
Absence of Circadian Rhythm in Fecal Microbiota of Laying Hens under Common Light

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Feeding
2.2. Feces sample Collection
2.3. DNA Extraction and 16S rRNA Amplification
2.4. Sequence Analysis
2.5. Diurnal Analysis of Microbiome Data Sets and MNTD Taxonomic β-Diversity Metrics
2.6. Construction of a Co-Occurrence Network
3. Results
3.1. Fecal Microbiota Structure in Laying Hens
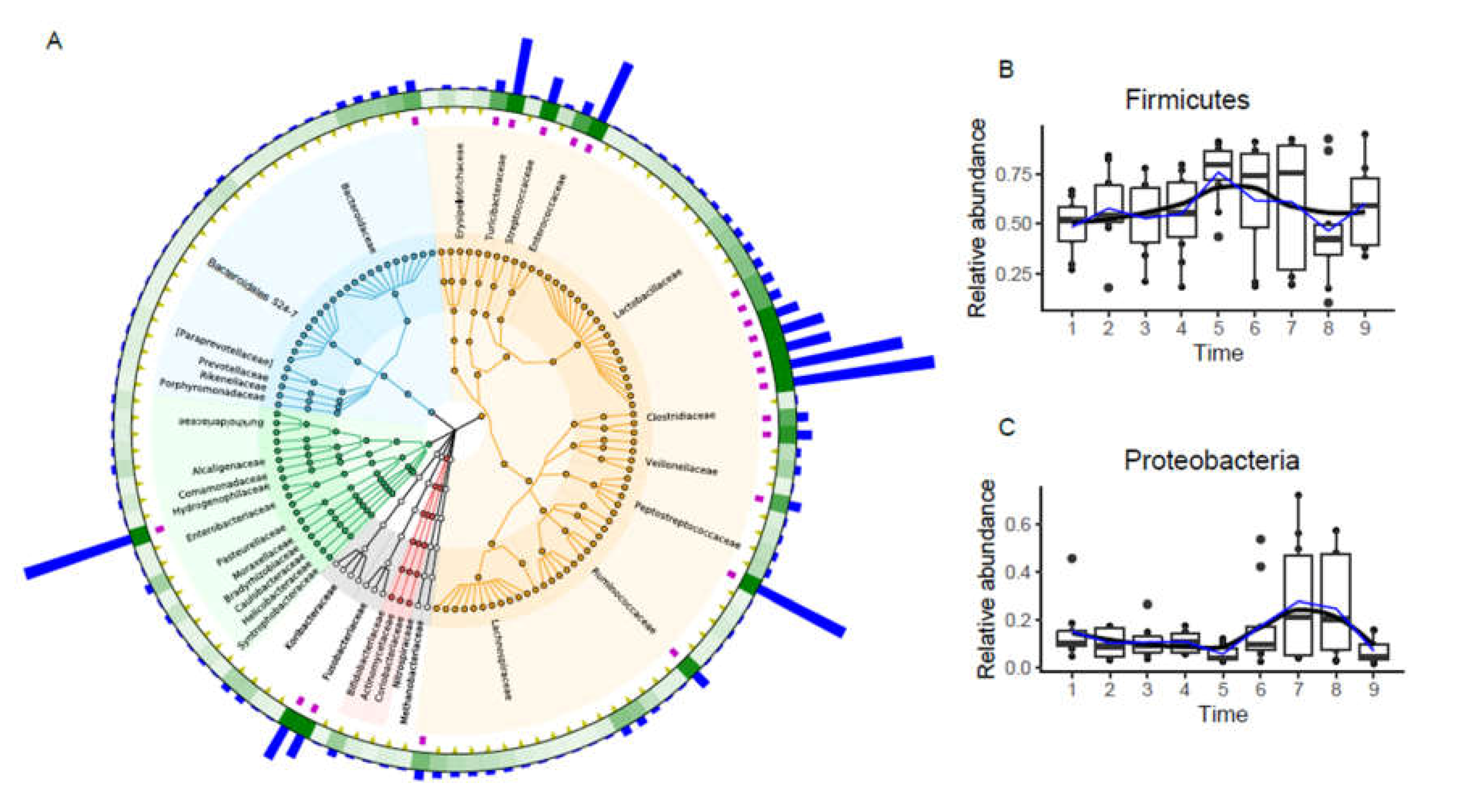
3.2. Fecal Microbiota Taxonomic Composition
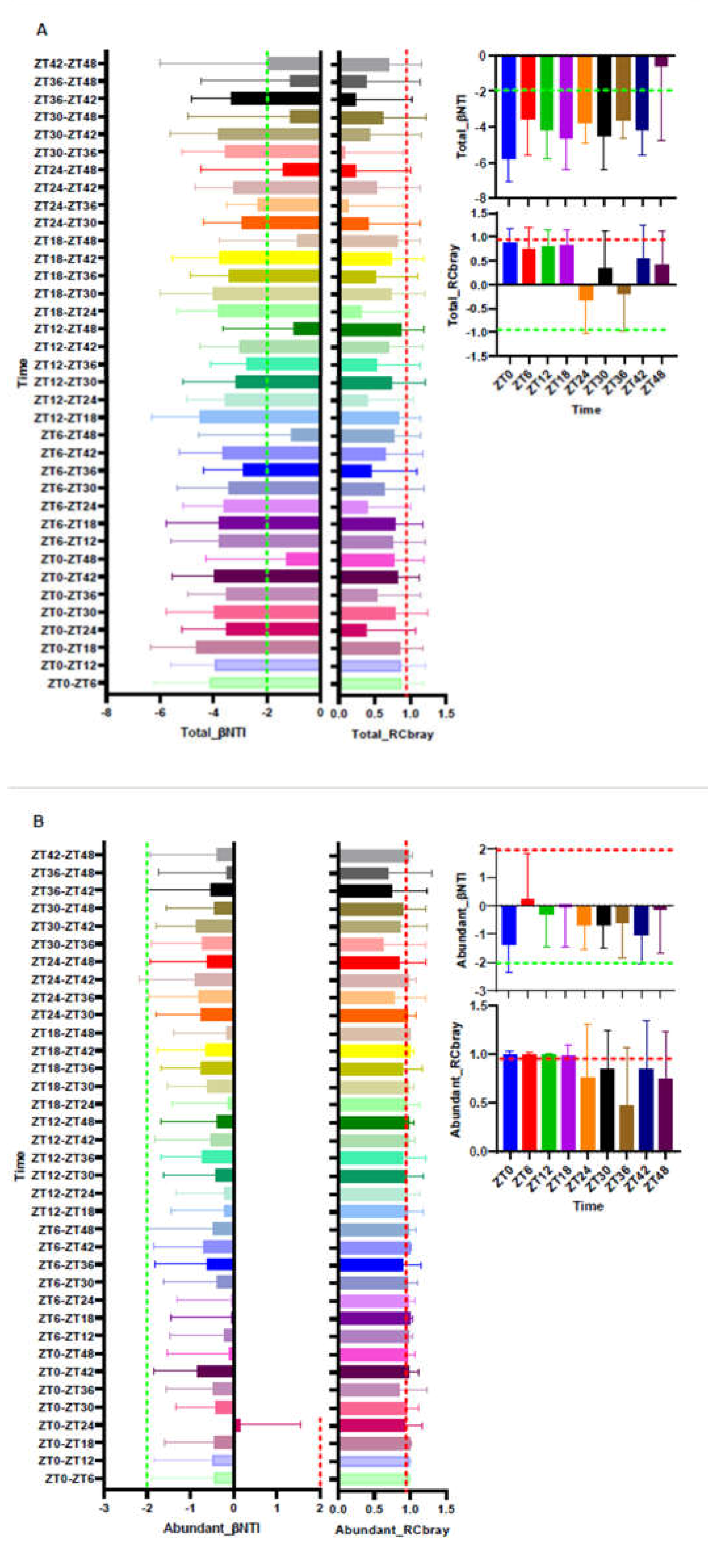
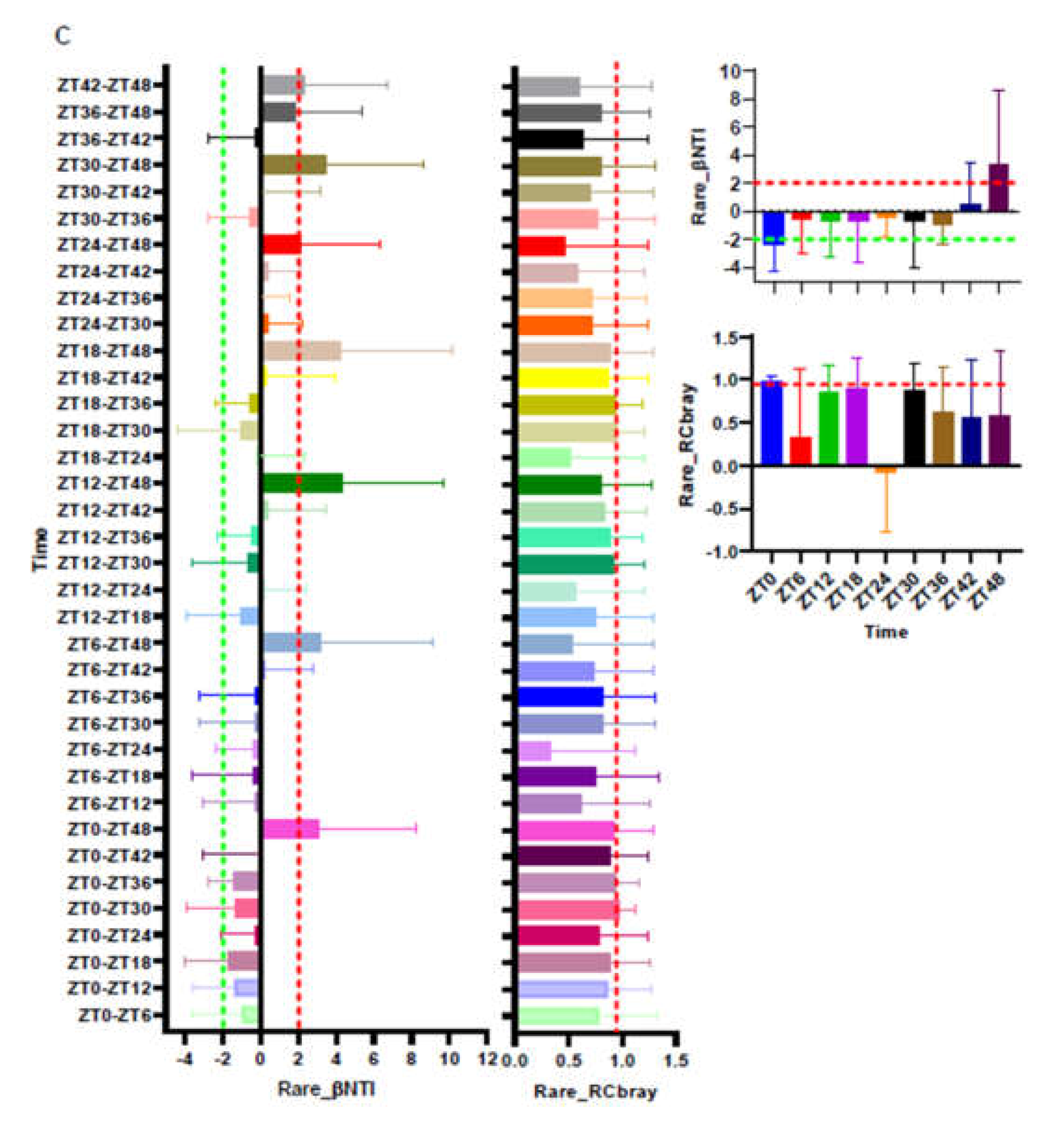
3.3. Turnover of the Total, Abundant, and Rare of ASV Fractions
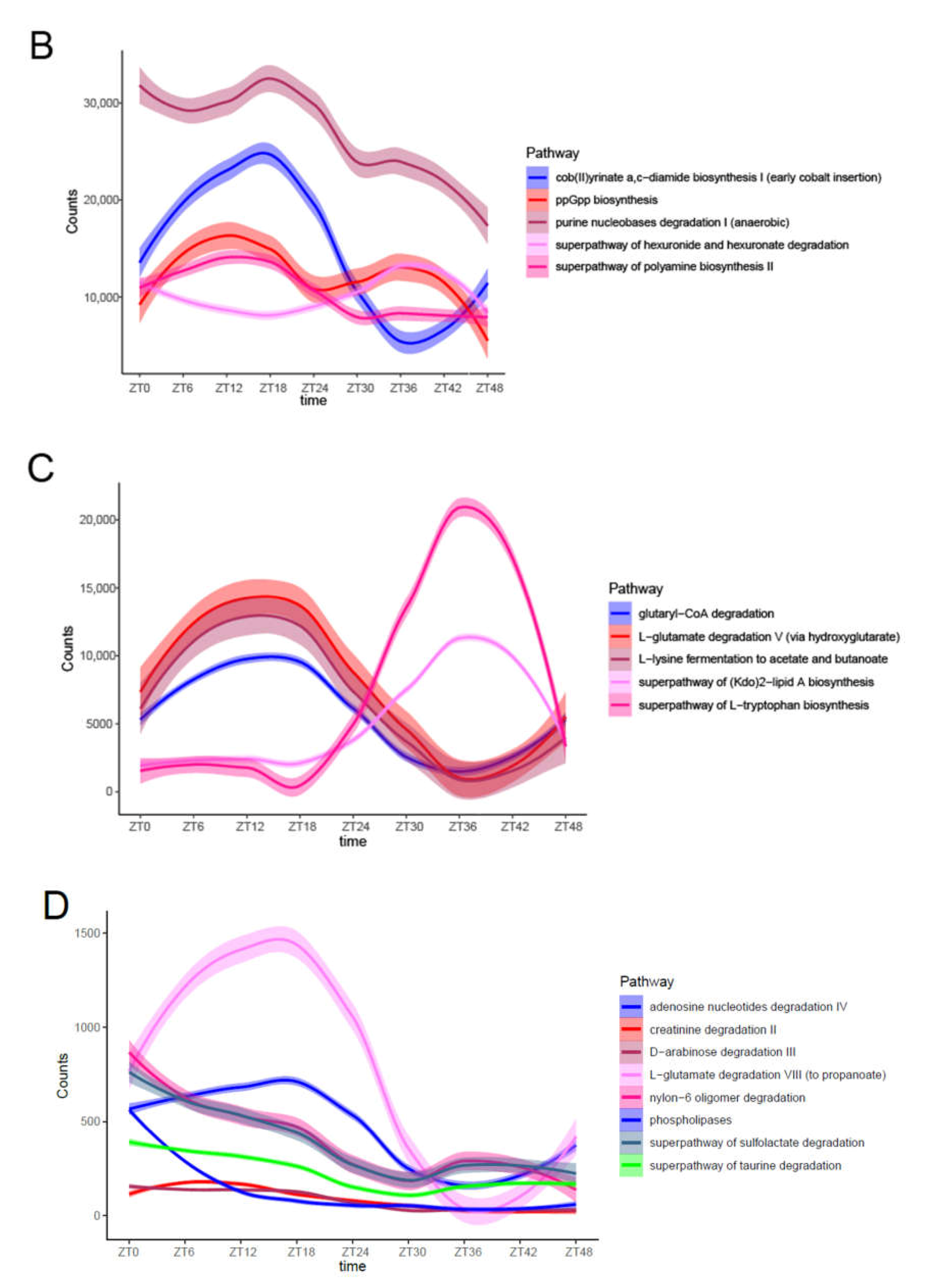
3.4. Predicted Molecular Functions of Fecal Microbiota
3.5. Cooccurrence Networks of Feces Bacteria
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Choi, K.Y.; Lee, T.K.; Sul, W.J. Metagenomic Analysis of Chicken Gut Microbiota for Improving Metabolism and Health of Chickens—A Review. Asian Australas. J. Anim. Sci. 2015, 28, 1217–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Ma, C.; Sun, Z.; Wang, L.; Huang, S.; Su, X.; Xu, J.; Zhang, H. Feed-additive probiotics accelerate yet antibiotics delay intestinal microbiota maturation in broiler chicken. Microbiome 2017, 5, 91. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cao, Z.H.; Yang, Y.T.; Chen, L.; Liu, J.P.; Lin, Q.Y.; Qiao, Y.Y.; Zhao, Z.Y.; An, Q.C.; Zhang, C.Y.; et al. Correlation between Jejunal Microbial Diversity and Muscle Fatty Acids Deposition in Broilers Reared at Different Ambient Temperatures. Sci. Rep. 2019, 9, 4. [Google Scholar] [CrossRef]
- Henchion, M.; McCarthy, M.; Resconi, V.C.; Troy, D. Meat consumption: Trends and quality matters. Meat Sci. 2014, 98, 561–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, Z.; Liu, R.; Wang, J.; Zheng, M.; Li, Q.; Cui, H.; Zhao, G.; Wen, J. Alteration of Hepatic Gene Expression along with the Inherited Phenotype of Acquired Fatty Liver in Chicken. Genes 2018, 9, 4. