
Characterization of RNA Editome in the Mammary Gland of Yaks during the Lactation and Dry Periods

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
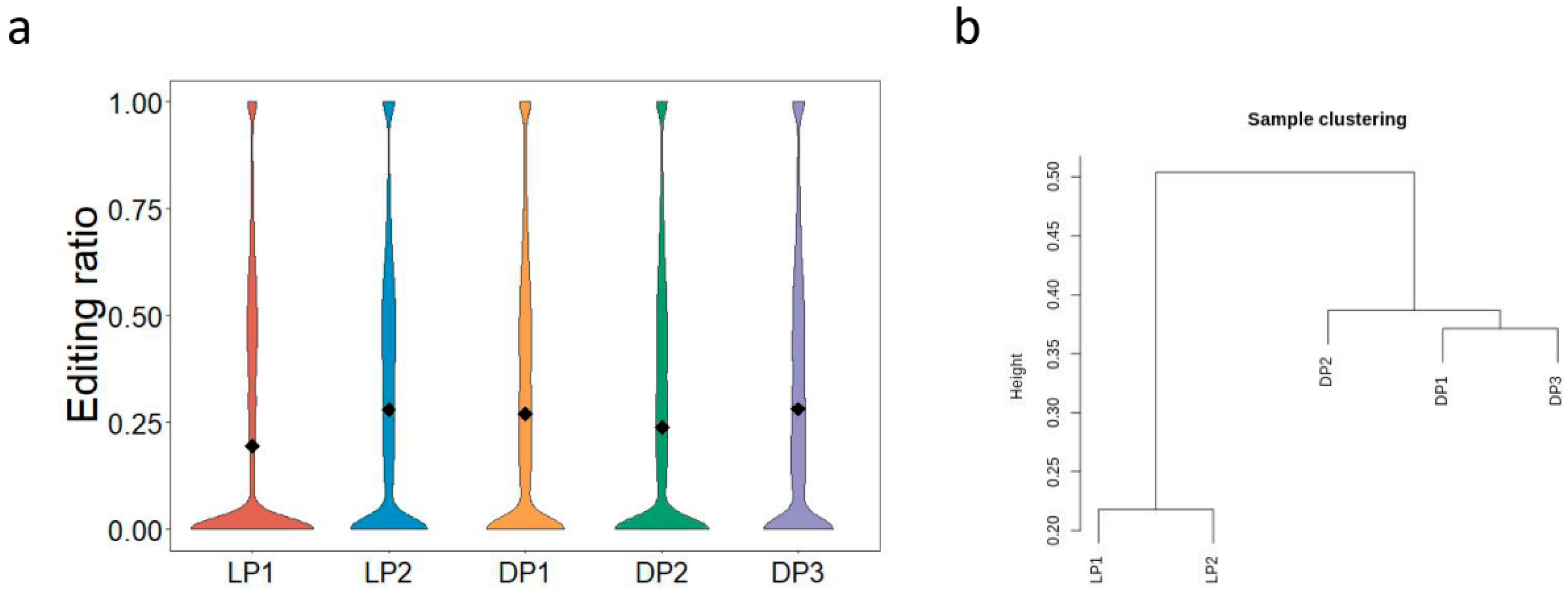{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Description of Datasets
2.2. Whole-Genome Sequencing
2.3. Read Mapping
2.4. Detection and Annotation of RNA Editing Sites
2.5. Conservation Analysis of RNA Editing Sites
2.6. Analysis of the RNA Editing Effects on miRNA Regulation
2.7. Pairwise Comparison of RNA Editing Sites between the Groups
2.8. Functional Analysis
3. Results
3.1. Identification of RNA Editing Sites in the Mammary Gland
3.2. Characterization of RNA Editing Sites in the Mammary Gland
3.3. Distribution of RNA Editing Sites across Different Genomic Regions
3.4. Effects of RNA Editing Sites on miRNA–mRNA Interactions
3.5. Cross-Species Analysis between Yak and Human
3.6. Distribution of RNA Editing Sites among Different Physiological Stages
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Farajollahi, S.; Maas, S. Molecular diversity through RNA editing: A balancing act. Trends Genet. 2010, 26, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Teoh, P.J.; Koh, M.Y.; Chng, W.J. ADARs, RNA editing and more in hematological malignancies. Leukemia 2020, 35, 346–359. [Google Scholar] [CrossRef]
- Hans, J.; Hajduk, S.L.; Madison-Antenucci, S. RNA-editing-associated protein 1 null mutant reveals link to mitochondrial RNA stability. RNA 2007, 13, 881–889. [Google Scholar] [CrossRef] [Green Version]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef] [Green Version]
- Salter, J.D.; Bennett, R.P.; Smith, H.C. The APOBEC Protein Family: United by Structure, Divergent in Function. Trends Biochem. Sci. 2016, 41, 578–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.H.; Li, Q.; Shanmugam, R.; Piskol, R.; Kohler, J.; Young, A.N.; Liu, K.I.; Zhang, R.; Ramaswami, G.; Ariyoshi, K.; et al. Dynamic landscape and regulation of RNA editing in mammals. Nature 2017, 550, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Bakhtiarizadeh, M.R.; Salehi, A.; Rivera, R.M. Genome-wide identification and analysis of A-to-I RNA editing events in bovine by transcriptome sequencing. PLoS ONE 2018, 13, e193316. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Shi, L.; Cao, M.; Shen, D.; Li, J.; Zhang, S.; Song, J. Pan-RNA editing analysis of the bovine genome. RNA Biol. 2021, 18, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Funkhouser, S.A.; Steibel, J.P.; Bates, R.O.; Raney, N.E.; Schenk, D.; Ernst, C.W. Evidence for transcriptome-wide RNA editing among Sus scrofa PRE-1 SINE elements. BMC Genom. 2017, 18, 360. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhu, M.; Fan, X.; Yao, Y.; Yan, J.; Tang, Y.; Liu, S.; Li, K.; Tang, Z. Developmental atlas of the RNA editome in Sus scrofa skeletal muscle. DNA Res. 2019, 26, 261–272. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, D.; Dong, X.; Wang, J.; Chen, J.; Yao, Y.; Darwish, H.Y.A.; Liu, W.; Deng, X. Genome-wide profiling of RNA editing sites in sheep. J. Anim. Sci. Biotechnol. 2019, 10, 31. [Google Scholar] [CrossRef]
