

Uncovering the Fecal Bacterial Communities of Sympatric Sika Deer (Cervus nippon) and Wapiti (Cervus canadensis)

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. DNA Extraction and Sequencing
2.2. Quality Control of the Raw Sequences
2.3. OTU Clustering and Species Classification Analysis
2.4. Complexity Analysis
2.5. Comparative Analysis
3. Results
3.1. Sequence Data Processing
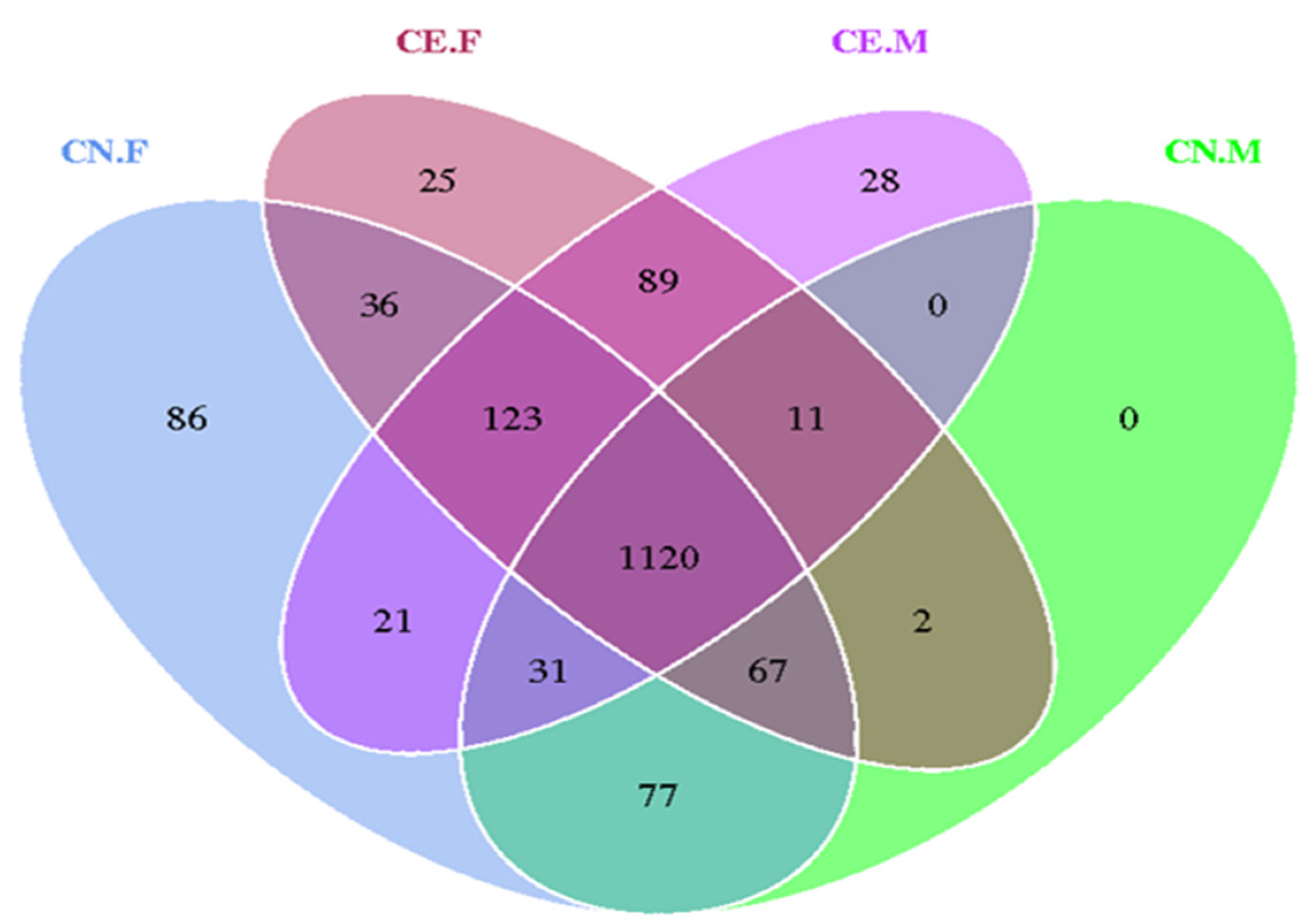
3.2. OTU Clustering and Species Annotation
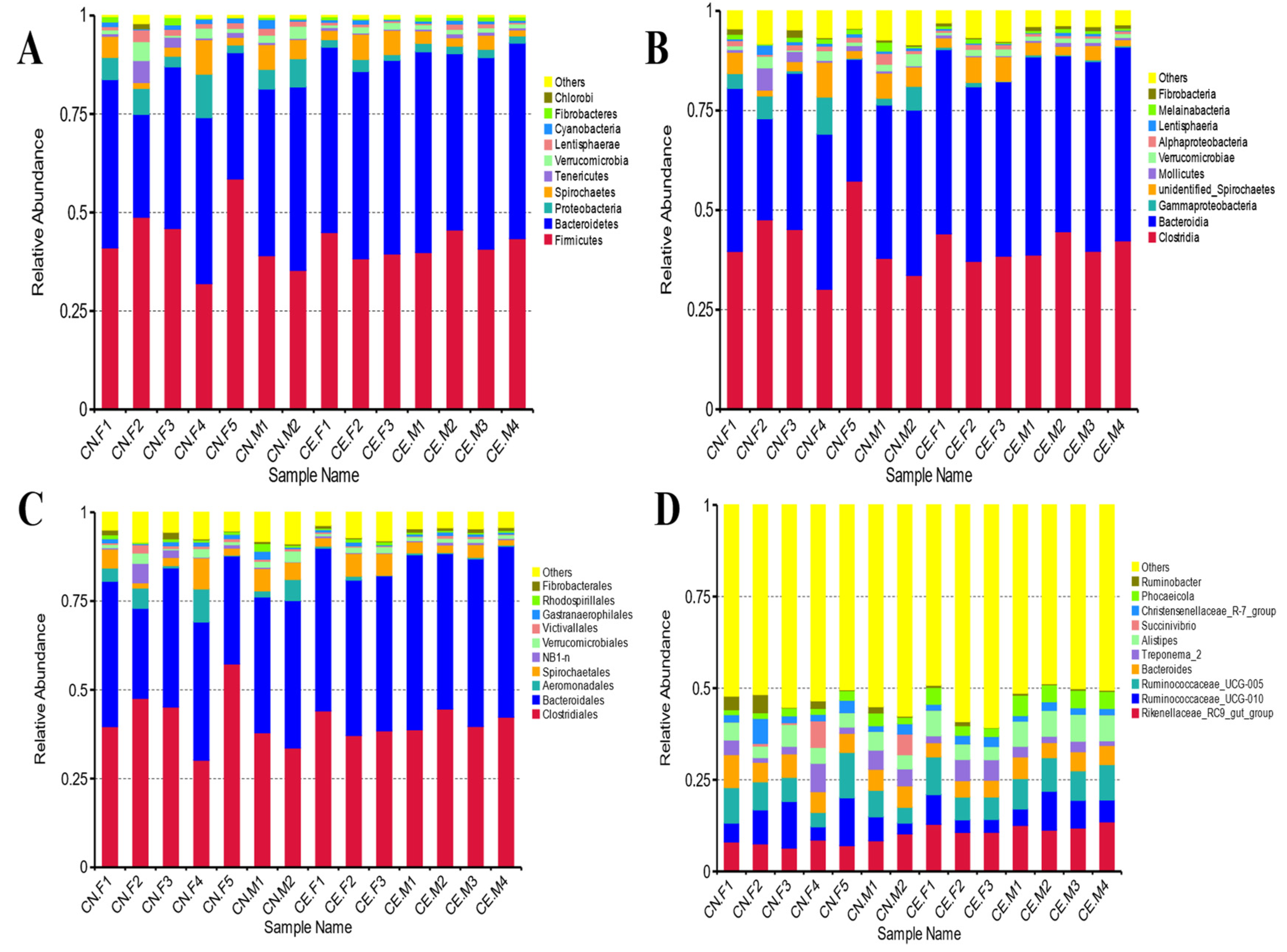
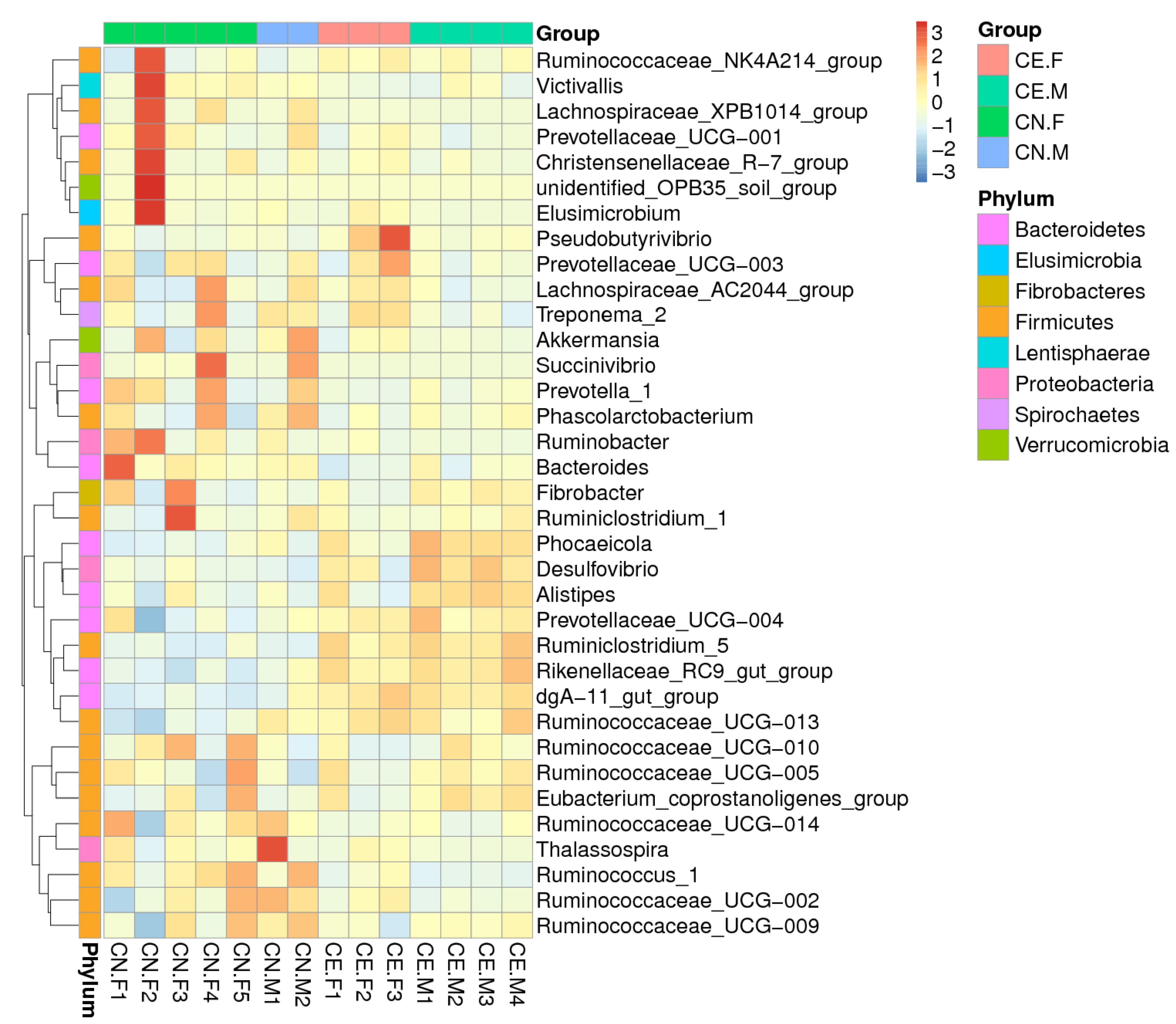
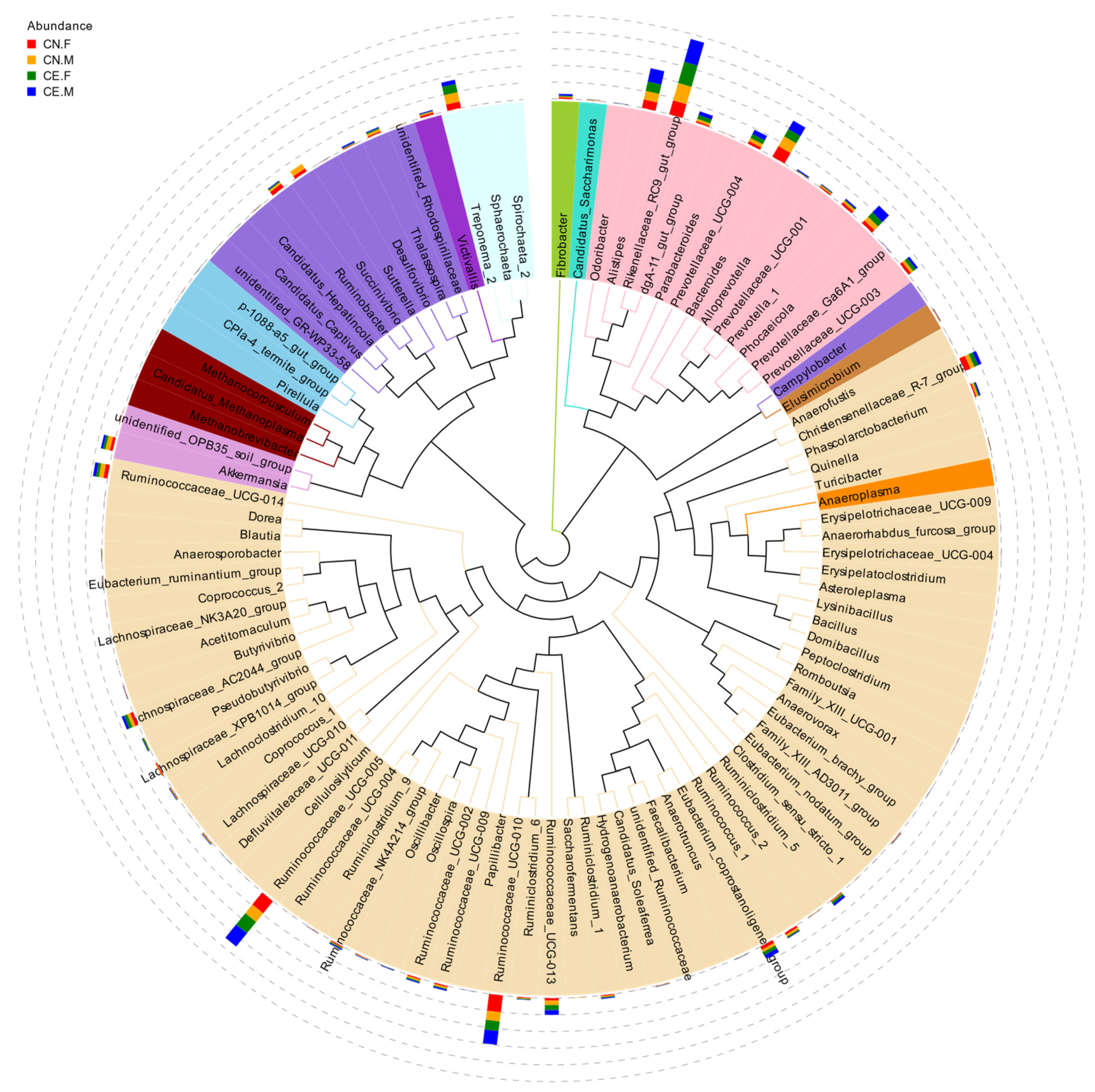
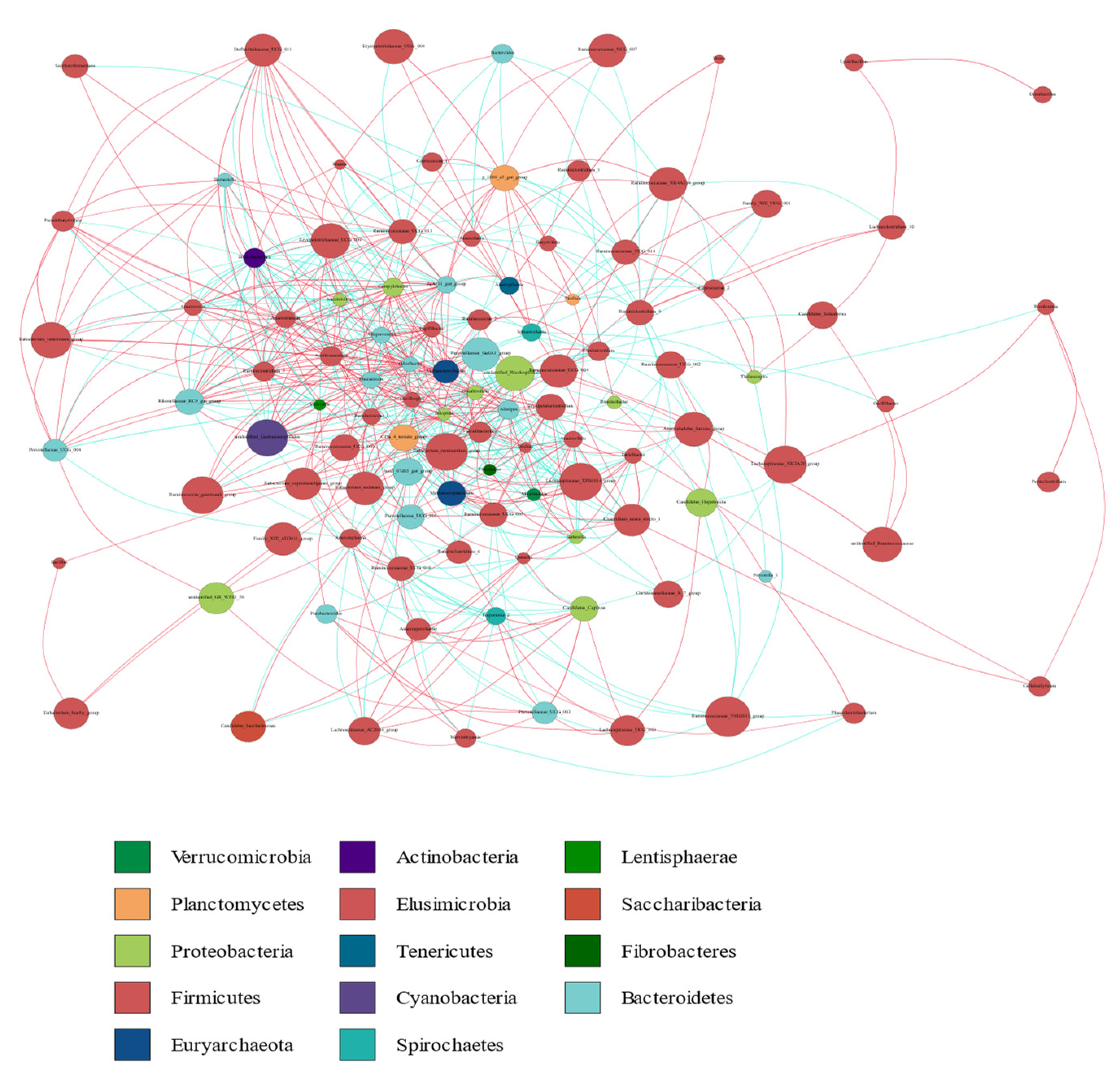
3.3. Distribution of Fecal Microbiota
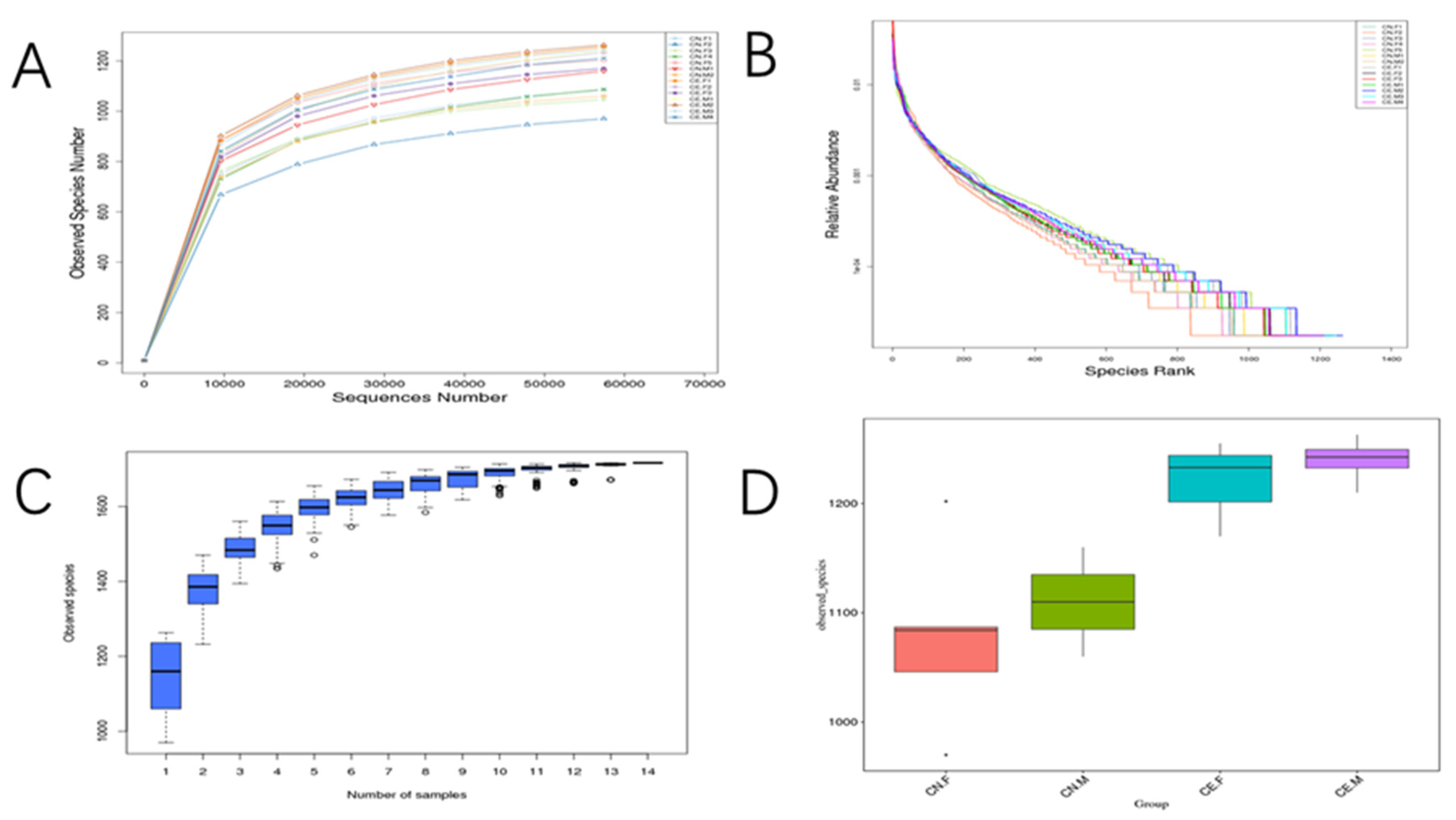
3.4. Alpha Diversity
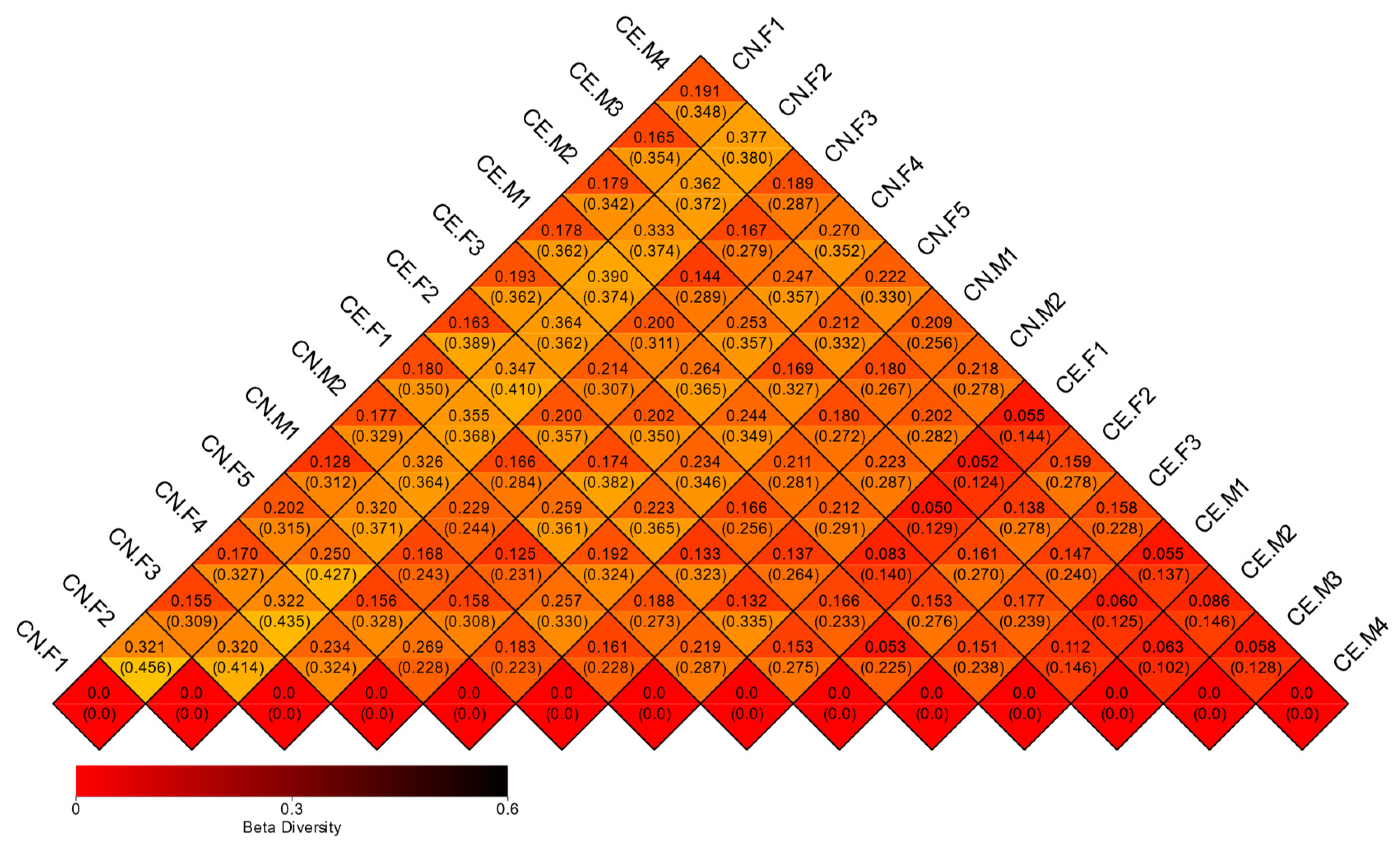
3.5. Beta Diversity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hassanin, A.; Douzery, E.J.P. Molecular and morphological phylogenies of ruminantia and the alternative position of the moschidae. Syst. Biol. 2003, 52, 206–228. [Google Scholar] [CrossRef] [PubMed]
- Meiri, M.; Lister, A.M.; Higham, T.F.G.; Stewart, J.R.; Straus, L.G.; Obermaier, H.; González Morales, M.R.; Marín-Arroyo, A.B.; Barnes, I. Late-glacial recolonization and phylogeography of european red deer (Cervus elaphus L.). Mol. Ecol. 2013, 22, 4711–4722. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.J.; Tamate, H.B.; Wilson, R.; Nagata, J.; Tatsuzawa, S.; Swanson, G.M.; Pemberton, J.M.; McCullough, D.R. Bottlenecks, drift and differentiation: The population structure and demographic history of sika deer (Cervus nippon) in the japanese archipelago. Mol. Ecol. 2001, 10, 1357–1370. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.T.; Yan, X.; Robert, S.H.; Darrin, L.; John, M.; Don, E.W.; Chris, W.W. A Guide to the Mammals of China; Princeton University Press: Princeton, NJ, USA, 2010. [Google Scholar]
- Won, C.; Smith, K.G. History and current status of mammals of the korean peninsula. Mammal Rev. 1999, 29, 3–36. [Google Scholar] [CrossRef]
- Harris, R.B. The IUCN Red List of Threatened Species. Available online: https://www.iucnredlist.org/ (accessed on 11 May 2021).
- Geist, V. The relation of social evolution and dispersal in ungulates during the pleistocene, with emphasis on the old world deer and the genus Bison. Quat. Res. 1971, 1, 285–315. [Google Scholar] [CrossRef]
- Krojerová-Prokešová, J.; Barančeková, M.; Koubek, P. Admixture of eastern and western european red deer lineages as a result of postglacial recolonization of the czech republic (Central Europe). J. Hered. 2015, 106, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Nichols, R.V.; Cromsigt, J.P.G.M.; Spong, G. Using edna to experimentally test ungulate browsing preferences. SpringerPlus 2015, 4, 489. [Google Scholar] [CrossRef] [PubMed]
- Jan, Z. Handbook of the Mammals of the World. J. Vertebr. Biol. 2020, 69, E2003.2001. [Google Scholar]