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhang, T.; Zhang, S.S.; Huang, J.X.; Zhang, G.X.; Xie, K.Z.; Wang, J.Y.; Wu, H.Q.; Dai, G.J. Identification of Long Non-Coding RNA-Associated Competing Endogenous RNA Network in the Differentiation of Chicken Preadipocytes. Genes 2019, 10, 795. [Google Scholar] [CrossRef] [Green Version]
- Geng, Y.; Ma, Q.; Wang, Z.; Guo, Y. Dietary vitamin D3 supplementation protects laying hens against lipopolysaccharide-induced immunological stress. Nutr. Metab. 2018, 15, 58. [Google Scholar] [CrossRef] [Green Version]
- Bestman, M.W.P.; Wagenaar, J.P. Farm level factors associated with feather pecking in organic laying hens. Livest. Prod. Sci. 2003, 80, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Birkl, P.; Bharwani, A.; Kjaer, J.B.; Kunze, W.; McBride, P.; Forsythe, P.; Harlander-Matauschek, A. Differences in cecal microbiome of selected high and low feather-pecking laying hens. Poultry Sci. 2018, 97, 3009–3014. [Google Scholar] [CrossRef]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.Y.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut Microbiota from Twins Discordant for Obesity Modulate Metabolism in Mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Walter, L.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ Brit. Med. J. 2018, 361, k2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Z.P.; Wang, X.Y.; Pang, Y.; Cheng, S.S.; Liu, J.Y. Biointerfacial self-assembly generates lipid membrane coated bacteria for enhanced oral delivery and treatment. Nat. Commun. 2019, 10, 5783. [Google Scholar] [CrossRef]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S.; et al. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar]