- Yang, L.; Li, L.; Kyei, B.; Guo, J.; Zhan, S.; Zhao, W.; Song, Y.; Zhong, T.; Wang, L.; Xu, L.; et al. Systematic analyses reveal RNA editing events involved in skeletal muscle development of goat (Capra hircus). Funct. Integr. Genom. 2020, 20, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Frésard, L.; Leroux, S.; Roux, P.F.; Klopp, C.; Fabre, S.; Esquerré, D.; Dehais, P.; Djari, A.; Gourichon, D.; Lagarrigue, S.; et al. Genome-wide characterization of RNA editing in chicken embryos reveals common features among vertebrates. PLoS ONE 2015, 10, e126776. [Google Scholar] [CrossRef] [Green Version]
- Shafiei, H.; Bakhtiarizadeh, M.R.; Salehi, A. Large-scale potential RNA editing profiling in different adult chicken tissues. Anim. Genet. 2019, 50, 460–474. [Google Scholar] [CrossRef] [PubMed]
- Wiener, G.; Han, J.; Long, R. The Yak, 2nd ed.; FAO Regional Office for Asia and the Pacific: Bangkok, Thailand, 2003; pp. 1–2. [Google Scholar]
- Ding, L.; Wang, Y.; Kreuzer, M.; Guo, X.; Mi, J.; Gou, Y.; Shang, Z.; Zhang, Y.; Zhou, J.; Wang, H.; et al. Seasonal variations in the fatty acid profile of milk from yaks grazing on the Qinghai-Tibetan plateau. J. Dairy Res. 2013, 80, 410–417. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Long, R.; Kreuzer, M.; Ding, L.; Shang, Z.; Zhang, Y.; Yang, Y.; Cui, G. Importance of Functional Ingredients in Yak Milk-Derived Food on Health of Tibetan Nomads Living Under High-Altitude Stress: A Review. Crit. Rev. Food Sci. Nutr. 2013, 54, 292–302. [Google Scholar] [CrossRef]
- Hurley, W. Mammary Gland Function During Involution. J. Dairy Sci. 1989, 72, 1637–1646. [Google Scholar] [CrossRef]
- Wu, J.; He, S.; Yu, Z.; Lan, D.; Xiong, X.; Li, Z. Transcriptomic study of yak mammary gland tissue during lactation. Anim. Biotechnol. 2020, 31, 1–8. [Google Scholar] [CrossRef]
- Fan, J.; Luo, Y.; Yu, S.; Cui, Y.; Xu, G.; Wang, L.; Pan, Y.; Honghong, H. Transcriptional profiling of two different physiological states of the yak mammary gland using RNA sequencing. PLoS ONE 2018, 13, e201628. [Google Scholar]
- Wu, X.; Zhou, X.; Xiong, L.; Pei, J.; Yao, X.; Liang, C.; Bao, P.; Chu, M.; Guo, X.; Yan, P. Transcriptome Analysis Reveals the Potential Role of Long Non-coding RNAs in Mammary Gland of Yak during Lactation and Dry Period. Front. Cell Dev. Biol. 2020, 8, 579708. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Picardi, E.; Pesole, G. REDItools: High-throughput RNA editing detection made easy. Bioinformatics 2013, 29, 1813–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picardi, E.; D’Erchia, A.M.; Montalvo, A.; Pesole, G. Using REDItools to Detect RNA Editing Events in NGS Datasets. Curr. Protoc. Bioinform. 