- Donadelli, R.A.; Aldrich, C.G. The effects on nutrient utilization and stool quality of beagle dogs fed diets with beet pulp, cellulose, and miscanthus grass. J. Anim. Sci. 2019, 97, 4134–4139. [Google Scholar] [CrossRef] [PubMed]
- Dearing, M.D.; Foley, W.J.; McLean, S. The influence of plant secondary metabolites on the nutritional ecology of herbivorous terrestrial vertebrates. Annu. Rev. Ecol. Evol. Syst. 2005, 36, 169–189. [Google Scholar] [CrossRef]
- Bo, T.-B.; Zhang, X.-Y.; Wen, J.; Deng, K.; Qin, X.-W.; Wang, D.-H. The microbiota–gut–brain interaction in regulating host metabolic adaptation to cold in male brandt’s voles (Lasiopodomys brandtii). ISME J. 2019, 13, 3037–3053. [Google Scholar] [CrossRef] [PubMed]
- Cui, K.; Qi, M.; Wang, S.; Diao, Q.; Zhang, N. Dietary energy and protein levels influenced the growth performance, ruminal morphology and fermentation and microbial diversity of lambs. Sci. Rep. 2019, 9, 16612. [Google Scholar] [CrossRef] [PubMed]
- Hanning, I.; Diaz-Sanchez, S. The functionality of the gastrointestinal microbiome in non-human animals. Microbiome 2015, 3, 51. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. Flash: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. Uchime improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16s rrna sequence formation and detection in sanger and 454-pyrosequenced pcr amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef]