- Estaki, M.; Pither, J.; Baumeister, P.; Little, J.P.; Gill, S.K.; Ghosh, S.; Ahmadi-Vand, Z.; Marsden, K.R.; Gibson, D.L. Cardiorespiratory fitness as a predictor of intestinal microbial diversity and distinct metagenomic functions. Microbiome 2016, 4, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkar, S.G.; Kalsbeek, A.; Cheeseman, J.F. Potential Role for the Gut Microbiota in Modulating Host Circadian Rhythms and Metabolic Health. Microorganisms 2019, 7, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, M.C.; Spieth, P.M.; Quinn, K.; Parotto, M.; Zhang, H.B.; Slutsky, A.S. Circadian rhythms: From basic mechanisms to the intensive care unit. Crit. Care Med. 2012, 40, 246–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaiss, C.A.; Zeevi, D.; Levy, M.; Segal, E.; Elinav, E. A day in the life of the meta-organism: Diurnal rhythms of the intestinal microbiome and its host. Gut Microbes 2015, 6, 137–142. [Google Scholar] [CrossRef]
- Thaiss, C.A.; Levy, M.; Korem, T.; Dohnalova, L.; Shapiro, H.; Jaitin, D.A.; David, E.; Winter, D.R.; Gury-BenAri, M.; Tatirovsky, E.; et al. Microbiota Diumal Rhythmicity Programs Host Transcriptome Oscillations. Cell 2016, 167, 1495. [Google Scholar] [CrossRef] [Green Version]
- Hut, R.A.; Beersma, D.G. Evolution of time-keeping mechanisms: Early emergence and adaptation to photoperiod. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 2141–2154. [Google Scholar] [CrossRef] [Green Version]
- Thaiss, C.A.; Zeevi, D.; Levy, M.; Zilberman-Schapira, G.; Suez, J.; Tengeler, A.C.; Abramson, L.; Katz, M.N.; Korem, T.; Zmora, N.; et al. Transkingdom Control of Microbiota Diurnal Oscillations Promotes Metabolic Homeostasis. Cell 2014, 159, 514–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cribbet, M.R.; Logan, R.W.; Edwards, M.D.; Hanlon, E.; Peek, C.B.; Stubblefield, J.J.; Vasudevan, S.; Ritchey, F.; Frank, E. Circadian rhythms and metabolism: From the brain to the gut and back again. Ann. Rep. 2016, 1385, 21–40. [Google Scholar] [CrossRef]
