
Genome-Wide Association Study on Reproduction-Related Body-Shape Traits of Chinese Holstein Cows

 ,
, 
 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals and Phenotypic Data
2.3. Adjustment of Phenotypes for Analysis
2.4. Genotypic Data
2.5. Linkage Disequilibrium Decay Analysis and Principal Component Analysis
2.6. Genome-Wide Association Studies
2.7. Annotation of Candidate Gene and Bioinformatic Analysis
3. Results
3.1. The Relationship between Phenotype Data and Reproductive Performance
3.2. Phenotypic Data and Genetic Parameters Estimation
3.3. SNP Data Statistics
3.4. Population Structure Analysis
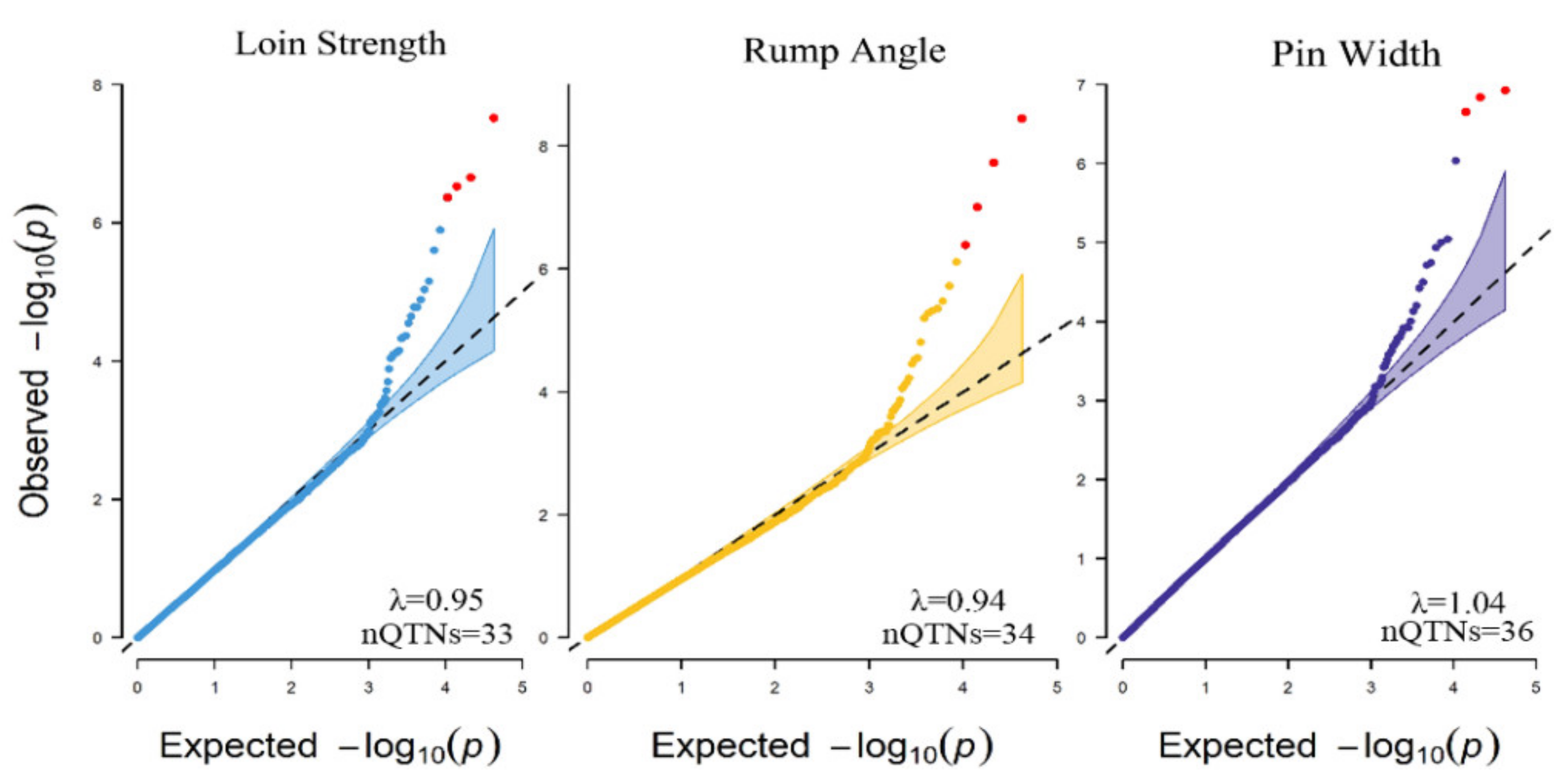
3.5. Genome-Wide Association Study
3.6. Enrichment Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berry, D.P.; Wall, E.; Pryce, J. Genetics and genomics of reproductive performance in dairy and beef cattle. Animal 2014, 8, 105–121. [Google Scholar] [CrossRef] [Green Version]
- Abdollahi-Arpanahi, R.; Carvalho, M.R.; Ribeiro, E.S.; Peñagaricano, F. Association of lipid-related genes implicated in conceptus elongation with female fertility traits in dairy cattle. J. Dairy Sci. 2019, 102, 10020–10029. [Google Scholar] [CrossRef]
- Rezende, F.M.; Haile-Mariam, M.; Pryce, J.E.; Peñagaricano, F. Across-country genomic prediction of bull fertility in Jersey dairy cattle. J. Dairy Sci. 2020, 103, 11618–11627. [Google Scholar] [CrossRef]
- Berry, D.P.; Evans, R.D. Genetics of reproductive performance in seasonal calving beef cows and its association with performance traits. J. Anim. Sci. 2014, 92, 1412–1422. [Google Scholar] [CrossRef] [Green Version]
- Miglior, F.; Fleming, A.; Malchiodi, F.; Brito, L.F.; Martin, P.; Baes, C.F. A 100-Year Review: Identification and genetic selection of economically important traits in dairy cattle. J. Dairy Sci. 2017, 100, 10251–10271. [Google Scholar] [CrossRef] [PubMed]
- Code of Practice of Type Classification in Chinese Holstein. Available online: https://www.holstein.org.cn/upload/fckupload/file/1527151112535606097363.pdf (accessed on 27 March 2021). (In Chinese).
- Pena, J. Genetic correlated traits for female fertility evaluations in Spanish Holsteins. Interbull Bull. 2006, 34, 31. [Google Scholar]
- Almeida, T.P.; Kern, E.L.; Daltro, D.D.S.; Neto, J.B.; McManus, C.; Neto, A.T.; Cobuci, J.A. Genetic associations between reproductive and linear-type traits of Holstein cows in Brazil. Rev. Bras. Zootec. 2017, 46, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Dadati, E.; Kennedy, B.; Burnside, E. Relationships Between Conformation and Reproduction in Holstein Cows: Type and Calving Performance. J. Dairy Sci. 1985, 68, 2639–2645. [Google Scholar] [CrossRef]
