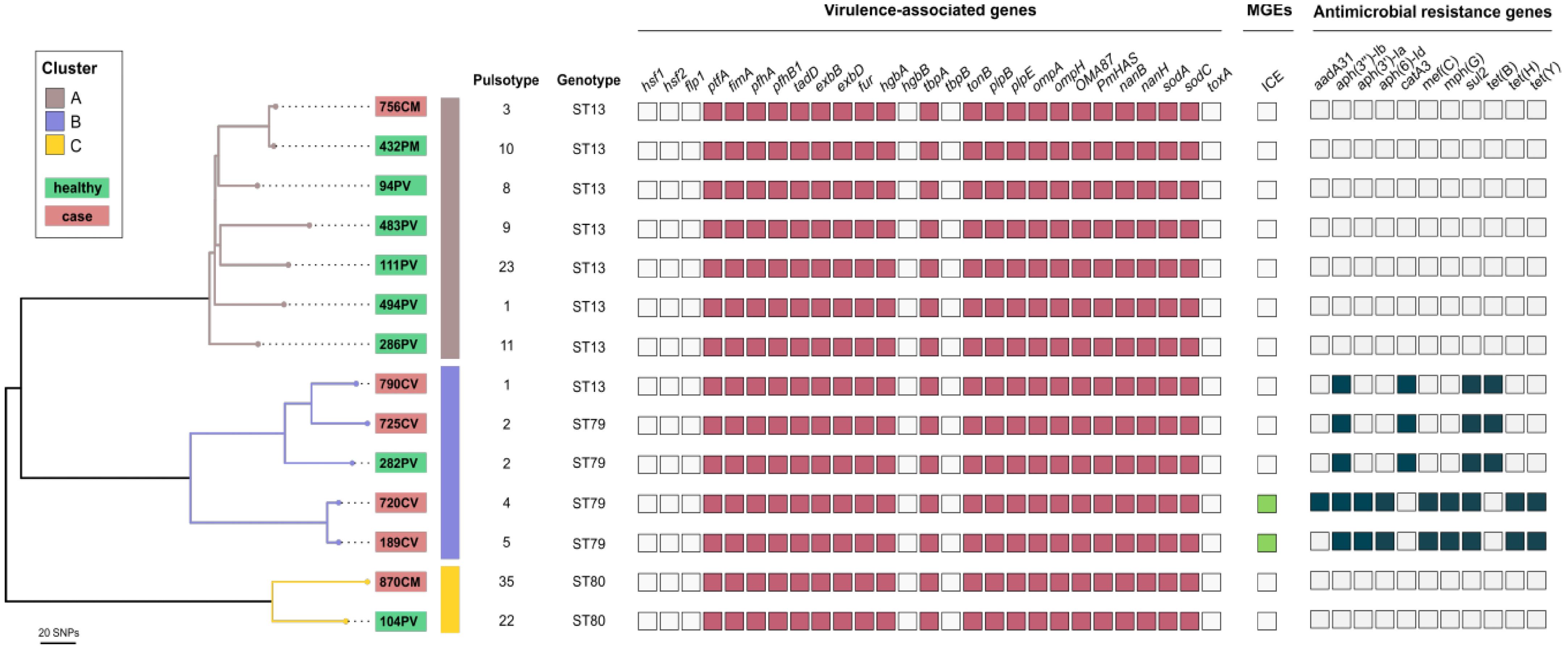
3.3. MLST Analysis and Screening of Virulence-Associated and Antimicrobial Resistance Genes and Mobile Genetic Elements in the Most Frequently Detected P. multocida Pulsotypes
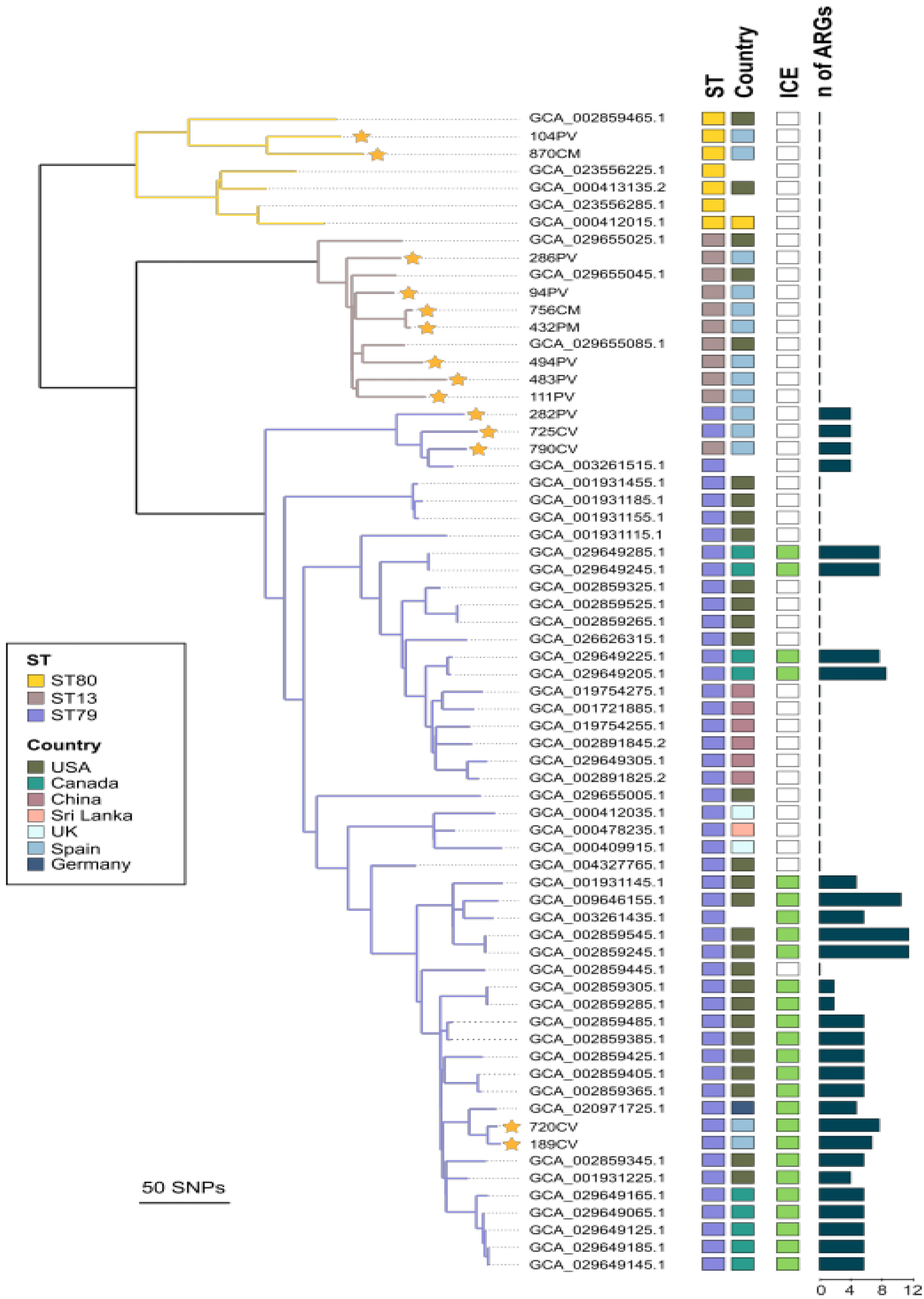
3.4. Phylogenetic Relatedness, Virulence-Associated Genes, Mobile Genetic Elements, and Acquired ARGs of Respiratory Bovine P. multocida Isolates of Sequence Types ST13, ST79, and ST80
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taylor, J.D.; Fulton, R.W.; Dabo, S.M.; Lehenbauer, T.W.; Confer, A.W. Comparison of genotypic and phenotypic characterization methods for Pasteurella multocida isolates from fatal cases of bovine respiratory disease J. Vet. Diagn. Investig. 2010, 22, 366–375. [Google Scholar] [CrossRef]
- Timsit, E.; Hallewell, J.; Booker, C.; Tison, N.; Amat, S.; Alexander, T.W. Prevalence and antimicrobial susceptibility of Mannheimia haemolytica, Pasteurella multocida, and Histophilus somni isolated from the lower respiratory tract of healthy feedlot cattle and those diagnosed with bovine respiratory disease. Vet. Microbiol. 2017, 208, 118–125. [Google Scholar] [CrossRef]
- Pardon, B.; Callens, J.; Maris, J.; Allais, L.; Van Praet, W.; Deprez, P.; Ribbens, S. Pathogen-specific risk factors in acute outbreaks of respiratory disease in calves. J. Dairy Sci. 2020, 103, 2556–2566. [Google Scholar] [CrossRef] [PubMed]
- Snyder, E.; Credille, B. Mannheimia haemolytica and Pasteurella multocida in bovine respiratory disease. How are they changing in response to efforts to control them? Vet. Clin. Food Anim. 2020, 36, 253–268. [Google Scholar] [CrossRef]
- Calderón Bernal, J.M.; Fernández, A.; Arnal, J.L.; Sanz Tejero, C.; Fernández-Garayzábal, J.F.; Vela, A.I.; Cid, D. Molecular Epidemiology of Pasteurella multocida associated with bovine respiratory disease outbreaks. Animals 2022, 13, 75. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, E.J.; Hodgson, J.C.; Schmitt-van de Leemput, E.; Dagleish, M.P.; Zadoks, R.N. Molecular epidemiology of Pasteurella multocida in dairy and beef calves. Vet. Microbiol. 2011, 151, 329–335. [Google Scholar] [CrossRef]