- Wang, J.; Mauvoisin, D.; Martin, E.; Atger, F.; Galindo, A.N.; Dayon, L.; Sizzano, F.; Palini, A.; Kussmann, M.; Waridel, P.; et al. Nuclear Proteomics Uncovers Diurnal Regulatory Landscapes in Mouse Liver. Cell Metab. 2017, 25, 102–117. [Google Scholar] [CrossRef] [Green Version]
- Leone, V.; Gibbons, S.M.; Martinez, K.; Hutchison, A.L.; Huang, E.Y.; Cham, C.M.; Pierre, J.F.; Heneghan, A.F.; Nadimpalli, A.; Hubert, N.; et al. Effects of Diurnal Variation of Gut Microbes and High-Fat Feeding on Host Circadian Clock Function and Metabolism. Cell Host Microbe 2015, 17, 681–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarrinpar, A.; Chaix, A.; Yooseph, S.; Panda, S. Diet and Feeding Pattern Affect the Diurnal Dynamics of the Gut Microbiome. Cell Metab. 2014, 20, 1006–1017. [Google Scholar] [CrossRef] [Green Version]
- Farghly, M.F.A.; Mahrose, K.M.; Rehman, Z.U.; Yu, S.Q.; Abdelfattah, M.G.; El-Garhy, O.H. Intermittent lighting regime as a tool to enhance egg production and eggshell thickness in Rhode Island Red laying hens. Poultry Sci. 2019, 98, 2459–2465. [Google Scholar] [CrossRef]
- Hieke, A.S.C.; Hubert, S.M.; Athrey, G. Circadian disruption and divergent microbiota acquisition under extended photoperiod regimens in chicken. PeerJ 2019, 7, 6592. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Joseph, C.M.L.; Allen, G.; Benson, A.K.; Mills, D.A. Next-Generation Sequencing Reveals Significant Bacterial Diversity of Botrytized Wine. PLoS ONE 2012, 7, 36357. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. The KEGG database. Novart. Fdn. Symp. 2002, 247, 91–103. [Google Scholar]
- Stegen, J.C.; Lin, X.; Konopka, A.E.; Fredrickson, J.K. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 2012, 6, 1653–1664. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Huang, H.; Tian, Y.; Yu, X.; Li, X. Stochasticity versus determinism: Microbial community assembly patterns under specific conditions in petrochemical activated sludge. J. Hazard. Mater. 2021, 407, 124372. [Google Scholar] [CrossRef]