2015, 49, 12.12.1–12.12.15. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Picardi, E.; D’Erchia, A.M.; Lo, G.C.; Pesole, G. REDIportal: A comprehensive database of A-to-I RNA editing events in hu-mans. Nucleic Acids Res. 2017, 45, D750–D757. [Google Scholar] [CrossRef] [Green Version]
- Mcgeary, S.E.; Lin, K.S.; Shi, C.Y.; Pham, T.M.; Bisaria, N.; Kelley, G.M.; Bartel, D.P. The biochemical basis of microRNA tar-geting efficacy. Science 2019, 366, eaav1741. [Google Scholar] [CrossRef] [PubMed]
- John, B.; Enright, A.; Aravin, A.A.; Tuschl, T.; Sander, C.; Marks, D.S. Human MicroRNA Targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrich-ment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for anno-tation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-Y.; Peng, Z.; Zhang, R.; Yang, X.-Z.; Tan, B.; Fang, H.; Liu, C.-J.; Shi, M.; Ye, Z.-Q.; Zhang, Y.E.; et al. RNA Editome in Rhesus Macaque Shaped by Purifying Selection. PLoS Genet. 2014, 10, e1004274. [Google Scholar] [CrossRef] [Green Version]
- Carmi, S.; Borukhov, I.; Levanon, E. Identification of Widespread Ultra-Edited Human RNAs. PLoS Genet. 2011, 7, e1002317. [Google Scholar] [CrossRef]
- Gu, T.; Buaas, F.W.; Simons, A.K.; Ackert-Bicknell, C.; Braun, R.E.; Hibbs, M.A. Canonical A-to-I and C-to-U RNA Editing Is Enriched at 3′UTRs and microRNA Target Sites in Multiple Mouse Tissues. PLoS ONE 2012, 7, e33720. [Google Scholar] [CrossRef]
- Gong, J.; Liu, C.-J.; Liu, W.; Xiang, Y.; Diao, L.; Guo, A.-Y.; Han, L. LNCediting: A database for functional effects of RNA editing in lncRNAs. Nucleic Acids Res. 2017, 45, D79–D84. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wagner, K.-U.; Larson, D.; Weaver, Z.; Li, C.; Ried, T.; Hennighausen, L.; Wynshaw-Boris, A.; Deng, C.-X. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat. Genet. 1999, 22, 37–43. [Google Scholar] [CrossRef]
- Smart, C.E.; Wronski, A.; French, J.D.; Edwards, S.; Asselin-Labat, M.-L.; Waddell, N.; Peters, K.; Brewster, B.L.; Brooks, K.; Simpson, K.; et al. Analysis of Brca1-deficient mouse mammary glands reveals reciprocal regulation of Brca1 and c-kit. Oncogene 2010, 30, 1597–1607. [Google Scholar] [CrossRef] [Green Version]
- Valenti, P.; Berlutti, F.; Conte, M.P.; Longhi, C.; Seganti, L. Lactoferrin functions: Current status and perspectives. J. Clin. Gastroenterol. 2004, 38, S127–S129. [Google Scholar] [CrossRef]
