
Genome-Wide Association Study of Body Weight Trait in Yaks


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Phenotyping
2.2. Sequencing and SNP Calling
2.3. Population Stratification and Heritability Estimation
2.4. Association Study
3. Results
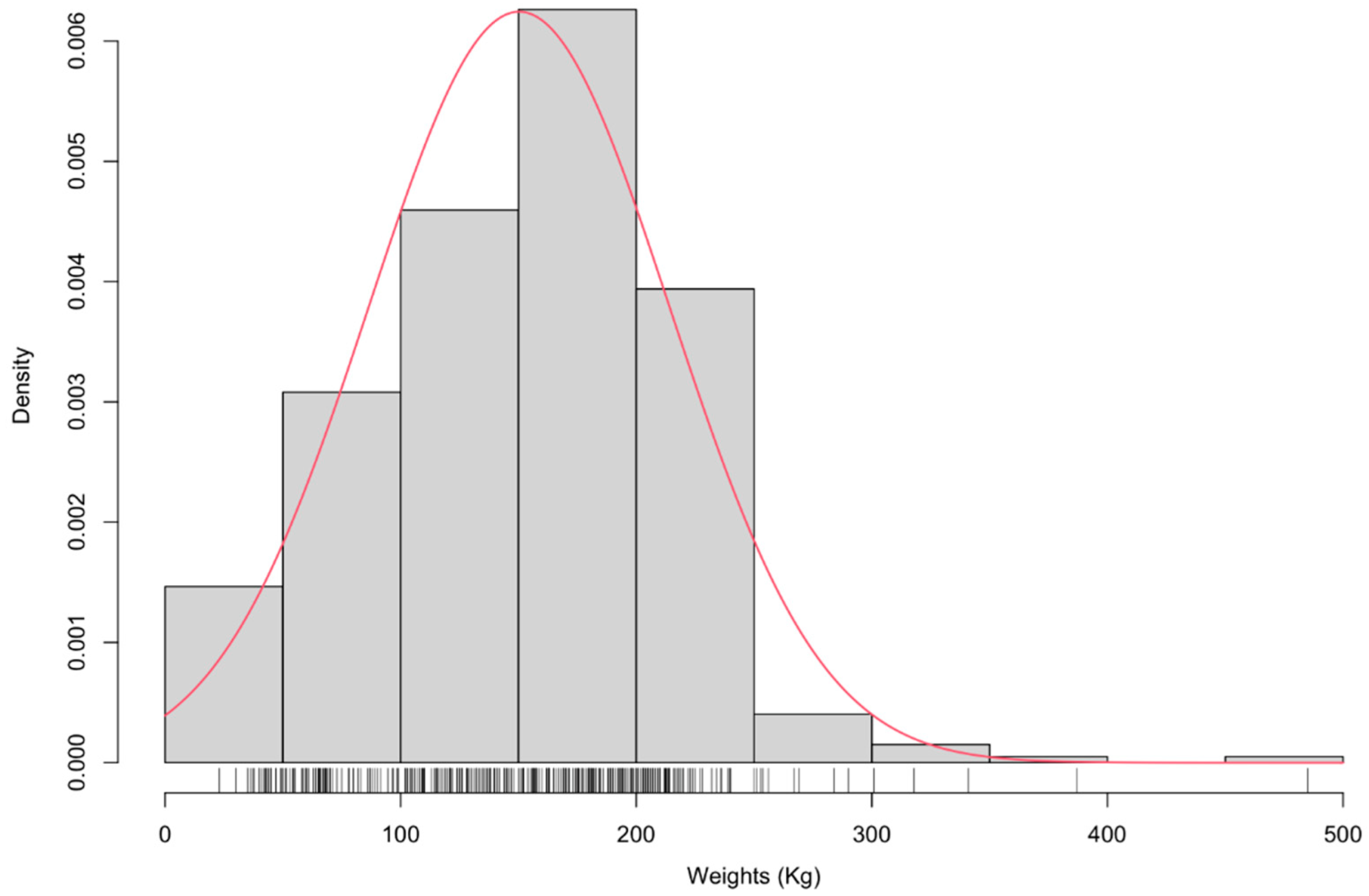
3.1. Phenotypic Distribution
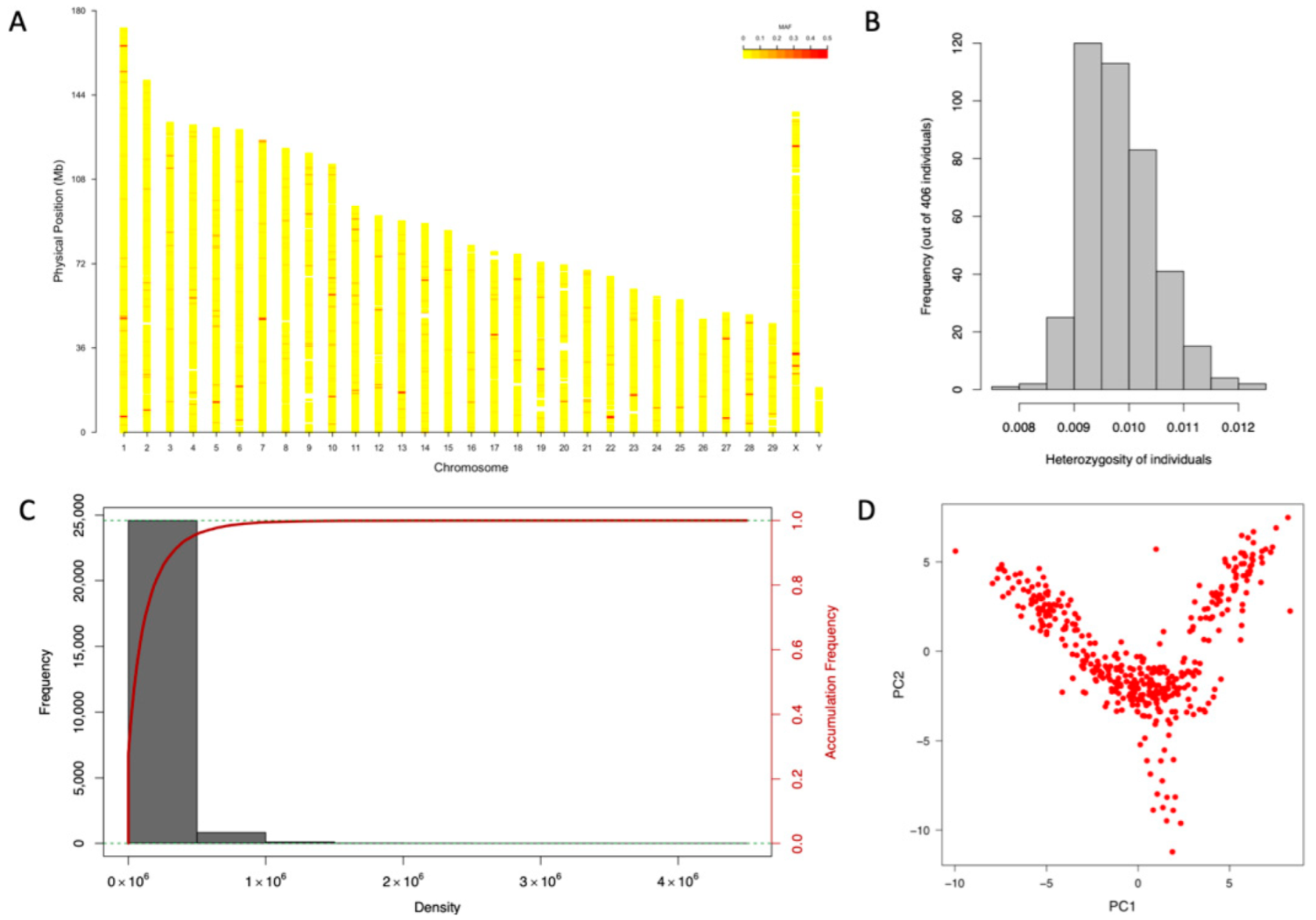
3.2. Genotype Calling and Marker Density
3.3. Population Structure and Heritability
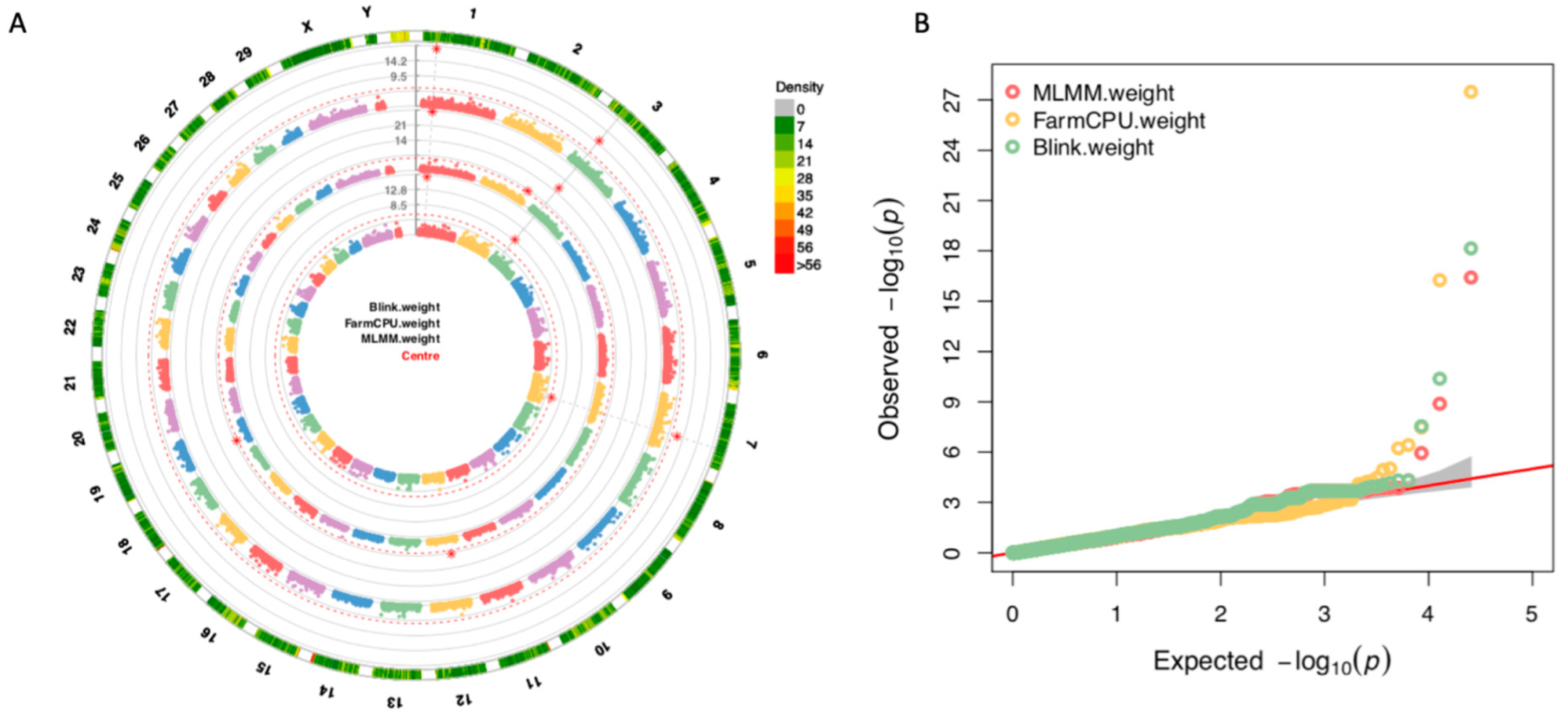
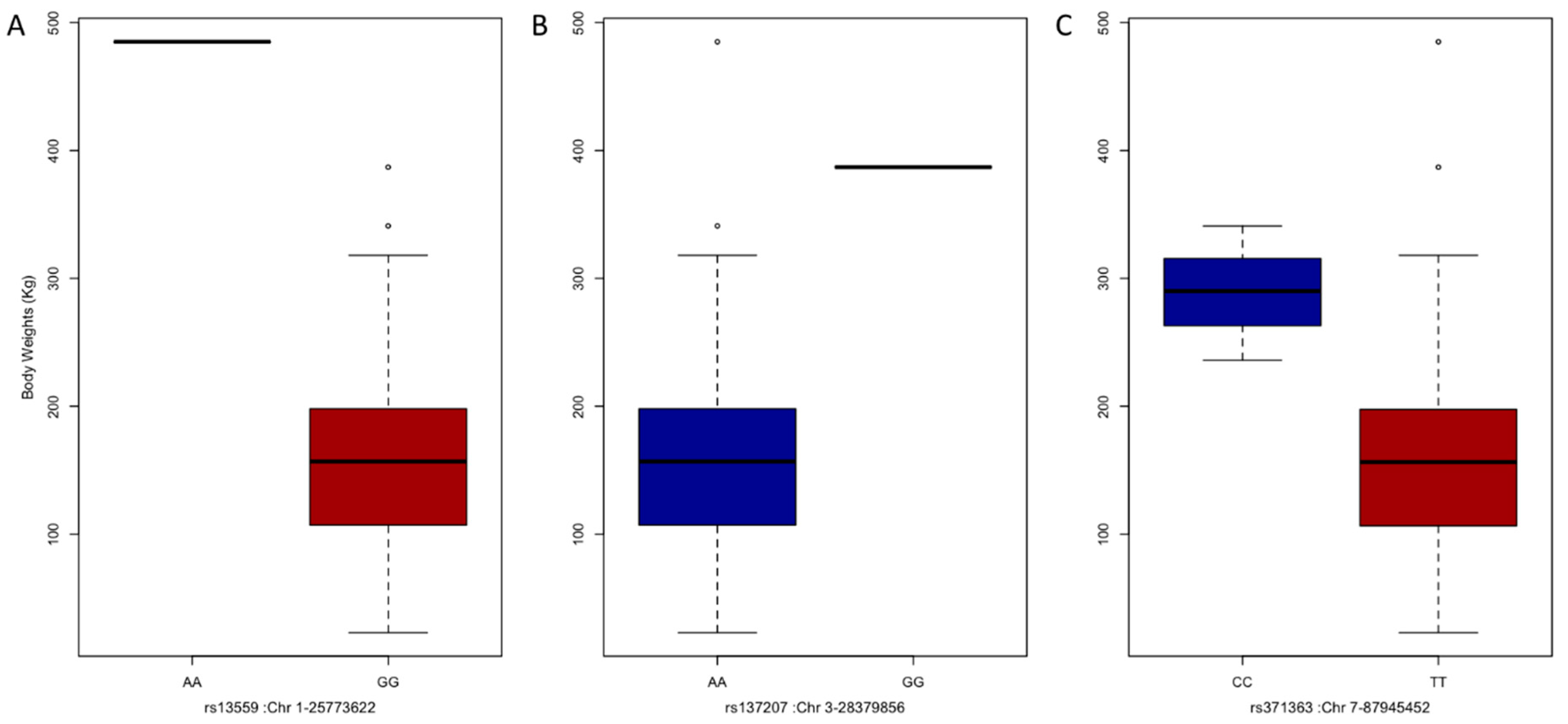
3.4. GWAS and Candidate Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, D.D.; Ding, X.D.; Wang, S.; Wójcik, J.M.; Zhang, Y.; Tokarska, M.; Li, Y.; Wang, M.S.; Faruque, O.; Nielsen, R.; et al. Pervasive Introgression Facilitated Domestication and Adaptation in the Bos Species Complex. Nat. Ecol. Evol. 2018, 2, 1139–1145. [Google Scholar] [CrossRef]
- Medugorac, I.; Graf, A.; Grohs, C.; Rothammer, S.; Zagdsuren, Y.; Gladyr, E.; Zinovieva, N.; Barbieri, J.; Seichter, D.; Russ, I.; et al. Whole-Genome Analysis of Introgressive Hybridization and Characterization of the Bovine Legacy of Mongolian Yaks. Nat. Genet. 2017, 49, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Xiao, S.; Wang, Q.; Wang, Z. Comparative Analysis of the GBLUP, EmBayesB, and GWAS Algorithms to Predict Genetic Values in Large Yellow Croaker (Larimichthys crocea). BMC Genom. 2016, 17, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ge, F.; Jia, C.; Chu, M.; Liang, C.; Yan, P. Copy Number Variation of the Cadm2 Gene and Its Association with Growth Traits in Yak. Animals 2019, 9, 1008. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Long, R.; Liang, J.B.; Guo, X.; Ding, L.; Shang, Z. Comparison of Nitrogen Metabolism in Yak (Bos Grunniens) and Indigenous Cattle (Bos Taurus) on the Qinghai-Tibetan Plateau. Asian-Australas. J. Anim. Sci. 2011, 24, 766–777. [Google Scholar] [CrossRef]
- Xue, B.; Zhao, X.Q.; Zhang, Y.S. Seasonal Changes in Weight and Body Composition of Yak Grazing on Alpine-Meadow Grassland in the Qinghai-Tibetan Plateau of China. J. Anim. Sci. 2005, 83, 1908–1913. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Wu, S.; Zhao, W.; Mipam, T.; Liu, J.; Liu, W.; Yi, C.; Shah, M.A.; Yu, S.; Cai, X. Differentially Expressed MicroRNAs between Cattleyak and Yak Testis. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Qiu, Q.; Zhang, G.; Ma, T.; Qian, W.; Ye, Z.; Cao, C.; Hu, Q.; Kim, J.; Larkin, D.M.; Auvil, L.; et al. The Yak Genome and Adaptation to Life at High Altitude. Nat. Genet. 2012, 44, 946–949. [Google Scholar] [CrossRef] [Green Version]
- Ji, Q.M.; Xin, J.W.; Chai, Z.X.; Zhang, C.F.; Dawa, Y.; Luo, S.; Zhang, Q.; Pingcuo, Z.; Peng, M.S.; Zhu, Y.; et al. A Chromosome-Scale Reference Genome and Genome-Wide Genetic Variations Elucidate Adaptation in Yak. Mol. Ecol. Resour. 2021, 21, 201–211. [Google Scholar] [CrossRef]