- Jiao, S.; Lu, Y. Soil pH and temperature regulate assembly processes of abundant and rare bacterial communities in agricultural ecosystems. Environ. Microbiol. 2020, 22, 1052–1065. [Google Scholar] [CrossRef] [PubMed]
- Goehry, B. Random forests for time-dependent processes. Esaim Probab. Stat. 2020, 24, 801–826. [Google Scholar] [CrossRef]
- Liang, X.; Bushman, F.D.; FitzGerald, G.A. Rhythmicity of the intestinal microbiota is regulated by gender and the host circadian clock. Proc. Natl. Acad. Sci. USA 2015, 112, 10479–10484. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.H.; Liao, R.R.; Wu, N.; Zhu, G.S.; Yang, C.S. Heat stress mediates changes in fecal microbiome and functional pathways of laying hens. Appl. Microbiol. Biot. 2019, 103, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Hold, G.L.; Harmsen, H.J.M.; Stewart, C.S.; Flint, H.J. Growth requirements and fermentation products of Fusobacterium prausnitzii, and a proposal to reclassify it as Faecalibacterium prausnitzii gen. nov., comb. nov. Int. J. Syst. Evol. Micr. 2002, 52, 2141–2146. [Google Scholar]
- Jousset, A.; Bienhold, C.; Chatzinotas, A.; Gallien, L.; Gobet, A.; Kurm, V.; Kusel, K.; Rillig, M.C.; Rivett, D.W.; Salles, J.F.; et al. Where less may be more: How the rare biosphere pulls ecosystems strings. ISME J. 2017, 11, 853–862. [Google Scholar] [CrossRef]
- Zhang, M.M.; Gao, C.; Guo, X.T.; Guo, S.T.; Kang, Z.Q.; Xiao, D.; Yan, J.X.; Tao, F.; Zhang, W.; Dong, W.Y.; et al. Increased glutarate production by blocking the glutaryl-CoA dehydrogenation pathway and a catabolic pathway involving L-2-hydroxyglutarate. Nat. Commun. 2018, 9, 2114. [Google Scholar] [CrossRef]
- Mazzoli, R.; Pessione, E. The Neuro-endocrinological Role of Microbial Glutamate and GABA Signaling. Front. Microbiol. 2016, 7, 1934. [Google Scholar] [CrossRef] [Green Version]
- Davies, L.C.; Rice, C.M.; Palmieri, E.M.; Taylor, P.R.; Kuhns, D.B.; McVicar, D.W. Peritoneal tissue-resident macrophages are metabolically poised to engage microbes using tissue-niche fuels. Nat. Commun. 2017, 8, 2074. [Google Scholar] [CrossRef] [PubMed]