- Edgar, R.C. Uparse: Highly accurate otu sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The silva ribosomal rna gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhang, X.; Guo, F.; Wu, W.; Zhang, T. Characterization of tetracycline resistant bacterial community in saline activated sludge using batch stress incubation with high-throughput sequencing analysis. Water Res. 2013, 47, 4207–4216. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, D.S.; Yourstone, S.; Mieczkowski, P.; Jones, C.D.; Dangl, J.L. Practical innovations for high-throughput amplicon sequencing. Nat. Methods 2013, 10, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Knight, R. Unifrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. Unifrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Asnicar, F.; Weingart, G.; Tickle, T.L.; Huttenhower, C.; Segata, N. Compact graphical representation of phylogenetic data and metadata with graphlan. PeerJ 2015, 3, e1029. [Google Scholar] [CrossRef]
- DeSantis, T.Z., Jr.; Hugenholtz, P.; Keller, K.; Brodie, E.L.; Larsen, N.; Piceno, Y.M.; Phan, R.; Andersen, G.L. Nast: A multiple sequence alignment server for comparative analysis of 16s rrna genes. Nucleic Acids Res. 2006, 34, W394–W399. [Google Scholar] [CrossRef] [PubMed]
- Lozupone Catherine, A.; Hamady, M.; Kelley Scott, T.; Knight, R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef]
- Phillips, C.D.; Phelan, G.; Dowd, S.E.; McDonough, M.M.; Ferguson, A.W.; Delton Hanson, J.; Siles, L.; OrdÓÑEz-Garza, N.; San Francisco, M.; Baker, R.J. Microbiome analysis among bats describes influences of host phylogeny, life history, physiology and geography. Mol. Ecol. 2012, 21, 2617–2627. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Vitorino, F.; Goldfarb, K.C.; Karaoz, U.; Leal, S.; Garcia-Amado, M.A.; Hugenholtz, P.; Tringe, S.G.; Brodie, E.L.; Dominguez-Bello, M.G. Comparative analyses of foregut and hindgut bacterial communities in hoatzins and cows. ISME J. 2012, 6, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Muegge Brian, D.; Kuczynski, J.; Knights, D.; Clemente Jose, C.; González, A.; Fontana, L.; Henrissat, B.; Knight, R.; Gordon Jeffrey, I. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 2011, 332, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Ruth, E.L.; Hamady, M.; Lozupone, C.; Peter, J.T.; Rob, R.R.; Bircher, J.S.; Michael, L.S.; Tammy, A.T.; Mark, D.S.; Knight, R.; et al. Evolution of mammals and their gut microbes. Science 2008, 320, 1647–1651. [Google Scholar]
- Henriques, S.F.; Dhakan, D.B.; Serra, L.; Francisco, A.P.; Carvalho-Santos, Z.; Baltazar, C.; Elias, A.P.; Anjos, M.; Zhang, T.; Maddocks, O.D.K.; et al. Metabolic cross-feeding in imbalanced diets allows gut microbes to improve reproduction and alter host behaviour. Nat. Commun. 2020, 11, 4236. [Google Scholar] [CrossRef] [PubMed]
- Youngblut, N.D.; Reischer, G.H.; Walters, W.; Schuster, N.; Walzer, C.; Stalder, G.; Ley, R.E.; Farnleitner, A.H. Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades. Nat. Commun. 2019, 10, 2200. [Google Scholar] [CrossRef]
- Abdul-Aziz, M.A.; Cooper, A.; Weyrich, L.S. Exploring relationships between host genome and microbiome: New insights from genome-wide association studies. Front. Microbiol. 2016, 7, 1611. [Google Scholar] [CrossRef]