- Pratelli, A.; Hodnik, A.; Cirone, F.; Capozza, P.; Trotta, A.; Corrente, M.; Balestrieri, A.; Buonavoglia, C. Bovine respiratory disease in beef calves supported long transport stress: An epidemiological study and strategies for control and prevention. Res. Vet. Sci. 2020, 135, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Wang, X.; Zhou, R.; Chen, H.; Wilson, B.A.; Wu, B. Pasteurella multocida: Genotypes and genomics. Microbiol. Mol. Biol. Rev. 2019, 83, e00014-19. [Google Scholar] [CrossRef] [PubMed]
- Townsend, K.M.; Boyce, J.D.; Chung, J.Y.; Frost, A.J.; Adler, B. Genetic organization of Pasteurella multocida cap loci and development of a multiplex capsular PCR typing system. J. Clin. Microbiol. 2001, 39, 924–929. [Google Scholar] [CrossRef]
- Harper, M.; John, M.; Turni, C.; Edmunds, M.; Michael, F.S.; Adler, B.; Blackall, P.J.; Cox, A.D.; Boyce, J.D. Development of a rapid multiplex PCR assay to genotype Pasteurella multocida strains by use of the lipopolysaccharide outer core biosynthesis locus. J. Clin. Microbiol. 2015, 53, 477–485. [Google Scholar] [CrossRef]
- Ewers, C.; Lübke-Becker, A.; Bethe, A.; Kießling, S.; Filter, M.; Wieler, L.H. Virulence genotype of Pasteurella multocida strains isolated from different hosts with various disease status. Vet. Microbiol. 2006, 114, 304–317. [Google Scholar] [CrossRef] [PubMed]
- García-Alvarez, A.; Vela, A.I.; San Martín, E.; Chaves, F.; Fernández-Garayzábal, J.F.; Domínguez, L.; Cid, D. Characterization of Pasteurella multocida associated with ovine pneumonia using multi-locus sequence typing (MLST) and virulence associated gene profile analysis and comparison with porcine isolates. Vet. Microbiol. 2017, 204, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Liang, W.; Wang, F.; Xu, Z.; Xie, Z.; Lian, Z.; Hua, L.; Zhou, R.; Chen, H.; Wu, B. Genetic and phylogenetic characteristics of Pasteurella multocida isolates from different host species. Front. Microbiol. 2018, 9, 1408. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Miller, E.; Aguayo, J.M.; Figueroa, C.F.; Nezworski, J.; Studniski, M.; Wileman, B.; Johnson, T. Genomic diversity and molecular epidemiology of Pasteurella multocida. PLoS ONE 2021, 16, e0249138. [Google Scholar] [CrossRef] [PubMed]