- Segers, A.; Desmet, L.; Thijs, T.; Verbeke, K.; Tack, J.; Depoortere, I. The circadian clock regulates the diurnal levels of microbial short-chain fatty acids and their rhythmic effects on colon contractility in mice. Acta Physiol. 2019, 225, 13193. [Google Scholar] [CrossRef]
- Hiippala, K.; Kainulainen, V.; Kalliomaki, M.; Arkkila, P.; Satokari, R. Mucosal Prevalence and Interactions with the Epithelium Indicate Commensalism of Sutterella spp. Front. Microbiol. 2016, 7, 1706. [Google Scholar] [CrossRef] [Green Version]
- Marco, M.L.; Heeney, D.; Binda, S.; Cifelli, C.J.; Cotter, P.D.; Foligne, B.; Ganzle, M.; Kort, R.; Pasin, G.; Pihlanto, A.; et al. Health benefits of fermented foods: Microbiota and beyond. Curr. Opin. Biotech. 2017, 44, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Patten, D.A.; Laws, A.P. Lactobacillus-produced exopolysaccharides and their potential health benefits: A review. Benef. Microbes 2015, 6, 457–471. [Google Scholar] [CrossRef]
- Shaufi, M.A.M.; Sieo, C.C.; Chong, C.W.; Gan, H.M.; Ho, Y.W. Deciphering chicken gut microbial dynamics based on high-throughput 16S rRNA metagenomics analyses. Gut Pathog. 2015, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagalingam, N.A.; Lynch, S.V. Role of the microbiota in inflammatory bowel diseases. Inflamm. Bowel Dis. 2012, 18, 968–980. [Google Scholar] [CrossRef]
- d’Herouel, A.F.; Wessner, F.; Halpern, D.; Ly-Vu, J.; Kennedy, S.P.; Serror, P.; Aurell, E.; Repoila, F. A simple and efficient method to search for selected primary transcripts: Non-coding and antisense RNAs in the human pathogen Enterococcus faecalis. Nucleic Acids Res. 2011, 39, e46. [Google Scholar] [CrossRef]
- Gilmore, M.S.; Ferretti, J.J. Microbiology: The thin line between gut commensal and pathogen. Science 2003, 299, 1999. [Google Scholar] [CrossRef] [Green Version]