- Ali, T.; Burnside, E.; Schaeffer, L. Relationship Between External Body Measurements and Calving Difficulties in Canadian Holstein-Friesian Cattle. J. Dairy Sci. 1984, 67, 3034–3044. [Google Scholar] [CrossRef]
- Huang, H.; Cao, J.; Hanif, Q.; Wang, Y.; Yu, Y.; Zhang, S.; Zhang, Y. Genome-wide association study identifies energy metabolism genes for resistance to ketosis in Chinese Holstein cattle. Anim. Genet. 2019, 50, 376–380. [Google Scholar] [CrossRef]
- Zhang, X.; Chu, Q.; Guo, G.; Dong, G.; Li, X.; Zhang, Q.; Zhang, S.; Zhang, Z.; Wang, Y. Genome-wide association studies identified multiple genetic loci for body size at four growth stages in Chinese Holstein cattle. PLoS ONE 2017, 12, e0175971. [Google Scholar] [CrossRef]
- Otto, P.I.; Guimarães, S.E.; Calus, M.P.; Vandenplas, J.; Machado, M.A.; Panetto, J.C.C.; Da Silva, M.V.G. Single-step genome-wide association studies (GWAS) and post-GWAS analyses to identify genomic regions and candidate genes for milk yield in Brazilian Girolando cattle. J. Dairy Sci. 2020, 103, 10347–10360. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, J.; Chen, C.; Zhang, J.; Wen, W.; Tian, J.; Zhang, Z.; Gu, Y. GWAS-Based Identification of New Loci for Milk Yield, Fat, and Protein in Holstein Cattle. Animals 2020, 10, 2048. [Google Scholar] [CrossRef] [PubMed]
- De Melo, T.P.; de Camargo, G.M.F.; Albuquerque, L.; Carvalheiro, R. Genome-wide association study provides strong evidence of genes affecting the reproductive performance of Nellore beef cows. PLoS ONE 2017, 12, e0178551. [Google Scholar] [CrossRef]
- Engle, B.N.; Herring, A.D.; Sawyer, J.; Riley, D.G.; O Sanders, J.; A Gill, C. Genome-wide association study for stayability measures in Nellore–Angus crossbred cows1. J. Anim. Sci. 2018, 96, 1205–1214. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, L.; Chen, C.J.; Zhang, M.; Lu, X.; Zhang, Z.; Huang, X.; Shi, Y. Genome-wide association study of milk and reproductive traits in dual-purpose Xinjiang Brown cattle. BMC Genom. 2019, 20, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atashi, H.; Salavati, M.; De Koster, J.; Crowe, M.A.; Opsomer, G.; Hostens, M.; the GplusE consortium. A Genome-Wide Association Study for Calving Interval in Holstein Dairy Cows Using Weighted Single-Step Genomic BLUP Approach. Animals 2020, 10, 500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadi, A.; Alijani, S.; Rafat, S.A.; Abdollahi-Arpanahi, R. Genome-Wide Association Study and Pathway Analysis for Female Fertility Traits in Iranian Holstein Cattle. Ann. Anim. Sci. 2020, 20, 825–851. [Google Scholar] [CrossRef]
- Madsen, P.; Jensen, J. A User’S Guide to DMU. A Package for Analysing Multivariate Mixed Models. Available online: https://dmu.ghpc.au.dk/dmu/DMU/Doc/Current/dmuv6_guide.5.2.pdf (accessed on 20 March 2021).