- Alhamami, T.; Chowdhury, P.R.; Venter, H.; Veltman, T.; Truswell, A.; Abraham, S.; Sapula, S.A.; Carr, M.; Djordjevic, S.P.; Trott, D.J. Genomic profiling of Pasteurella multocida isolated from feedlot cases of bovine respiratory disease. Vet. Microbiol. 2023, 283, 109773. [Google Scholar] [CrossRef]
- Pérez-Sancho, M.; Cerdá, I.; Fernández-Bravo, A.; Domínguez, L.; Figueras, M.J.; Fernández-Garayzábal, J.F.; Vela, A.I. Limited performance of MALDI-TOF for identification of fish Aeromonas isolates at species level. J. Fish. Dis. 2018, 41, 1485–1493. [Google Scholar] [CrossRef]
- Townsend, K.M.; Frost, A.J.; Lee, C.W.; Papadimitriou, J.M.; Dawkins, H.J.S. Development of PCR assays for species-and type-specific identification of Pasteurella multocida isolates. J. Clin. Microbiol. 1998, 36, 1096–1100. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Schwengers, O.; Jelonek, L.; Dieckmann, M.A.; Beyvers, S.; Blom, J.; Goesmann, A. Bakta: Rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microbial. Genom. 2021, 7, 000685. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef]
- Verma, S.; Sharma, M.; Katoch, S.; Verma, L.; Kumar, S.; Dogra, V.; Chahota, R.; Dhar, P.; Singh, G. Profiling of virulence associated genes of Pasteurella multocida isolated from cattle. Vet. Res. Commun. 2013, 37, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Katsuda, K.; Hoshinoo, K.; Ueno, Y.; Kohmoto, M.; Mikami, O. Virulence genes and antimicrobial susceptibility in Pasteurella multocida isolates from calves. Vet. Microbiol. 2013, 167, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [PubMed]
- Michael, G.B.; Kadlec, K.; Sweeney, M.T.; Brzuszkiewicz, E.; Liesegang, H.; Daniel, R.; Murray, R.W.; Watts, J.L.; Schwarz, S. ICEPmu1, an integrative conjugative element (ICE) of Pasteurella multocida: Structure and transfer. J. Antimicrob. Chemother. 2012, 67, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Schink, A.K.; Hanke, D.; Semmler, T.; Brombach, J.; Bethe, A.; Lübke-Becker, A.; Teske, K.; Müller, K.E.; Schwarz, S. Novel multiresistance-mediating integrative and conjugative elements carrying unusual antimicrobial resistance genes in Mannheimia haemolytica and Pasteurella multocida. J. Antimicrob. Chemother. 2022, 77, 2033–2035. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.A.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Didelot, X.; Wilson, D.J. ClonalFrameML: Efficient inference of recombination in whole bacterial genomes. PLoS Comp. Biol. 2015, 11, e1004041. [Google Scholar] [CrossRef]