- Trait, G.C.; Gcta, A.; Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. Genome-Wide Association Studies and Genomic Prediction. In Methods in Molecular Biology; Gondro, C., van der Werf, J., Hayes, B., Eds.; Humana Press: Totowa, NJ, USA, 2013; Volume 1019, pp. 215–236. ISBN 978-1-62703-446-3. [Google Scholar]
- Yano, K.; Yamamoto, E.; Aya, K.; Takeuchi, H.; Lo, P.C.; Hu, L.; Yamasaki, M.; Yoshida, S.; Kitano, H.; Hirano, K.; et al. Genome-Wide Association Study Using Whole-Genome Sequencing Rapidly Identifies New Genes Influencing Agronomic Traits in Rice. Nat. Genet. 2016, 48, 927–934. [Google Scholar] [CrossRef]
- Svishcheva, G.R.; Axenovich, T.I.; Belonogova, N.M.; Van Duijn, C.M.; Aulchenko, Y.S. Rapid Variance Components-Based Method for Whole-Genome Association Analysis. Nat. Genet. 2012, 44, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Z. GAPIT Version 3: Boosting Power and Accuracy for Genomic Association and Prediction. Genom. Proteom. Bioinform. 2021, 19, 629–640. [Google Scholar] [CrossRef]
- Jia, C.; Li, C.; Fu, D.; Chu, M.; Zan, L.; Wang, H.; Liang, C.; Yan, P. Identification of Genetic Loci Associated with Growth Traits at Weaning in Yak through a Genome-Wide Association Study. Anim. Genet. 2020, 51, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [PubMed]
- Zhang, S.; Liu, W.; Liu, X.; Du, X.; Zhang, K.; Zhang, Y.; Song, Y.; Zi, Y.; Qiu, Q.; Lenstra, J.A.; et al. Structural Variants Selected during Yak Domestication Inferred from Long-Read Whole-Genome Sequencing. Mol. Biol. Evol. 2021, 38, 3676–3680. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Yu, X.; Sun, S. Comparing a Few SNP Calling Algorithms Using Low-Coverage Sequencing Data. BMC Bioinform. 2013, 14. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Islam, T.; Hae, C.; Iwata, H.; Shimono, H.; Kimura, A. A Deep Learning Method to Impute Missing Values and Compress Genome-Wide Polymorphism Data in Rice. InBioinformatics 2021, 3, 101–109. [Google Scholar] [CrossRef]
- Ayres, D.L.; Darling, A.; Zwickl, D.J.; Beerli, P.; Holder, M.T.; Lewis, P.O.; Huelsenbeck, J.P.; Ronquist, F.; Swofford, D.L.; Cummings, M.P.; et al. BEAGLE: An Application Programming Interface and High-Performance Computing Library for Statistical Phylogenetics. Syst. Biol. 2012, 61, 170–173. [Google Scholar] [CrossRef]
- Kang, H.M.; Zaitlen, N.A.; Wade, C.M.; Kirby, A.; Heckerman, D.; Daly, M.J.; Eskin, E. Efficient Control of Population Structure in Model Organism Association Mapping. Genetics 2008, 178, 1709–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Buckler, E.S.; Casstevens, T.M.; Bradbury, P.J. Software Engineering the Mixed Model for Genome-Wide Association Studies on Large Samples. Brief. Bioinform. 2009, 10, 664–675. [Google Scholar] [PubMed] [Green Version]
- Neves, H.H.R.; Carvalheiro, R.; Queiroz, S.A. A Comparison of Statistical Methods for Genomic Selection in a Mice Population. BMC Genet. 2012, 13, 100. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef]
- Huang, M.; Liu, X.; Zhou, Y.; Summers, R.M.; Zhang, Z. BLINK: A Package for the next Level of Genome-Wide Association Studies with Both Individuals and Markers in the Millions. GigaScience 2018, 91, 399–404. [Google Scholar] [CrossRef]