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Accounting for sex in the genome. Nat. Med. 2017, 23, 1243. [CrossRef]
- Sayres, M.A.W. Genetic Diversity on the Sex Chromosomes. Genome Biol. Evol. 2018, 10, 1064–1078. [Google Scholar] [CrossRef]
- Wise, A.L.; Gyi, L.; Manolio, T.A. eXclusion: Toward Integrating the X Chromosome in Genome-wide Association Analyses. Am. J. Hum. Genet. 2013, 92, 643–647. [Google Scholar] [CrossRef] [Green Version]
- Happle, R. X-chromosome inactivation: Role in skin disease expression. Acta Paediatr. 2006, 95, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Chen, Q.; Sun, X. Association of skewed X chromosome inactivation and idiopathic recurrent spontaneous abortion: A systematic review and meta-analysis. Reprod. Biomed. Online 2015, 31, 140–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: AnRPackage for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Kahle, D.; Wickham, H. ggmap: Spatial Visualization with ggplot2. R J. 2013, 5, 144–161. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Tian, F.; Pan, Y.; Buckler, E.S.; Zhang, Z. A SUPER Powerful Method for Genome Wide Association Study. PLoS ONE 2014, 9, e107684. [Google Scholar] [CrossRef] [PubMed]
- Bland, J.M.; Altman, D.G. Statistics notes: Multiple significance tests: The Bonferroni method. BMJ 1995, 310, 170. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Hale, M.L.; Thapa, I.; Ghersi, D. FunSet: An open-source software and web server for performing and displaying Gene Ontology enrichment analysis. BMC Bioinform. 2019, 20, 1–8. [Google Scholar] [CrossRef]
- Roche, J.; Friggens, N.; Kay, J.K.; Fisher, M.W.; Stafford, K.J.; Berry, D. Invited review: Body condition score and its association with dairy cow productivity, health, and welfare. J. Dairy Sci. 2009, 92, 5769–5801. [Google Scholar] [CrossRef] [Green Version]
- Corredor, F.-A.; Sanglard, L.P.; Ross, J.W.; Keating, A.F.; Leach, R.J.; Serão, N.V.L. Phenotypic and genomic relationships between vulva score categories and reproductive performance in first-parity sows. J. Anim. Sci. Biotechnol. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Ogawa, S.; Ohnishi, C.; Ishii, K.; Uemoto, Y.; Satoh, M. Genetic relationship between litter size traits at birth and body measurement and production traits in purebred Duroc pigs. Anim. Sci. J. 2020, 91, e13497. [Google Scholar] [CrossRef] [PubMed]
- Thekkoot, D.M.; Kemp, R.A.; Rothschild, M.F.; Plastow, G.; Dekkers, J.C.M. Estimation of genetic parameters for traits associated with reproduction, lactation, and efficiency in sows1. J. Anim. Sci. 2016, 94, 4516–4529. [Google Scholar] [CrossRef] [PubMed]