- Page, A.J.; Taylor, B.; Delaney, A.J.; Soares, J.; Seemann, T.; Keane, J.A.; Harris, S.R. SNP-sites: Rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genom. 2016, 2, e000056. [Google Scholar] [CrossRef]
- Yu, G. Using ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef]
- Lopez-Canovas, L.; Martinez Benitez, M.B.; Herrera Isidron, J.A.; Flores Soto, E. Pulsed Field Gel Electrophoresis: Past, present, and future. Anal. Biochem. 2019, 573, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, C.; Timsit, E.; Uddin, M.S.; Guan, L.L.; Alexander, T.W. Comparison of pathogenic bacteria in the upper and lower respiratory tracts of cattle either directly transported to a feedlot or co-mingled at auction markets prior to feedlot placement. Front. Vet. Sci. 2023, 9, 1026470. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Zhao, Z.; Wu, X.; Duan, L.; Li, N.; Fang, R.; Li, P.; Peng, Y. Transcriptomic Analysis of High- and Low-Virulence Bovine Pasteurella multocida in vitro and in vivo. Front. Vet. Sci. 2021, 8, 616774. [Google Scholar] [CrossRef] [PubMed]
- Constable, P.D.; Hinchchliff, K.W.; Done, S.H.; Walter, G. Practical Antimicrobial Therapeutics. In Veterinary Medicine: A Text Book of the Diseases of Cattle, Horses, Sheep Pigs and Goats, 11th ed.; Elsevier: St. Louis, MO, USA, 2017; Volume one, pp. 153–174. [Google Scholar]
- Michael, G.B.; Freitag, C.; Wendlandt, S.; Eidam, C.; Feßler, A.T.; Lopes, G.V.; Kadlec, K.; Schwarz, S. Emerging issues in antimicrobial resistance of bacteria from food-producing animals. Future Microbiol. 2015, 10, 427–443. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Feedlot | Feedlot Location | Batch | Feedlot Entry Date | Animal Source | No. of Animals |
|---|---|---|---|---|---|
| 1 | Valencia | LV1 | 13 April 2021 | Purchased | 80 |
| LV2 | 15 June 2021 | Purchased | 87 | ||
| LV3 | 15 September 2021 | Purchased | 80 | ||
| LV4 | 2 November 2021 | Purchased | 83 | ||
| LV5 | 5 May 2022 | Purchased | 80 | ||
| LV6 | 14 June 2022 | Purchased | 85 | ||
| LV7 | 13 January 2022 | Purchased | 80 | ||
| LV8 | 26 January 2022 | Purchased | 75 | ||