- El-Domany, W.B.; Radwan, H.A.; Ateya, A.I.; Ramadan, H.H.; Marghani, B.H.; Nasr, S.M. Genetic Polymorphisms in LTF/EcoRI and TLR4/AluI loci as candidates for milk and reproductive performance assessment in Holstein cattle. Reprod. Domest. Anim. 2019, 54, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Nautiyal, J.; Steel, J.H.; Mane, M.R.; Oduwole, O.; Poliandri, A.; Alexi, X.; Wood, N.; Poutanen, M.; Zwart, W.; Stingl, J.; et al. The transcriptional co-factor RIP140 regulates mammary gland development by promoting the generation of key mitogenic signals. Development 2013, 140, 1079–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.-Z.; Shi, K.; Wu, X.-H.; Xue, M.-Y.; Wei, Z.-H.; Liu, J.-X.; Liu, H.-Y. Lactation-related metabolic mechanism investigated based on mammary gland metabolomics and 4 biofluids’ metabolomics relationships in dairy cows. BMC Genom. 2017, 18, 936. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Liu, Y.; Long, J.; Zhou, X.; Zeng, S.; Li, T.; Yin, Y. Maternal serine supply from late pregnancy to lactation improves offspring performance through modulation of metabolic pathways. Food Funct. 2020, 11, 8089–8098. [Google Scholar] [CrossRef]
- Do, D.; Bissonnette, N.; Lacasse, P.; Miglior, F.; Sargolzaei, M.; Zhao, X.; Ibeagha-Awemu, E. Genome-wide association analysis and pathways enrichment for lactation persistency in Canadian Holstein cattle. J. Dairy Sci. 2017, 100, 1955–1970. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.W.; Wehde, B.L.; Sakamoto, K.; Triplett, A.A.; Anderson, S.M.; Tsichlis, P.N.; Leone, G.; Wagner, K.U. Stat5 regulates the phosphatidylinositol 3-kinase/Akt1 pathway during mammary gland development and tumorigenesis. Mol. Cell. Biol. 2014, 34, 1363–1377. [Google Scholar] [CrossRef] [Green Version]
- Hadsell, D.; Bonnette, S.; Lee, A. Genetic Manipulation of the IGF-I Axis to Regulate Mammary Gland Development and Function. J. Dairy Sci. 2002, 85, 365–377. [Google Scholar] [CrossRef]
- Szewczuk, M.; Zych, S.; Czerniawska-Pia¸tkowska, E. Association between IGF1/TasI polymorphism and milk traits of Polish HolsteinFriesian cows. Arch. Anim. Breed. 2011, 54, 10–17. [Google Scholar] [CrossRef]
- Huebner, R.J.; Neumann, N.; Ewald, A.J. Mammary epithelial tubes elongate through MAPK-dependent coordination of cell migration. Development 2016, 143, 983–993. [Google Scholar] [CrossRef] [Green Version]
- Do, D.N.; Li, R.; Dudemaine, P.L.; Ibeagha-Awemu, E.M. MicroRNA roles in signalling during lactation: An insight from differential expression, time course and pathway analyses of deep sequence data. Sci. Rep. 2017, 7, 44605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hao, Z.; Hu, J.; Liu, X.; Li, S.; Shen, J.; Song, Y.; Ke, N.; Luo, Y. Small RNA deep sequencing reveals the expressions of microRNAs in ovine mammary gland development at peak-lactation and during the non-lactating period. Genomics 2020, 113, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Feng, S.; Wang, X.; Qazi, I.H.; Long, K.; Luo, Y.; Li, G.; Ning, C.; Wang, Y.; Hu, S.; et al. Exploration of exosomal mi-croRNA expression profiles in pigeon ‘Milk’ during the lactation period. BMC Genom. 2018, 19, 828. [Google Scholar] [CrossRef] [Green Version]