- Daw, K.; Baghdayan, A.S.; Awasthi, S.; Shankar, N. Biofilm and planktonic Enterococcus faecalis elicit different responses from host phagocytes in vitro. FEMS Immunol. Med. Mic. 2012, 65, 270–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.; Zhang, Y.; Xiao, K.P.; Jiang, F.; Wang, H.C.; Tang, D.Z.; Liu, D.; Liu, B.; Liu, Y.S.; He, X.; et al. The chicken gut metagenome and the modulatory effects of plant-derived benzylisoquinoline alkaloids. Microbiome 2018, 6, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandit, R.J.; Hinsu, A.T.; Patel, N.V.; Koringa, P.G.; Jakhesara, S.J.; Thakkar, J.R.; Shah, T.M.; Limon, G.; Psifidi, A.; Guitian, J.; et al. Microbial diversity and community composition of caecal microbiota in commercial and indigenous Indian chickens determined using 16s rDNA amplicon sequencing. Microbiome 2018, 6, 115. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Ijaz, U.Z.; Kelly, C.; Linton, M.; Sloan, W.T.; Green, B.D.; Lavery, U.; Dorrell, N.; Wren, B.W.; Richmond, A.; et al. Impact of industrial production system parameters on chicken microbiomes: Mechanisms to improve performance and reduce Campylobacter. Microbiome 2020, 8, 1. [Google Scholar] [CrossRef]
- Glendinning, L.; Stewart, R.D.; Pallen, M.J.; Watson, K.A.; Watson, M. Assembly of hundreds of novel bacterial genomes from the chicken caecum (vol 21, 34, 2020). Genome Biol. 2021, 22, 34. [Google Scholar] [CrossRef] [PubMed]
- Dyar, K.A.; Lutter, D.; Artati, A.; Ceglia, N.J.; Liu, Y.; Armenta, D.; Jastroch, M.; Schneider, S.; de Mateo, S.; Cervantes, M.; et al. Atlas of Circadian Metabolism Reveals System-wide Coordination and Communication between Clocks. Cell 2018, 174, 1571. [Google Scholar] [CrossRef] [Green Version]