- Zheng, Y.; Guan, J.; Wang, L.; Luo, X.; Zhang, X. Comparative Proteomic Analysis of Spleen Reveals Key Immune-Related Proteins in the Yak (Bos Grunniens) at Different Growth Stages. Comp. Biochem. Physiol. Part D Genom. Proteom. 2022, 42, 100968. [Google Scholar] [CrossRef]
- Cai, X.; Mipam, T.D.; Zhao, F.F.; Sun, L. SNPs Detected in the Yak MC4R Gene and Their Association with Growth Traits. Animal 2015, 9, 1097–1103. [Google Scholar] [CrossRef] [Green Version]
- Ge, F.; Jia, C.; Bao, P.; Wu, X.; Liang, C.; Yan, P. Accuracies of Genomic Prediction for Growth Traits at Weaning and Yearling Ages in Yak. Animals 2020, 10, 1793. [Google Scholar] [CrossRef]
- Liu, J.J.; Liang, A.X.; Campanile, G.; Plastow, G.; Zhang, C.; Wang, Z.; Salzano, A.; Gasparrini, B.; Cassandro, M.; Yang, L.G. Genome-Wide Association Studies to Identify Quantitative Trait Loci Affecting Milk Production Traits in Water Buffalo. J. Dairy Sci. 2018, 101, 433–444. [Google Scholar] [CrossRef] [Green Version]
- Medicine, N. Mfsd14a (Hiat1) Gene Disruption Causes Globozoospermia and Infertility in Male Mice. 2. Reproduction 2016, 152, 91–99. [Google Scholar]
- Fan, B.; Onteru, S.K.; Du, Z.-Q.; Garrick, D.J.; Stalder, K.J.; Rothschild, M.F. Genome-Wide Association Study Identifies Loci for Body Composition and Structural Soundness Traits in Pigs. PLoS ONE 2011, 6, e14726. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Duan, X.; An, B.; Chang, T.; Liang, M.; Xu, L.; Zhang, L. Genome-Wide Association Study Based on Random Regression Model Reveals Candidate Genes Associated with Longitudinal Data in Chinese Simmental Beef Cattle. Animals 2021, 11, 1–16. [Google Scholar]
- Zhou, Z.; Jiang, Y.; Wang, Z.; Gou, Z.; Lyu, J.; Li, W.; Yu, Y.; Shu, L.; Zhao, Y.; Ma, Y.; et al. Resequencing 302 Wild and Cultivated Accessions Identifies Genes Related to Domestication and Improvement in Soybean. Nat. Biotechnol. 2015, 33, 408–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP No. | Chromosome | Position (bp) | Gene Name |
|---|---|---|---|
| rs13559 | 1 | 25,773,622 | ENSBGRG00000000052-ENSBGRG00000000053 |
| rs137207 | 3 | 28,379,856 | MFSD14A, SASS6, TRMT13, LRRC39, DBT |
| rs371363 | 7 | 87,945,452 | U1 |
| rs10942 | 1 | 22,314,135 | NCAM2 |
| rs118493 | 2 | 140,542,613 | TFPI |
| rs815163 | 19 | 16,238,800 | BPTF, KPNA2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Li, X.; Peng, W.; Zhong, J.; Jiang, M. Genome-Wide Association Study of Body Weight Trait in Yaks. Animals 2022, 12, 1855. https://doi.org/10.3390/ani12141855
Wang J, Li X, Peng W, Zhong J, Jiang M. Genome-Wide Association Study of Body Weight Trait in Yaks. Animals. 2022; 12(14):1855. https://doi.org/10.3390/ani12141855
Chicago/Turabian StyleWang, Jiabo, Xiaowei Li, Wei Peng, Jincheng Zhong, and Mingfeng Jiang. 2022. "Genome-Wide Association Study of Body Weight Trait in Yaks" Animals 12, no. 14: 1855. https://doi.org/10.3390/ani12141855