| LV9 | 18 February 2022 | Purchased | 74 | ||
| LV10 | 24 March 2022 | Purchased | 87 | ||
| LV11 | 31 March 2022 | Purchased | 85 | ||
| LV12 | 9 June 2022 | Purchased | 98 | ||
| 2 | Madrid | LM1 | 18 January 2021 | Own farm | 26 |
| LM2 | 11 April 2021 | Own farm | 24 | ||
| LM3 | 25 May 2021 | Own farm | 28 | ||
| 3 | Madrid | LM4 | 10 October 2021 | Own farm | 30 |
| 4 | Madrid | LM5 | 20 September 2022 | Purchased | 28 |
| 5 | Madrid | LM6 | 23 May 2022 | Own farm | 20 |
| 6 | Madrid | LM7 | 11 May 2022 | Own farm | 14 |
| 7 | Madrid | LM8 | 12 May 2022 | Own farm | 18 |
| Class | No. of Isolates | % | Genes |
|---|---|---|---|
| Aminoglycosides | 30 | 44.7 | aadA25; aadA31; ant(2″)-Ia; aph(3″)-Ib; aph(3′)-Ia; aph(6)-Id |
| Beta-lactams | 4 | 5.9 | blaOXA-2 |
| Lincosamides | 5 | 7.0 | erm42 |
| Macrolides | 11 | 16.4 | mefC; mphE; msrE; erm42 |
| Phenicols | 10 | 14.9 | catA3; floR; msrE |
| Streptogramins | 9 | 13.4 | msrE; erm42 |
| Sulfonamides | 25 | 37.3 | sul2 |
| Tetracyclines | 30 | 44.7 | tetB; tetH; tetY |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calderón Bernal, J.M.; Serna, C.; García Muñoz, Á.; Díez Guerrier, A.; Domínguez, L.; Fernández-Garayzábal, J.F.; Vela, A.I.; Cid, D. Genotypic Comparison of Pasteurella multocida from Healthy Animals at Entry to the Feedlots with That and from Bovine Respiratory Disease-Affected Animals during the Fattening Period. Animals 2023, 13, 2687. https://doi.org/10.3390/ani13172687
Calderón Bernal JM, Serna C, García Muñoz Á, Díez Guerrier A, Domínguez L, Fernández-Garayzábal JF, Vela AI, Cid D. Genotypic Comparison of Pasteurella multocida from Healthy Animals at Entry to the Feedlots with That and from Bovine Respiratory Disease-Affected Animals during the Fattening Period. Animals. 2023; 13(17):2687. https://doi.org/10.3390/ani13172687
Chicago/Turabian StyleCalderón Bernal, Johan Manuel, Carlos Serna, Ángel García Muñoz, Alberto Díez Guerrier, Lucas Domínguez, José Francisco Fernández-Garayzábal, Ana Isabel Vela, and Dolores Cid. 2023. "Genotypic Comparison of Pasteurella multocida from Healthy Animals at Entry to the Feedlots with That and from Bovine Respiratory Disease-Affected Animals during the Fattening Period" Animals 13, no. 17: 2687. https://doi.org/10.3390/ani13172687