- Jílek, F.; Pytloun, P.; Kubešová, M.; Štípková, M.; Bouska, J.; Volek, J.; Frelich, J.; Rajmon, R. Relationships among body condition score, milk yield and reproduction in Czech Fleckvieh cows. Czech J. Anim. Sci. 2008, 53, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; He, D.; Norton, T. Automatic estimation of dairy cattle body condition score from depth image using ensemble model. Biosyst. Eng. 2020, 194, 16–27. [Google Scholar] [CrossRef]
- Oikonomou, G.; Arsenos, G.; Valergakis, G.E.; Tsiaras, A.; Zygoyiannis, D.; Banos, G. Genetic Relationship of Body Energy and Blood Metabolites with Reproduction in Holstein Cows. J. Dairy Sci. 2008, 91, 4323–4332. [Google Scholar] [CrossRef] [Green Version]
- Kul, E.; Erdem, H. Relationships between milk insulin-like growth factor-I (IGF-I) concentration and body condition score with reproductive performance and milk yield in Jersey cows. Large Anim. Rev. 2018, 24, 65–70. [Google Scholar]
- Wolcott, M.L.; Johnston, D.J.; Barwick, S.A. Genetic relationships of female reproduction with growth, body composition, maternal weaning weight and tropical adaptation in two tropical beef genotypes. Anim. Prod. Sci. 2014, 54, 60–73. [Google Scholar] [CrossRef]
- Silva, R.; Espigolan, R.; Berton, M.; Stafuzza, N.B.; Santos, F.; Negreiros, M.; Schuchmann, R.; Rodriguez, J.; Lôbo, R.; Banchero, G.; et al. Genetic parameters and genomic regions associated with calving ease in primiparous Nellore heifers. Livest. Sci. 2020, 240, 104183. [Google Scholar] [CrossRef]
- Yamazaki, T.; Yamaguchi, S.; Takeda, H.; Osawa, T.; Hagiya, K. Genetic parameters for conception rate and milk production traits within and across Holstein herds with different housing types and feeding systems during the first 3 lactations. J. Dairy Sci. 2020, 103, 10361–10373. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Satoh, M. Random Regression Analysis of Calving Interval of Japanese Black Cows. Animals 2021, 11, 202. [Google Scholar] [CrossRef]
- Sanchez, M.-P.; Govignon-Gion, A.; Croiseau, P.; Fritz, S.; Hozé, C.; Miranda, G.; Martin, P.; Barbat-Leterrier, A.; Letaïef, R.; Rocha, D.; et al. Within-breed and multi-breed GWAS on imputed whole-genome sequence variants reveal candidate mutations affecting milk protein composition in dairy cattle. Genet. Sel. Evol. 2017, 49, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mota, L.F.M.; Lopes, F.B.; Júnior, G.A.F.; Rosa, G.J.M.; Magalhães, A.F.B.; Carvalheiro, R.; Albuquerque, L.G. Genome-wide scan highlights the role of candidate genes on phenotypic plasticity for age at first calving in Nellore heifers. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Mokhber, M.; Shahrbabak, M.M.; Sadeghi, M.; Shahrbabak, H.M.; Stella, A.; Nicolzzi, E.; Williams, J.L. Study of whole genome linkage disequilibrium patterns of Iranian water buffalo breeds using the Axiom Buffalo Genotyping 90K Array. PLoS ONE 2019, 14, e0217687. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Sul, J.H.; Service, S.K.; A Zaitlen, N.; Kong, S.-Y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voorman, A.; Lumley, T.; McKnight, B.; Rice, K. Behavior of QQ-Plots and Genomic Control in Studies of Gene-Environment Interaction. PLoS ONE 2011, 6, e19416. [Google Scholar] [CrossRef] [Green Version]