- Bolnick, D.I.; Snowberg, L.K.; Hirsch, P.E.; Lauber, C.L.; Org, E.; Parks, B.; Lusis, A.J.; Knight, R.; Caporaso, J.G.; Svanbäck, R. Individual diet has sex-dependent effects on vertebrate gut microbiota. Nat. Commun. 2014, 5, 4500. [Google Scholar] [CrossRef]
- Wu, X.; Shang, Y.; Wei, Q.; Chen, J.; Zhang, H.; Chen, Y.; Gao, X.; Wang, Z.; Zhang, H. Gut microbiota in dholes during estrus. Front. Microbiol. 2020, 11, 575731. [Google Scholar] [CrossRef]
- Shinkai, T.; Ueki, T.; Kobayashi, Y. Detection and identification of rumen bacteria constituting a fibrolytic consortium dominated by Fibrobacter succinogenes. Anim. Sci. J. 2010, 81, 72–79. [Google Scholar] [CrossRef]
- Edrington, T.S.; Dowd, S.E.; Farrow, R.F.; Hagevoort, G.R.; Callaway, T.R.; Anderson, R.C.; Nisbet, D.J. Development of colonic microflora as assessed by pyrosequencing in dairy calves fed waste milk. J. Dairy Sci. 2012, 95, 4519–4525. [Google Scholar] [CrossRef]
- Orso, C.; Stefanello, T.B.; Franceschi, C.H.; Mann, M.B.; Varela, A.P.M.; Castro, I.M.S.; Frazzon, J.; Frazzon, A.P.G.; Andretta, I.; Ribeiro, A.M.L. Changes in the ceca microbiota of broilers vaccinated for coccidiosis or supplemented with salinomycin. Poult. Sci. 2021, 100, 100969. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Species | Sex | Raw PE | Raw Tags | Effective Tags | Base (nt) | Q20 (%) |
|---|---|---|---|---|---|---|---|
| CE.F1 | Wapiti | Female | 215,245 | 141,222 | 134,475 | 60,715,588 | 97.58 |