- Lü, L.-M.; Li, Q.-Z.; Huang, J.-G.; Gao, X.-J. Proteomic and Functional Analyses Reveal MAPK1 Regulates Milk Protein Synthesis. Molecules 2012, 18, 263–275. [Google Scholar] [CrossRef]
- Feramisco, J.D.; Goldstein, J.L.; Brown, M.S. Membrane Topology of Human Insig-1, a Protein Regulator of Lipid Synthesis. J. Biol. Chem. 2004, 279, 8487–8496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Luo, J.; Zhang, T.; Tian, H.; Ma, Y.; Xu, H.; Yao, D.; Loor, J.J. MicroRNA-26a/b and their host genes synergistically regulate triacylglycerol synthesis by targeting the INSIG1 gene. RNA Biol. 2016, 13, 500–510. [Google Scholar] [CrossRef] [Green Version]
- Chapman, R.S.; Lourenco, P.C.; Tonner, E.; Flint, D.J.; Selbert, S.; Takeda, K.; Akira, S.; Clarke, A.R.; Watson, C.J. Suppression of epithelial apoptosis and delayed mammary gland involution in mice with a conditional knockout of Stat3. Genes Dev. 1999, 13, 2604–2616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, D.N.; Dudemaine, P.-L.; Li, R.; Ibeagha-Awemu, E.M. Co-Expression Network and Pathway Analyses Reveal Important Modules of miRNAs Regulating Milk Yield and Component Traits. Int. J. Mol. Sci. 2017, 18, 1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brock, M.; Trenkmann, M.; Gay, R.E.; Michel, B.A.; Gay, S.; Fischler, M.; Ulrich, S.; Speich, R.; Huber, L.C. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ. Res. 2009, 104, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Xuan, R.; Chao, T.; Wang, A.; Zhang, F.; Sun, P.; Liu, S.; Guo, M.; Wang, G.; Ji, Z.; Wang, J.; et al. Characterization of mi-croRNA profiles in the mammary gland tissue of dairy goats at the late lactation, dry period and late gestation stages. PLoS ONE 2020, 15, e234427. [Google Scholar] [CrossRef]
- Rivetti, S.; Chen, C.; Chen, C.; Bellusci, S. Fgf10/Fgfr2b signaling in mammary gland development, homeostasis, and cancer. Front Cell Dev. Biol. 2020, 8, 415. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Ayalew, W.; Chu, M.; Pei, J.; Liang, C.; Bao, P.; Guo, X.; Yan, P. Characterization of RNA Editome in the Mammary Gland of Yaks during the Lactation and Dry Periods. Animals 2022, 12, 207. https://doi.org/10.3390/ani12020207
Wu X, Ayalew W, Chu M, Pei J, Liang C, Bao P, Guo X, Yan P. Characterization of RNA Editome in the Mammary Gland of Yaks during the Lactation and Dry Periods. Animals. 2022; 12(2):207. https://doi.org/10.3390/ani12020207
Chicago/Turabian StyleWu, Xiaoyun, Wondossen Ayalew, Min Chu, Jie Pei, Chunnian Liang, Pengjia Bao, Xian Guo, and Ping Yan. 2022. "Characterization of RNA Editome in the Mammary Gland of Yaks during the Lactation and Dry Periods" Animals 12, no. 2: 207. https://doi.org/10.3390/ani12020207