- Price, A.L.; Zaitlen, N.A.; Reich, D.; Patterson, N. New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet. 2010, 11, 459–463. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Buckler, E.S.; Casstevens, T.M.; Bradbury, P.J. Software engineering the mixed model for genome-wide association studies on large samples. Briefings Bioinform. 2009, 10, 664–675. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Rosenberg, N.A.; Donnelly, P. Association Mapping in Structured Populations. Am. J. Hum. Genet. 2000, 67, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Todhunter, R.J.; Buckler, E.; Van Vleck, L.D. Technical note: Use of marker-based relationships with multiple-trait derivative-free restricted maximal likelihood. J. Anim. Sci. 2007, 85, 881–885. [Google Scholar] [CrossRef]
- Pirinen, M.; Donnelly, P.; A Spencer, C.C. Including known covariates can reduce power to detect genetic effects in case-control studies. Nat. Genet. 2012, 44, 848–851. [Google Scholar] [CrossRef]
- Yang, Y.; Chai, Y.; Zhang, X.; Lu, S.; Zhao, Z.; Wei, D.; Chen, L.; Hu, Y.-G. Multi-Locus GWAS of Quality Traits in Bread Wheat: Mining More Candidate Genes and Possible Regulatory Network. Front. Plant Sci. 2020, 11, 1091. [Google Scholar] [CrossRef]
- Zhang, H.; Yu, J.-Q.; Xin-Yang, Z.; Kramer, L.M.; Zhang, X.-Y.; Na, W.; Reecy, J.M.; Li-Li, Y. Identification of genome-wide SNP-SNP interactions associated with important traits in chicken. BMC Genom. 2017, 18, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Bellucco, F.T.; De Oliveira-Júnior, H.R.; Guilherme, R.S.; Bragagnolo, S.; Perez, A.B.A.; Meloni, V.A.; Melaragno, M.I. Deletion of Chromosome 13 due to Different Rearrangements and Impact on Phenotype. Mol. Syndr. 2019, 10, 139–146. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Kim, D.-H. dRAGging amino Acid-mTORC1 signaling by SH3BP4. Mol. Cells 2012, 35, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, E.; Haas, A.K.; Spooner, R.A.; Yoshimura, S.-I.; Lord, J.M.; Barr, F.A. Specific Rab GTPase-activating proteins define the Shiga toxin and epidermal growth factor uptake pathways. J. Cell Biol. 2007, 177, 1133–1143. [Google Scholar] [CrossRef]
- Rodríguez-Barrientos, C.-A.; Trevino, V.; Zavala, J.; Montalvo-Parra, M.-D.; Guerrero-Ramírez, G.-I.; Aguirre-Gamboa, R.; Valdez-Garcia, J. Arresting proliferation improves the cell identity of corneal endothelial cells in the New Zealand rabbit. Molecular Vision 2019, 25, 745–755. [Google Scholar]