| CE.F2 | Wapiti | Female | 312,835 | 198,233 | 187,510 | 84,818,640 | 97.52 |
| CE.F3 | Wapiti | Female | 166,116 | 90,359 | 84,310 | 38,127,557 | 97.19 |
| CE.M1 | Wapiti | Male | 405,347 | 259,034 | 243,872 | 110,341,845 | 97.53 |
| CE.M2 | Wapiti | Male | 259,462 | 161,632 | 152,476 | 68,796,462 | 97.5 |
| CE.M3 | Wapiti | Male | 372,031 | 231,191 | 216,808 | 98,053,905 | 97.53 |
| CE.M4 | Wapiti | Male | 253,677 | 164,855 | 156,897 | 70,878,325 | 97.56 |
| CN.F1 | Sika | Female | 262,154 | 169,901 | 158,383 | 71,622,503 | 97.6 |
| CN.F2 | Sika | Female | 268,349 | 157,103 | 149,495 | 67,308,187 | 97.43 |
| CN.F3 | Sika | Female | 182,780 | 91,628 | 85,071 | 38,336,278 | 97.14 |
| CN.F4 | Sika | Female | 223,971 | 128,275 | 116,926 | 52,878,708 | 97.28 |
| CN.F5 | Sika | Female | 127,220 | 66,761 | 62,767 | 28,159,699 | 97.12 |
| CN.M1 | Sika | Male | 133,959 | 69,757 | 64,502 | 29,144,342 | 97.15 |
| CN.M2 | Sika | Male | 162,926 | 89,577 | 82,270 | 37,163,066 | 97.2 |
| Sample | Species | Sex | Observed_Species | Shannon | Simpson | Chao1 | ACE | Goods_Coverage |
|---|---|---|---|---|---|---|---|---|
| CN.F1 | Wapiti | Female | 1084 | 7.974 | 0.991 | 1148.024 | 1156.249 | 0.998 |
| CN.F2 | Wapiti | Female | 970 | 7.703 | 0.99 | 1044.882 | 1059.369 | 0.998 |
| CN.F3 | Wapiti | Female | 1046 | 8.227 | 0.993 | 1100.301 | 1103.797 | 0.998 |
| CN.F4 | Wapiti | Male | 1087 | 7.675 | 0.986 | 1190.5 | 1195.605 | 0.997 |