- Weger, B.D.; Gobet, C.; Yeung, J.; Martin, E.; Jimenez, S.; Betrisey, B.; Foata, F.; Berger, B.; Balvay, A.; Foussier, A.; et al. The Mouse Microbiome Is Required for Sex-Specific Diurnal Rhythms of Gene Expression and Metabolism. Cell Metab. 2019, 29, 362. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.L.A.; Vieira-Silva, S.; Liston, A.; Raes, J. How informative is the mouse for human gut microbiota research? Dis. Model Mech. 2015, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.J.; Wang, Y.P.; Liu, S.Y.; Huang, J.J.; Zhai, Z.X.; He, C.; Ding, J.M.; Wang, J.; Wang, H.J.; Fan, W.B.; et al. The Dynamic Distribution of Porcine Microbiota across Different Ages and Gastrointestinal Tract Segments. PLoS ONE 2015, 10, e0117441. [Google Scholar] [CrossRef] [Green Version]
- Nobs, S.P.; Tuganbaev, T.; Elinav, E. Microbiome diurnal rhythmicity and its impact on host physiology and disease risk. Embo Rep. 2019, 20, e47129. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Huang, M.Q.; You, X.; Zhao, J.Y.; Chen, L.L.; Wang, L.; Luo, Y.J.; Chen, Y. Gut microbiota mediates the anti-obesity effect of calorie restriction in mice. Sci. Rep. 2018, 8, 13037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaix, A.; Lin, T.; Le, H.D.; Chang, M.W.; Panda, S. Time-Restricted Feeding Prevents Obesity and Metabolic Syndrome in Mice Lacking a Circadian Clock. Cell Metab. 2019, 29, 303. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Sun, L.; Zhu, R.; Zhang, S.; Liu, S.; Wang, Y.; Wu, Y.; Liao, X.; Mi, J. Absence of Circadian Rhythm in Fecal Microbiota of Laying Hens under Common Light. Animals 2021, 11, 2065. https://doi.org/10.3390/ani11072065
Zhang Y, Sun L, Zhu R, Zhang S, Liu S, Wang Y, Wu Y, Liao X, Mi J. Absence of Circadian Rhythm in Fecal Microbiota of Laying Hens under Common Light. Animals. 2021; 11(7):2065. https://doi.org/10.3390/ani11072065
Chicago/Turabian StyleZhang, Yu, Lan Sun, Run Zhu, Shiyu Zhang, Shuo Liu, Yan Wang, Yinbao Wu, Xindi Liao, and Jiandui Mi. 2021. "Absence of Circadian Rhythm in Fecal Microbiota of Laying Hens under Common Light" Animals 11, no. 7: 2065. https://doi.org/10.3390/ani11072065