- Guarini, A.; Lourenco, D.; Brito, L.; Sargolzaei, M.; Baes, C.; Miglior, F.; Misztal, I.; Schenkel, F. Genetics and genomics of reproductive disorders in Canadian Holstein cattle. J. Dairy Sci. 2019, 102, 1341–1353. [Google Scholar] [CrossRef] [Green Version]
- Hastie, P.; Ulbrich, M.H.; Wang, H.-L.; Arant, R.J.; Lau, A.G.; Zhang, Z.; Isacoff, E.Y.; Chen, L. AMPA receptor/TARP stoichiometry visualized by single-molecule subunit counting. Proc. Natl. Acad. Sci. USA 2013, 110, 5163–5168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummins, S.; Lonergan, P.; Evans, A.; Butler, S. Genetic merit for fertility traits in Holstein cows: II. Ovarian follicular and corpus luteum dynamics, reproductive hormones, and estrus behavior. J. Dairy Sci. 2012, 95, 3698–3710. [Google Scholar] [CrossRef] [PubMed]
- Surlis, C.; Cormican, P.; Waters, S.M.; Lonergan, P.; Keogh, K.; Doyle, D.N.; Kenny, D.A. Effects of dietary n-3-PUFA supplementation, post-insemination plane of nutrition and pregnancy status on the endometrial transcriptome of beef heifers. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Rezende, F.M.; Rodriguez, E.; Leal-Gutiérrez, J.D.; Elzo, M.A.; Johnson, D.D.; Carr, C.; Mateescu, R.G. Genomic Approaches Reveal Pleiotropic Effects in Crossbred Beef Cattle. Front. Genet. 2021, 12. [Google Scholar] [CrossRef]
- Hao, D.; Wang, X.; Wang, X.; Thomsen, B.; Kadarmideen, H.N.; Lan, X.; Huang, Y.; Chen, H. Transcriptomic changes in bovine skeletal muscle cells after resveratrol treatment. Gene 2020, 754, 144849. [Google Scholar] [CrossRef] [PubMed]
- Ge, F.; Jia, C.; Chu, M.; Liang, C.; Yan, P. Copy Number Variation of the CADM2 Gene and Its Association with Growth Traits in Yak. Animals 2019, 9, 1008. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Liu, X.; Bai, X.; Xiao, C.; Dong, Y. Identification and Characterization of circRNA in Longissimus Dorsi of Different Breeds of Cattle. Front. Genet. 2020, 11, 11. [Google Scholar] [CrossRef]
- Silva-Vignato, B.; Coutinho, L.L.; Cesar, A.S.M.; Poleti, M.D.; Regitano, L.C.A.; Balieiro, J.C.C. Comparative muscle transcriptome associated with carcass traits of Nellore cattle. BMC Genom. 2017, 18, 506. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Van Der Werf, J.; Lee, S.H.; Lim, D.J.; Park, E.W.; Gondro, C.; Yoon, D.; Oh, S.J.; Kim, O.H.; Gibson, J.; et al. Genome wide QTL mapping to identify candidate genes for carcass traits in Hanwoo (Korean Cattle). Genes Genom. 2012, 34, 43–49. [Google Scholar] [CrossRef]
- Liao, W.-J.; Tsao, K.-C.; Yang, R.-B. Electrostatics and N-glycan-mediated membrane tethering of SCUBE1 is critical for promoting bone morphogenetic protein signalling. Biochem. J. 2016, 473, 661–672. [Google Scholar] [CrossRef]