| CN.F5 | Wapiti | Male | 1202 | 8.526 | 0.994 | 1251.5 | 1249.636 | 0.998 |
| CN.M1 | Wapiti | Male | 1160 | 8.172 | 0.992 | 1294.384 | 1287.833 | 0.997 |
| CN.M2 | Wapiti | Male | 1060 | 7.686 | 0.986 | 1113.509 | 1118.624 | 0.998 |
| CE.F1 | Sika | Female | 1255 | 8.313 | 0.993 | 1325.522 | 1334.977 | 0.998 |
| CE.F2 | Sika | Female | 1233 | 8.168 | 0.992 | 1365.026 | 1340.944 | 0.997 |
| CE.F3 | Sika | Female | 1170 | 8.053 | 0.99 | 1232.046 | 1236.775 | 0.998 |
| CE.M1 | Sika | Female | 1240 | 8.201 | 0.993 | 1390.976 | 1369.852 | 0.997 |
| CE.M2 | Sika | Female | 1263 | 8.408 | 0.993 | 1322.049 | 1335.561 | 0.998 |
| CE.M3 | Sika | Male | 1245 | 8.328 | 0.993 | 1319.773 | 1321.592 | 0.998 |
| CE.M4 | Sika | Male | 1210 | 8.27 | 0.993 | 1331.454 | 1298.882 | 0.997 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, J.; Wu, X.; Wang, X.; Shang, Y.; Zhang, H. Uncovering the Fecal Bacterial Communities of Sympatric Sika Deer (Cervus nippon) and Wapiti (Cervus canadensis). Animals 2022, 12, 2468. https://doi.org/10.3390/ani12182468
Yan J, Wu X, Wang X, Shang Y, Zhang H. Uncovering the Fecal Bacterial Communities of Sympatric Sika Deer (Cervus nippon) and Wapiti (Cervus canadensis). Animals. 2022; 12(18):2468. https://doi.org/10.3390/ani12182468
Chicago/Turabian StyleYan, Jiakuo, Xiaoyang Wu, Xibao Wang, Yongquan Shang, and Honghai Zhang. 2022. "Uncovering the Fecal Bacterial Communities of Sympatric Sika Deer (Cervus nippon) and Wapiti (Cervus canadensis)" Animals 12, no. 18: 2468. https://doi.org/10.3390/ani12182468