- Kirby, D.J.; Young, M.F. Isolation, production, and analysis of small leucine-rich proteoglycans in bone. In Methods in Cell Biology; Elsevier BV: Amsterdam, The Netherlands, 2018; Volume 143, pp. 281–296. [Google Scholar]
- Du, M.; Zhao, J.X.; Yan, X.; Huang, Y.; Nicodemus, L.V.; Yue, W.; McCormick, R.J.; Zhu, M.J. Fetal muscle development, mesenchymal multipotent cell differentiation, and associated signaling pathways1,2. J. Anim. Sci. 2011, 89, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wan, S.; Li, Q.; Dong, X.; Diao, J.; Liao, Q.; Wang, G.-Y.; Gao, Z.-X. Genome-Wide Integrated Analysis Revealed Functions of lncRNA–miRNA–mRNA Interaction in Growth of Intermuscular Bones in Megalobrama amblycephala. Front. Cell Dev. Biol. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Cinque, L.; Forrester, A.; Bartolomeo, R.; Svelto, M.; Venditti, R.; Montefusco, S.; Polishchuk, E.; Nusco, E.; Rossi, A.; Medina, D.L.; et al. FGF signalling regulates bone growth through autophagy. Nat. Cell Biol. 2015, 528, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, Y.; Liu, Q.; Gao, T.; Feng, J.Q.; Dechow, P.; D’Souza, R.; Qin, C.; Liu, X. Biomimetic Engineering of Nanofibrous Gelatin Scaffolds with Noncollagenous Proteins for Enhanced Bone Regeneration. Tissue Eng. Part A 2013, 19, 1754–1763. [Google Scholar] [CrossRef] [Green Version]
- Wade-Gueye, N.M.; Delissen, O.; Gourmelon, P.; Aigueperse, J.; Dublineau, I.; Souidi, M. Chronic exposure to natural uranium via drinking water affects bone in growing rats. Biochim. Biophys. Acta BBA Gen. Subj. 2012, 1820, 1121–1127. [Google Scholar] [CrossRef]
- Tachibana, R.; Tatehara, S.; Kumasaka, S.; Tokuyama, R.; Satomura, K. Effect of Melatonin on Human Dental Papilla Cells. Int. J. Mol. Sci. 2014, 15, 17304–17317. [Google Scholar] [CrossRef] [Green Version]
- Zoidis, E.; Ghirlanda-Keller, C.; Schmid, C. Triiodothyronine stimulates glucose transport in bone cells. Endocrine 2012, 41, 501–511. [Google Scholar] [CrossRef]
- Timmons, J.A.; Atherton, P.J.; Larsson, O.; Sood, S.; O Blokhin, I.; Brogan, R.J.; Volmar, C.-H.; Josse, A.; Slentz, C.; Wahlestedt, C.; et al. A coding and non-coding transcriptomic perspective on the genomics of human metabolic disease. Nucleic Acids Res. 2018, 46, 7772–7792. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.K.; Key, C.-C.C.; Civelek, M.; Wabitsch, M.; Comeau, M.E.; Langefeld, C.D.; Parks, J.S.; Das, S.K. Genetic Regulation of Enoyl-CoA Hydratase Domain-Containing 3 in Adipose Tissue Determines Insulin Sensitivity in African Americans and Europeans. Diabetes 2019, 68, 1508–1522. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Arithmetic Mean | Minimum | Maximum | SD | CV (%) | Kurtosis | Skewness | h2 (SE) |
|---|---|---|---|---|---|---|---|---|
| LS | −0.08 | −5.78 | 4.91 | 1.53 | −18.19 | 3.84 | −0.27 | 0.38 (0.05) |
| RA | −0.04 | −4.95 | 3.58 | 1.37 | −38.49 | 3.51 | −0.35 | 0.22 (0.02) |
| PW | 0.05 | −5.45 | 2.56 | 1.09 | 23.67 | 3.56 | −0.44 | 0.20 (0.02) |
| Trait | SNP | CHR | Position | Nearest Gene | Distance | MAF | Effect | EVG | p-Value |
|---|---|---|---|---|---|---|---|---|---|
| LS | rs42946768 | 20 | 51505605 | CDH12 | Within (intronic) | 0.373476 | −0.31 | 1.30% | 3.08 × 10−8 |
| rs109073659 | 12 | 40319808 | PCDH9 | Within (intronic) | 0.489837 | 0.29 | 1.22% | 2.23 × 10−7 | |
| rs43162548 | 4 | 50163217 | TARP | 5 Kb | 0.172764 | 0.33 | 0.97% | 2.99 × 10−7 | |
| rs133475777 | 6 | 55719468 | DTHD1 | Within (intronic) | 0.272358 | 0.29 | 0.91% | 4.29 × 10−7 | |
| RA | rs43486059 | 6 | 102570596 | LOC781835 | Within (intronic) | 0.489329 | −0.29 | 1.38% | 3.61 × 10−9 |
| rs137244035 | 7 | 45115020 | FSTL4 | Within (intronic) | 0.455285 | −0.28 | 1.32% | 1.88 × 10−8 | |
| rs43352090 | 3 | 82508654 | ATG4C | 200 kb | 0.365854 | −0.29 | 1.05% | 9.91 × 10−8 | |
| rs43366267 | 3 | 114684449 | SH3BP4 | 50 Kb | 0.318089 | 0.27 | 0.93% | 4.10 × 10−7 | |
| PW | rs109578471 | 13 | 12679178 | USP6NL | Within (intronic) | 0.315041 | −0.18 | 0.96% | 1.18 × 10−7 |
| rs42051017 | 29 | 3370134 | LOC101907665 | 200 Kb | 0.21748 | −0.20 | 0.87% | 1.45 × 10−7 | |
| rs43430205 | 22 | 26807183 | CNTN3 | 200 Kb | 0.272358 | −0.18 | 0.87% | 2.24 × 10−7 |
| Traits | Pathway | Description | Gene Name | p-Value |
|---|---|---|---|---|
| LS | bta04144 | Endocytosis | ARAP2 | 0.0279 |
| RA | bta00511 | Other glycan degradation | LOC781835, LOC523503 | 0.0003 |
| bta04512 | ECM-receptor interaction | DSPP, DMP1 | 0.0041 | |
| bta04136 | Autophagy—other | ATG4C | 0.0361 | |
| PW | bta03015 | mRNA surveillance pathway | UPF2 | 0.0113 |
| bta03013 | RNA transport | UPF2 | 0.0210 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, X.; Abdalla, I.M.; Nazar, M.; Fan, Y.; Zhang, Z.; Wu, X.; Xu, T.; Yang, Z. Genome-Wide Association Study on Reproduction-Related Body-Shape Traits of Chinese Holstein Cows. Animals 2021, 11, 1927. https://doi.org/10.3390/ani11071927
Lu X, Abdalla IM, Nazar M, Fan Y, Zhang Z, Wu X, Xu T, Yang Z. Genome-Wide Association Study on Reproduction-Related Body-Shape Traits of Chinese Holstein Cows. Animals. 2021; 11(7):1927. https://doi.org/10.3390/ani11071927
Chicago/Turabian StyleLu, Xubin, Ismail Mohamed Abdalla, Mudasir Nazar, Yongliang Fan, Zhipeng Zhang, Xinyue Wu, Tianle Xu, and Zhangping Yang. 2021. "Genome-Wide Association Study on Reproduction-Related Body-Shape Traits of Chinese Holstein Cows" Animals 11, no. 7: 1927. https://doi.org/10.3390/ani11071927